
Abstract
Ribosome biogenesis and protein synthesis are fundamental rate-limiting steps for cell growth and proliferation. The ribosomal proteins (RPs), comprising the structural parts of the ribosome, are essential for ribosome assembly and function. In addition to their canonical ribosomal functions, multiple RPs have extra-ribosomal functions including activation of p53-dependent or p53-independent pathways in response to stress, resulting in cell cycle arrest and apoptosis. Defects in ribosome biogenesis, translation, and the functions of individual RPs, including mutations in RPs have been linked to a diverse range of human congenital disorders termed ribosomopathies. Ribosomopathies are characterized by tissue-specific phenotypic abnormalities and higher cancer risk later in life. Recent discoveries of somatic mutations in RPs in multiple tumor types reinforce the connections between ribosomal defects and cancer. In this article, we review the most recent advances in understanding the molecular consequences of RP mutations and ribosomal defects in ribosomopathies and cancer. We particularly discuss the molecular basis of the transition from hypo- to hyper-proliferation in ribosomopathies with elevated cancer risk, a paradox termed “Dameshek’s riddle.” Furthermore, we review the current treatments for ribosomopathies and prospective therapies targeting ribosomal defects. We also highlight recent advances in ribosome stress-based cancer therapeutics. Importantly, insights into the mechanisms of resistance to therapies targeting ribosome biogenesis bring new perspectives into the molecular basis of cancer susceptibility in ribosomopathies and new clinical implications for cancer therapy.
Similar content being viewed by others


Introduction
Cell growth and proliferation are two distinct but coupled biological processes that are directly dependent on the tight coordination of protein synthesis and metabolic activity. These biological capabilities are essential characteristics that enable cancer cells to sustain uncontrolled proliferation.1 The biosynthesis of ribosomes, the molecular machines that translate messenger RNA (mRNA) into proteins,2 is a fundamental biological process that is intimately linked to cell growth and proliferation and is considered to be one of the most energy-consuming processes in proliferating mammalian cells.3,4,5,6
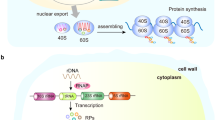
Ribosome biogenesis is a highly dynamic and coordinated process, in which ribosomal RNA (rRNA) is synthesized, modified and assembled with RPs to form mature ribosomes. Ribosome biogenesis takes place within specialized subnuclear compartments known as the nucleoli7,8 and depends on the coordinated regulation of the three DNA-dependent RNA polymerases (Pol I, Pol II, and Pol III), as well as the involvement of a plethora of transcription factors, small nucleolar RNAs (snoRNAs) and non-RPs that cooperatively promote the transcription, modification and processing of rRNAs, synthesis of RPs and ribosome assembly2,9,10,11,12 (Fig. 1a). This process is exquisitely regulated by multiple signaling pathways in response to growth factors, energy and nutrients that together modulate protein synthesis and thereby cell growth rate and proliferation.10,13,14,15,16

A schematic representation of ribosome biogenesis in mammalian cells. a Ribosome biogenesis is a tightly coordinated process involving all three RNA polymerases (Pol I, Pol II, and Pol III). RNA Pol I transcribes the ribosomal RNA (rRNA) genes (rDNA) to produce the 47S precursor rRNA (47S pre-rRNA) transcript in the nucleolus. Pol I transcription initiation involves binding of the upstream binding factor (UBF) to the core promoter region (core) and upstream control element (UCE) of the rDNA promoters and facilitating the recruitment and binding of the selectivity factor 1 (SL-1) complex. SL-1 is composed of the TATA-box-binding protein (TBP) and five Pol I-specific TATA-box-associated factors (TAFs). This complex in turn recruits the Pol I-specific initiation factor RRN3, which associates with DNA topoisomerase IIα (TOPIIα) and Pol I to complete assembly of a transcriptionally-competent Pol I complex. Following transcription, the 47S pre-rRNA is subsequently cleaved and processed into the mature 18S, 5.8S, and 28S rRNA species. These molecules are then assembled along with ribosomal proteins and the 5S rRNA produced by Pol II and III, respectively, to form the major catalytic and architectural components of the small (40S) and the large (60S) ribosomal subunits. Once assembled, ribosomal complexes are exported from the nucleolus to the cytoplasm, where they form the mature (80S) ribosome required to initiate mRNA translation and thus protein synthesis. b A diverse range of anticancer drugs target ribosome biogenesis by inhibiting Pol I transcription and/or pre-rRNA processing
Defects in ribosome biogenesis and function account for the pathogenesis of a heterogeneous group of diseases called ribosomopathies. Ribosomopathies are generally defined as diseases caused by mutations in RPs or factors associated with Pol I transcription and rRNA processing, resulting in the disruption of ribosome production or assembly.17,18,19,20,21 There is a growing list of diseases that have been classified as ribosomopathies; however, the extent to which defects in ribosome synthesis contribute to clinical phenotypes remains to be defined. In Treacher Collins Syndrome, Diamond–Blackfan anemia and Shwachman-Diamond syndrome, the defects in ribosome synthesis have been causally linked to disease pathotypes. However, in other putative ribosomopathies such as X-linked-dyskeratosis congenita and cartilage–hair hypoplasia–anauxetic dysplasia, Blooms and Werner syndrome and cohesinopathies, it is likely that stress responses associated with ribosomal defects and/or altered mRNA translation contribute to a component of their disease phenotype and impact on disease severity.20 Ribosomopathies are characterized by tissue-specific phenotypic abnormalities. For example, the hematopoietic system is frequently affected, which is thought to be due to tissue-specific sensitivity to p53 pathway activation in response to ribosome stress.22 Ribosomopathy patients also have a higher risk of developing cancer.23 The paradoxical transition from early symptoms due to hypo-proliferative phenotypes to an increased cancer risk later in life, was first reported by Dameshek in 1967 and referred to as Dameshek’s riddle.24 The mechanisms by which genetically compromised ribosome biogenesis leads to hyper-proliferative cancer phenotypes in patients with ribosomopathies remain a mystery.
The recent discoveries of somatic mutations in RP genes in hematological cancers (such as T-cell acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL) and multiple myeloma) and solid tumors (such as breast cancer and melanoma) emphasize that defects in ribosome biogenesis could potentially promote oncogenic transformation.23 In fact, deletions in RP genes are common events across human cancers, particularly in concert with TP53 mutations. This is in contrast to mutation in RPs (without concurrent TP53 mutation) that are primarily associated with ribosomopathies.25 Here, we review the current knowledge and new perspectives with respect to diseases linked to mutations or deletions in RP genes and the molecular mechanisms driving cancer susceptibility. We highlight how translational rewiring associated with chronic ribosome stress provides mechanistic insights into cancer development and new clinical implications for cancer therapy.
In contrast to RP mutations-associated ribosomal defects in cancers, oncogene-driven cancers are associated with hyperactivation of ribosome biogenesis that promote uncontrolled growth and proliferation and altered metabolism.26,27,28,29,30,31 Indeed, hyperactivation of Pol I transcription of the 47S rRNA precursor and the increase in production of ribosomes are necessary to support the increased rate of protein synthesis required to sustain unrestricted cell growth.26,32,33 Intriguingly, many classical chemotherapeutic agents (e.g., oxaliplatin, cisplatin, actinomycin D and 5-fluorouracil (5-FU)) have been discovered to act through distinct mechanisms of action that include inhibition of rRNA synthesis, rRNA processing or ribosome biogenesis34,35,36,37,38 (Fig. 1b). The development of a number of less genotoxic drugs that selectively target Pol I transcription and ribosome biogenesis has established a new paradigm for cancer therapy.36,39,40,41,42 These compounds are active against subsets of tumors but their therapeutic response can be more potent in cancers with deregulated ribosome biogenesis.43,44,45 In this review, we discuss the rationale for targeting ribosome biogenesis as a treatment strategy to combat cancer and the current understanding of the therapeutic potential of the first-in-class inhibitor of Pol I transcription CX-5461. Importantly, recent studies of the key molecular mechanisms of acquired resistance to selective inhibition of ribosome biogenesis provide a new conceptual framework to expand on the understanding that specific rewiring of translation in response to chronic ribosome stress promotes cancer progression. We highlight the potential of this research for the development of novel treatments for human diseases linked to mutations in RPs and deregulated ribosome biogenesis.
Ribosome biogenesis: a key regulator of cell growth and proliferation
All eukaryotic ribosomes sediment at 80S (S: the Svedberg unit for sedimentation coefficients) and are divided into two distinct subunits of unequal size. The small (40S) subunit functions as a “decoding site,” interacting with the anticodon-containing ends of complementary tRNAs so as to translate the codon information contained in mRNA into its corresponding sequence of amino acids.46,47,48 The large (60S) subunit contains peptidyl transferase activity and is responsible for linking the amino acids into a polypeptide chain.8,46,47,48 Each ribosome subunit forms a ribonucleoprotein particle such that the small 40S subunit of the eukaryotic ribosome contains one 18S rRNA chain and 33 RPs, while the large 60S subunit consists of three rRNA molecules (28S, 5.8S, and 5S) and 48 RPs.49
The nucleolus is the largest subnuclear structure, forms around active clusters of the 47S rRNA genes to establish the site of Pol I transcription and is therefore known as the site of ribosome biogenesis. The nucleolar morphological structure is compartmentalized into a fibrillar center (FC), a dense fibrillar component (DFC), and a granular component (GC).50 In mammals, the majority of the 47S precursor rRNA is thought to be synthesized at the boundary between the FC and the DFC although a smaller proportion can be transcribed in the FC.51 The precursor-rRNA is processed and post-transcriptionally modified in the DFC, where small nucleolar RNAs (snoRNAs) of two different classes of non-coding RNAs catalyze covalent modifications (2‘-O-ribose methylation and pseudouridylation) of the rRNA molecules to form the mature 18S, 5.8S, and 28S rRNAs.8,9,11,51,52 These mature rRNAs, in conjunction with the 5S rRNA synthesized by RNA Pol III in the nucleoplasm are then assembled in the GC of the nucleolus with RPs encoded by Pol II to form the major catalytic and architectural components of the small (40S) and the large (60S) ribosomal subunits.11,51,53 These subunits are then exported to the cytoplasm where they form the functional (80S) ribosome after the final maturation steps.51,54,55
Eukaryotic cells have multiple copies of tandemly repeated 47S rRNA genes per haploid genome, termed rDNA repeats.56,57 These rDNA clusters of 70–80 repeats are arranged as head-to-tail arrays on the short arms of the five human acrocentric chromosomes.58,59,60 Each repeating rDNA unit possesses the pre-rRNA coding region, encoding the 18S, 5.8S and 28S rRNAs, as well as external (ETS) and internal (ITS) transcribed and non-transcribed regions, separated by intergenic spacer sequences.13,61 Although present at high copy number, only a proportion of rDNA repeats are actively transcribed by Pol I at any given time in metabolically active cells.61,62,63 Recent studies indicate that the rDNA repeats are regulated by their distinct chromatin states and epigenetic modifications, and primarily exist in four functional states: (i) silent, (ii) pseudo-silent, (iii) transcriptionally competent but poised, or (iv) transcriptionally active rDNA.61,64,65 In general, an open chromatin structure correlates with transcriptional competency and is characterized by DNA hypomethylation, acetylated histones as well as the binding of the upstream binding factor (UBF), which is essential for recruiting Pol I to the rDNA promoter and maintaining the active rDNA state.62,66 Inactive rRNA genes, on the other hand, are not bound by UBF and are characterized by repressive histone modifications and CpG hypermethylation at the rDNA promoter and thus are stably silenced, or they can be non-CpG methylated and hence are in a “pseudo-silent” state.62 While the production of the 47S pre-rRNA has long been thought to be controlled by modulating the rate of Pol I transcription in response to growth stimuli or cell cycle cues,16,67,68,69,70 alterations in rDNA chromatin states have also been shown to contribute to the long-term regulation of Pol I transcription such as during cellular differentiation and transformation.62,71,72,73,74
Dysregulated Pol I transcription is linked to ribosomopathies and cancer. Mutations in factors closely associated with Pol I transcription and deregulation of Pol I transcription are linked to the etiology of a number of ribosomopathies.21 In contrast to ribosomopathies, altered Pol I transcription in cancer is largely a result of dysregulated oncogenic pathways upstream of Pol I transcription or due to direct modulation of the Pol I transcription apparatus by oncoproteins or tumor suppressors.21 Moreover, due to the repetitive nature and high transcription rate of the rDNA repeats as well as their challenging DNA replication, the rDNA loci are inherently unstable and have been shown to be increasingly susceptible to DNA damage and chromosomal recombination events resulting in large copy number variations.75,76 Variation in rDNA copy number is correlated with the expression of functionally coherent genes involved in ribosome biogenesis and this association has been proposed as a mechanism for cellular homeostasis including a rapid and reversible source of adaptation to coordinate ribosome biogenesis.77 Genomic instability of the rDNA loci has been reported in congenital diseases characterized as putative ribosomopathies with a high cancer risk, such as Bloom and Werner syndromes.21,78
Canonical and extra-ribosomal functions of RPs
In the context of ribosome assembly and function, the RPs are involved in the stabilization of the small and large subunit structures, rRNA processing and stabilization of secondary structures in the rRNA, pre-ribosome transport, RNA folding and/ or interaction with auxiliary factors required for ribosome assembly and mRNA translation.79 This review is focused on the cytoplasmic RPs as opposed to the 75 mitochondrial RPs that assemble into the mitochondrial ribosome. We refer readers to other excellent reviews on mitochondrial RPs and their role in mitochondrial protein synthesis.80,81
In addition to their structural and regulatory roles in the assembly of the ribosome, RPs perform other “moonlighting” extra-ribosomal functions including the regulation of cell growth, proliferation and differentiation, immune signaling, DNA repair and apoptosis. These functions are defined based on specific interactions between RPs with non-ribosomal cellular components independent of the ribosome.82,83 This review is focused on the extra-ribosomal roles of RPs in development and tumorigenesis. For further reading on RPs extra-ribosomal functions, we refer readers to other reviews.82,83,84,85,86
Nucleolar stress response
In addition to ribosome production, the nucleolus plays a critical role as a central hub in coordinating cellular response to stress by integrating various stress response pathways including activation of the p53 pathway.84,87 Central to this activity is the nucleolar stress response (also known as the impaired ribosome biogenesis checkpoint),28 whereby perturbations in ribosome biogenesis, such as inactivation of Pol I transcription, impaired rRNA processing, ribosome assembly and transport are established mechanisms of nucleolar stress that induces p53 pathway activation, leading to cell cycle arrest, senescence, autophagy, and apoptosis87,88,89 (Fig. 2).

p53-mediated nucleolar stress response. Cell growth and proliferation remain under constant nucleolar surveillance. Under normal growth conditions, levels of the tumor suppressor p53 are suppressed by the binding of the E3 ubiquitin ligase mouse double minute 2 (Mdm2) and its homolog Mdm4, leading to ubiquitination and degradation of p53. When ribosome biogenesis is disrupted at the level of rRNA synthesis, processing or ribosome assembly, free ribosomal proteins (RPs) (primarily RPL5 and RPL11 and RPL23) and the 5S rRNA are released from the nucleolus to the nucleoplasm where they bind and sequester Mdm2/Mdm4. This in turn prevents the poly-ubiquitination and proteasome-mediated degradation of p53, thereby mediating its stabilization. The RPs (indicated) have been shown to regulate the Mdm2/p53 axis through various mechanisms including binding Mdm2 and its homolog and binding partner Mdm4. Additional mechanisms of nucleolar stress response include ribosome stress-mediated increase in RPL11 mRNA translation, which leads to enhanced interaction between RPL11 and Mdm2 and subsequent accumulation of p53. Following nucleolar stress, p53 can also be activated by RPL26 binding to p53 mRNA and enhancing its translation. Upon activation, p53 transactivates several downstream targets, leading to cell cycle arrest, apoptosis, autophagy or senescence
In response to nucleolar stress, p53 stabilization can be achieved via different mechanisms including post-translational modifications, protein-protein interactions and increases in the translation rate of p53 mRNA. One of the best documented mechanisms of p53 induction upon nucleolar stress involves the binding of RPs to Mdm2 (mouse double minute 2; also referred to as HDM2 in human), leading to p53 stabilization.90 In proliferating cells, p53 activity is kept repressed by Mdm2 via two complementary mechanisms: (i) Mdm2 acts as an E3 ubiquitin ligase that directly transfers ubiquitin onto p53 thereby targeting it for 26S proteasomal degradation;91,92 and (ii) the direct binding of Mdm2 to the N-terminal domain of p53 inhibits the transcriptional activity of p53 by preventing its interaction with the Pol II transcription machinery.93,94 This Mdm2-dependent surveillance of p53 activity is regulated by distinct and independent mechanisms to those involving replicative stress and the DNA damage response (DDR) where phosphorylation of either Mdm2 or p53 prevents their interaction, leading to p53 stabilization. p53 stabilization by the nucleolar stress response requires RPs.90,95
Furthermore, an increasing number of RPs including RPL5,96 RPL6,97 RPL11,98,99 RPL22,100 RPL23,101,102 RPL26,103 RPL37,104 RPS3,105 RPS7,106,107 RPS14,108 RPS15,104 RPS19,109 RPS20,104 RPS25,110 RPS26,111 RPS27,112 RPS27A,113 and RPS27L112,114 have been shown to regulate the Mdm2/ Mdm4 (Mdm2’s homolog and partner also known as MdmX in mice and HDMX in human)/p53 axis, consequently suppressing cell proliferation. A number of different models have been proposed for how these various interactions might regulate p53 (Fig. 2). In the “riding the ribosome” model, the interaction of p53 and/or Mdm2 with ribosomal subunits facilitates p53/Mdm2 transport from the nucleolus to the cytoplasm thereby preventing p53 from activating its target genes and promoting its ubiquitin-mediated degradation in the cytoplasm.84,86 Conversely, impairment of ribosome production and export of ribosome subunits is predicted to decrease p53/Mdm2 transport to the cytoplasm, thus allowing p53 to interact with its target genes in the nucleoplasm.84 A recently characterized alternative model described that inhibition of rRNA transcription or ribosome synthesis and assembly in the nucleolus creates a pool of free RPs that directly interact and sequester Mdm2. For instance, inhibition of Pol I transcription by a range of treatments including low doses of actinomycin D and oxaliplatin; the reduction of particular RPs; serum starvation; increases in cell confluence and nucleotides depletion83,86,88,89,115,116,117 have all been shown to induce the release of RPL5, RPL11 and RPL23 into the nucleoplasm where they can interact with the central acidic domain of Mdm2, suppressing p53 ubiquitination.84,86 Although most of the RPs interact with Mdm2 directly, some of them such as RPS7,107 RPS15,104 RPS20,104 RPS25,110 and RPL37104 have also been shown to bind to Mdm4. These RPs were shown to employ different mechanisms in regulating the Mdm2-p53-Mdm4 network.104,107
RPL5 and RPL11 can associate with each other and 5S rRNA and this pre-ribosomal complex is essential for p53 activation upon impairment of ribosome biogenesis.118,119 Intriguingly, depleting RPL5 and RPL11 but not other RPs (e.g., RPS7 and RPL23) is sufficient to reverse cell cycle arrest induced by defects in ribosome biogenesis.117,120 However, a synergistic suppression of Mdm2 activity through cooperation of RPL11 and RPL5 has been observed, suggesting they have distinct roles in inhibiting Mdm2 function.119 The cancer-associated Mdm2 C305F mutation, which disrupts the interaction between Mdm2 and RPL5 and RPL11 prevents p53 activation in response to nucleolar stress.121 A knock-in mouse model with the Mdm2 C305F mutation displayed accelerated Myc-induced lymphomagenesis,95 indicating a tumor-suppressive role for the RPL11–RPL5–Mdm2–p53 against tumorigenesis. Mdm2 can also be inhibited by other nucleolar proteins such as p19ARF, nucleophosmin and nucleostemin in response to nucleolar stress.122,123
The RPS7/RPL26 association with Mdm2 has a different regulatory function as they are both reported to be substrates for Mdm2 ubiquitination. In turn, an RPS7-ubiquitin fusion protein selectively inhibits Mdm2 degradation of p53 and promotes apoptosis. This indicates that Mdm2 ubiquitination of RPS7 is involved in sustaining the p53 response.107 RPL26 is unique in its ability to bind the 5’untranslated region of the p53 mRNA to promote its translation. Its interaction with Mdm2 triggers its own ubiquitination and degradation, which in turn causes downregulation of Tp53 mRNA translation124 (Fig. 2). Further support for the diverse roles of RPs in the regulation of the Mdm2-p53 pathway stems from the finding that knockdown of RPS6 reduces 40S ribosome biogenesis but increases RPL11 mRNA translation. This enhances the interaction between RPL11 and Mdm2, leading to p53 activation125 (Fig. 2). Since multiple RPs have distinct mechanisms for activating p53, it is plausible that they may sense and integrate different types of signals, leading to activation of nucleolar stress pathways.90 Upregulation of p53 as a consequence of defective ribosome biosynthesis and subsequent activation of the nucleolar stress response is linked to a wide spectrum of hypo-proliferative phenotypes displayed by ribosomopathy patients.18,126,127
P53-independent RP-mediated responses to nucleolar stress
In addition to activating p53; RPL5, RPL11 and RPS14 have been shown to bind to the p53 homolog p73 to prevent Mdm2 from binding to p73 at target gene promoters, such as those of p21 and Puma, leading to p73 activation and p73-dependent apoptosis.128 Intriguingly, simultaneous knockdown of p73 and either RPL5 or RPL11 was required to rescue 5-FU-induced apoptosis of p53-null tumor cells,128 supporting the essential role of RPL5 and RPL11 in p73-mediated apoptosis.
In addition to activating p53 and p73, several RPs can also inactivate oncoproteins, such as c-Myc. RPL11 specifically binds to the Myc box II domain of c-Myc and inhibits its transcriptional activity.129 RPL11 was also shown to bind c-Myc mRNA and promote its degradation in response to nucleolar stress.130 Moreover, both RPL5 and RPL11 co-resided on c-Myc mRNA and suppressed c-Myc expression through a RNA-induced silencing complex-mediated miRNA targeting mechanim.131 Similarly, RPS14 has also been found to suppress c-Myc transcriptional activity and promote its mRNA turnover.132 This reveals a negative feedback regulatory loop of RPs as MYC also activates expression of RPs.133 The auto-regulatory feedback regulation of RPs and MYC may act as a sensor of abnormal ribosome biogenesis that consequently limits MYC activity.133
Other studies have shown that RPL41 facilitates the shuttling of activating transcription factor 4 (ATF4), a regulator of tumor cell survival, from the nucleus to the cytoplasm to be degraded, consequently sensitizing tumor cells to chemotherapy.134 Additionally, RPS3 can induce apoptosis by interacting and collaborating with E2F1, and Akt-mediated phosphorylation of RPS3 attenuates apoptosis by abrogating the RPS3–E2F1 interaction.135 Phospho-RPS3 was also shown to shuttle to the nucleus and upregulate pro-survival gene expression via association with NF-κB in non-small cell lung cancer cells.136 These studies thus reveal the ribosome-independent functions of RPs and provide further understanding of the extra-ribosomal roles of RPs in tumorigenesis.
RP mutations and human diseases
Mutations of RP-encoding genes are highly associated with genetic diseases such as ribosomopathies and cancer (Table 1). Large-scale sequencing studies and various experimental models have provided in-depth insights into the ribosomal and extra-ribosomal functions of RPs, which we review in the following sections. In addition, emerging evidence suggests post-translational modifications of RPs, for example, phosphorylation and ubiquitylation, may functionally influence translational control and are linked to human diseases (reviewed in refs. 137,138)
Ribosomopathies
Mutations in RPs or other factors involved in the process of ribosome synthesis and assembly account for the pathogenesis of a heterogeneous group of diseases called ribosomopathies.15,16,17 This review focus on Diamond–Blackfan anemia (DBA) and Myelodysplastic syndrome (MDS) with chromosome 5q deletion (del(5q) MDS) in which RP mutations have been linked to the disease etiology (Table 1), and on diseases associated with mutations in other ribosome biogenesis factors including Shwachman-Diamond syndrome (SDS), X-linked-dyskeratosis congenita (XL-DC), cartilage–hair hypoplasia–anauxetic dysplasia (CHH-AD) and Treacher Collins Syndrome (TCS).
Ribosomopathies associated with mutations in RPs
Diamond–Blackfan anemia (DBA)
DBA is an autosomal dominant genetic disorder characterized by erythroid aplasia in association with a wide spectrum of congenital anomalies, including craniofacial, limb, genitourinary, and heart malformations, as well as an increased susceptibility to cancer. Since mutations in RPS19 were identified as the first causal genetic lesions for DBA in 1999,139 mutations in nineteen of the eighty-one RP-encoding genes have been identified, with RPS19 (25%), RPL5 (7%), RPS26 (6.6%) and RPL11(5%) being the most frequently mutated genes in DBA140,141 (Table 2). Approximately 70% of DBA cases are caused by loss-of-function mutations in a single copy of RP genes.142 In addition to RPs, several non-RP genes also contribute to the pathogenesis of DBA. Reduced translation of the key erythroid transcription factor GATA1,143 increased degradation of the GATA1 chaperone HSP70,144 or mutation in the RPS26 chaperone protein TSR2,145 impairs erythroid lineage commitment, resulting in specific defects in erythropoiesis in DBA.
Myelodysplastic syndrome (MDS) with chromosome 5q deletion (del(5q) MDS)
Del(5q) MDS is a distinct subtype of myelodysplastic syndrome (MDS). The disease is characterized by erythroid hypoplasia, macrocytic anemia, hypolobated megakaryocytes with <5% bone marrow myeloblasts, <1% circulating peripheral blasts and absence of Auer rods as well as the presence of chromosome 5q deletion.146 Haploinsufficiency of the genes located in two distinct commonly deleted regions (CDR) of chromosome 5q (5q32 and 5q31) accounts for the pathobiology of this disease.147 Among the 40 genes identified in 5q32, haploinsufficiency of RPS14 is a critical molecular event responsible for the erythroid differentiation defect in 5q-syndrome.148,149 Alternatively, heterozygous loss of other genes in the CDR including HSPA9 (Heat shock protein family A Hsp70 member 9),150 CSNK1A1151 encoding for Casein Kinase 1 alpha 1, a component of the beta-catenin complex regulating Wnt/ beta-catenin and p53 signaling pathways, and miR-145 and miR-146a also account for the hematologic features of the Del(5q) MDS.152
Ribosomopathies associated with mutations in other ribosome biogenesis factors
Shwachman-Diamond syndrome (SDS)
SDS is an autosomal recessive disorder associated with bone marrow failure featured by neutropenia or multilineage cytopenias.153 It is also characterized by multiple developmental anomalies such as exocrine pancreatic dysfunction and impaired bone development. More than 90% of SDS patients display biallelic inactivating mutations in the SBDS ribosome maturation factor gene (SBDS).154 SBDS acts in concert with Elongation Factor Like GTPase 1 (EFL1) in removing a ribosomal anti-association factor Eukaryotic Translation Initiation Factor 6 (EIF6) from pre-60S subunits, which is necessary for ribosome maturation.155,156,157 EIF6 is involved in both pre-rRNA processing and export of the pre-60S subunit to the cytoplasm, and thus removal of EIF6 is critical for the association of the nascent 60S subunit with the 40S subunit to form a translationally competent ribosome. In SDS patients, mutation of SBDS stalls 60S maturation and impairs ribosome assembly, resulting in a reduced number of 80S ribosomes.157 Recently, the identification of mutations in another 60S ribosome assembly factors DnaJ Heat Protein Family Member C21 (DNAJC21) and EFL1 support that impaired 60S ribosome maturation as a consequence of defective EIF6 eviction is the primary cause of SDS pathogenesis.158,159
X-linked-Dyskeratosis Congenita (XL-DC)
DC is a genetic disorder characterized by bone marrow failure usually associated with skin hyperpigmentation, nail dystrophy, mucosal leukoplakia and pulmonary fibrosis. The X-linked variant is associated with mutations in DKC1, encoding for a pseudouridine synthase Dyskerin.160 Dyskerin catalyzes rRNA pseudouridylation by binding to a group of snoRNA containing H-box (ANANNA) and ACA box (ACA) sequence motifs in rRNA, a process important for ribosome production and function. In addition to rRNA modification, Dyskerin also bind to H/ACA sequence in the nascent telomerase RNA component (TERC). The role of Dyskerin in the regulation of telomere function is in line with other gene mutations identified in DC such as TERC and TERT (telomerase reverse transcriptase).161 Thus, telomere dysfunction rather than ribosomal defects has been considered to directly contribute to pathogenesis of DC.
Cartilage hair hypoplasia–anauxetic dysplasia (CHH-AD)
CHH-AD is an autosomal recessive disease presented with short stature, bone marrow failure, hair hypoplasia and immunodeficiency. It is caused by mutations in RMRP (RNA Component of Mitochondrial RNA Processing Endoribonuclease), a long non-coding RNA component of the ribonuclease MRP complex involved in pre-rRNA processing, to give rise to the mature 18S and 5.8S rRNAs.162 In addition to its role in ribosome biogenesis, the MRP complex has pleotropic effects on the processing of mitochondrial RNA and cell cycle-associated mRNAs.
Treacher Collins Syndrome (TCS)
Distinct from DBA, SDS, X-linked DC, and CHH-AD, which are classified as inherited bone marrow failure syndrome, TCS is a rare congenital disorder characterized by various craniofacial malformations. It is primarily associated with autosomal dominant mutations in TCOF1, a gene encoding for a 144 kDa protein called Treacle.163 Treacle is responsible for pre-rRNA transcription and methylation.164,165 Furthermore, defects in pre-rRNA synthesis by TCOF1 depletion are associated with the delocalization of the nucleolar DExD-Box Helicase 21 (DDX21), a DEAD box RNA helicase involved in rRNA synthesis and processing, to the nucleoplasm, leading to inhibition of ribosome biogenesis.166 In addition to its function in rRNA synthesis and modification, a recent study revealed a critical role for TCOF1 in DNA repair by the formation of a component of the MDC1-RAD50-NBS1-MRE11 complex. Haploinsufficiency of Tcof1 perturbs the DNA damage response and causes the extensive apoptosis of neuroepithelial cells associated with the pathogenesis of TCS.167 In addition to TCOF1, mutations in POLR1C and POLR1D, which encode subunits of both RNA polymerase I and III, are also associated with TCS.168,169 Like TCOF1 mutations, decreased pre-rRNA transcription and number of functional 80S ribosomes were found in both polr1c- and polr1d-mutant zebrafish models of TCS.170,171 These findings highlight a causal link between ribosomal defects and the pathogenesis of TCS.
The extent to which ribosomal defects in ribosomopathies contribute to clinical phenotypes remains to be defined. As discussed above, in TCS, DBA and SDS, the defects in ribosome production and function have been causally linked to clinical phenotypes. However, in other putative ribosomopathies such as XL-DC and CHH-AD, it is likely that the ribosomal defects contribute to a component of their disease features and impact on disease severity.20
A prominent characteristic of ribosomopathies is tissue-specific defects. Despite the ubiquitous requirement for ribosomes in all cell types, patients frequently exhibit symptoms arising from tissue-specific growth arrest, including bone marrow failure, anemia or other tissue developmental defects.172 Nevertheless, these diseases collectively possess many overlapping clinical features. For instance, the patients commonly present with symptoms of aging including deafness, cataracts and loss of subcutaneous fat. Patients with DBA and SDS have decreased numbers of hematopoietic stem cells (HSCs), with a significant subpopulation of their HSCs showing senescence markers including G1/S cell cycle arrest, SA-β-galactosidase and γH2AX staining, suggesting that ribosomopathies are associated with premature senescence.173 As disruption of ribosome biogenesis at various steps can promote cell cycle arrest and senescence, cellular senescence may underpin the common defects observed in ribosomopathies. A detailed description of the molecular mechanism underpinning ribosomopathy phenotypes is presented below. While senescence is an early feature of ribosomopathies, in the long-term, cancer susceptibility has been observed in patients with ribosomopathies except TCS. In this review, we also summarize and discuss the current understanding of the mechanisms underpinning an increased cancer risk in ribosomopathies.
The molecular basis of ribosomopathies
The nucleolar stress response
A critical role for p53 activation in the pathogenesis of ribosomopathies has been well established in zebrafish, murine or human disease models, including DBA,174,175,176 del (5q) MDS,148 SDS,153TCS177, and XL-DC.178 The nucleolar stress response triggered by ribosomal defects is the primary cause of p53 stabilization and activation. In DBA, RP haploinsufficiency results in impaired rRNA processing and disruption of the cognate 40S or 60S subunit biosynthesis,179 leading to accumulation of free RPs, including RPS3, RPS7, RPL5, RPL11, and RPL23 in the nucleoplasm. As noted above RPL5 and RPL11 together with 5S rRNA play a central role in the activation of the p53-dependent nucleolar stress signaling pathway180 (Fig. 2). In TCS, apoptotic elimination of neuroepithelial cells and neural crest cells upon p53 activation mediated by the nucleolar stress response has been considered to be the primary cause of craniofacial anomalies in TCS.177 Genetical or pharmacological inactivation of p53 can rescue disease-associated phenotypes, strongly supporting that p53 is a key molecular mediator of the hypo-proliferative clinical symptoms of ribosomopathies.18,19,126,127
Altered mRNA translation by ribosomal defects
As ribosomes are essential for translating mRNA into proteins, defects in ribosome synthesis and function alter the translation capacity and efficiency of ribosomes (Fig. 3). Genome-wide translational profiling of cellular models of ribosomopathies has revealed that translation of subsets of mRNAs, particularly mRNAs encoding proteins involved in cell fate decisions, are specifically impaired under the limited availability of functional ribosomes.23 Particularly, the specific reduction in translation of GATA1, a master regulator of hematopoiesis, contributes to bone marrow erythroid hypoplasia in DBA patients.143,181,182 Selective translation defects have also been described in some mRNAs containing an internal ribosome entry site (IRES). Deficiencies in RPL11 or RPS19 reduced IRES-mediated translation of the erythroblast proliferation and differentiation factors BAG1 and CSDE1 in DBA murine models and patient samples.183 In addition, rRNA pseudouridylation defects in XL-DC caused a defect in ribosome binding to IRES elements, resulting in reduced translational fidelity and decreased translation of several IRES-containing mRNAs, including the tumor suppressor p27 and the antiapoptotic factors XIAP and Bcl-xL. These factors are linked to two specific pathological features of XL-DC: susceptibility to cancer and bone marrow failure.184

Altered mRNA translation in cells with ribosome biogenesis defects. a In normal cells, functional mature ribosomes (80S) comprise the small (40S) subunit and the large (60S) subunit. The small subunit interacts with the anticodon-containing ends of complementary tRNAs so as to translate the codon information contained in mRNA into its corresponding sequence of amino acids. The large subunit contains peptidyl transferase activity and is responsible for linking the amino acids into a polypeptide chain. b In ribosomopathies such as DBA (Diamond–Blackfan anemia), mutations in RPS19 can cause a decrease in the number of functional ribosomes, which may lead to a competition for ribosomes among cellular mRNAs, leading to changes in the translation efficiency of subsets of mRNAs, including reduced translation of GATA1 mRNA. Reduced levels of GATA1, a key erythroid transcription factor impairs erythroid lineage commitment and results in specific defects in erythropoiesis in DBA. c Ribosome defects due to RP mutations and variation in RP composition may generate heterogeneous ribosomes with reduced translation fidelity, resulting in altered translation patterns. In DBA patients, deficiencies in RPL11 or RPS19 due to mutations can reduce the translation of IRES-containing mRNAs BAG1 and CSDE1, which encode erythroblast proliferation and differentiation factors. In X-linked-Dyskeratosis Congenita, defects in rRNA pseudouridylation can impair the binding of ribosomes to IRES elements, resulting in reduced translational fidelity and decreased translation of several IRES-containing mRNAs, including p27, XIAP, and Bcl-xL and enhanced bone marrow failure and cancer susceptibility
The altered translatome is thought to contribute to the tissue specificity of ribosomopathies. Two hypotheses have been proposed including the “altered/heterogenous ribosome” hypothesis and the “ribosome concentration” hypothesis to explain the selective mRNA translation in determination of disease pathogenesis19 (Fig. 3). The altered/ heterogenous ribosome hypothesis argues that variations in rRNA sequence, RP composition or ribosome-associated proteins can result in heterogeneous ribosomes with differential interactions with mRNAs, resulting in the preferential translation of particular mRNAs.185 On the other hand, the ribosome concentration hypothesis argues that a limited number of translationally competent ribosomes may cause competition for ribosomes among cellular mRNAs, leading to changes in the translation efficiency of subsets of mRNAs.182 mRNAs with long, highly structured 5’UTRs are speculated to be more affected by an insufficiency of ribosomes. This is supported by a study showing that reduction of GATA1 mRNA translation in DBA patients is attributed to its highly structured long 5’UTR.143 Paradoxically, recent reports have shown that reduction of ribosome abundance impairs the translation of mRNAs that are normally highly translated and have short/unstructured 5’UTRs.181,182 These contradictory results indicate that further defining the effects of structural features of mRNAs on translation efficiency will improve our understanding of translation specificity regulated by ribosome availability and their contribution to the pathological features of ribosomopathies.
While p53 activation and mRNA translation reprogramming as a result of impaired ribosome synthesis and function contribute to the disease phenotypes associated with RP haploinsufficiency, these two mechanisms have long been considered to be mutually exclusive in ribosomopathies. Intriguingly, a recent study of a DBA embryonic mouse model has demonstrated that Rps6 haploinsufficiency leads to selective limb phenotypes, mediated by deregulation of translation. However, the majority of mRNAs undergoing differential translational changes were rescued upon the loss of p53,186 indicating an intimate link between translational control and p53 activation upon ribosome perturbation in ribosomopathies. They further demonstrated that p53 transcriptionally induces the expression of 4E-BP1, a negative regulator of eIF4E-mediated cap-dependent translation and that the p53-4E-BP1-eIF4E axis contribute to the selective changes in cap-dependent translation.186 Therefore, an altered translatome within specific cellular and/or tissue contexts in response to p53 activation may contribute, in part, to the tissue-specific phenotypes of ribosomopathies.
Oxidative stress and deregulated protein degradation
It is becoming clear that fully functional ribosomes are required for cellular redox homeostasis. Increased reactive oxygen species (ROS) levels were detected in murine DBA cells with RPL5 and RPS19 deficiency187 and in lymphocytes from DC patients.188 Reduction of ROS levels by antioxidant treatment rescued the growth defect in cells with RPL10 mutation or with SBDS inactivation, reinforcing a strong link between oxidative stress and ribosomal defects.189
Excessive ROS causes oxidative damage to all macromolecules, including nucleic acid, proteins, and lipids. The high abundance of rRNA and RPs render them susceptible to chemical modification by ROS, leading to further disruptions in ribosome assembly and function.190,191 Indeed, Tcof1+/− embryos exhibited a high level of ROS in the neuroepithelium and the impaired DNA repair capacity resulted in increased sensitivity of neuroepithelial cells to oxidative stress and extensive apoptosis.192 Oxidative stress also causes mitochondrial dysfunction and suppresses oxidative phosphorylation and ATP production, which contributes to the hypo-proliferative phenotype observed in ribosomopathies.193
While the significance of oxidative stress in the pathogenesis of ribosomopathies is increasingly being recognized, the mechanisms by which defective ribosome biogenesis causes ROS accumulation remain unclear. Two recent studies suggested that selective defects in Globin mRNA translation lead to the imbalance of heme-globin, resulting in excess free heme, which contributes to ROS production followed by apoptosis and delayed erythroid differentiation in DBA patients.194 In addition, p53 activation in the mesenchymal cells of the hematopoietic niche with Sbds deletion led to secretion of the inflammatory molecule S100 A8/A9, which subsequently induced oxidative stress and the DNA damage response in the surrounding wild-type hematopoietic stem and progenitor cells, contributing to impaired hematopoiesis in SDS.195 Therefore, the oxidative stress that occurs in ribosomopathies is a downstream effect of either intracellular ribosomal defects or microenvironmental assaults.
While ribosomes function to synthesize proteins, recent studies suggest a potential interaction between ribosomes and the protein degradation machinery in maintaining protein homeostasis.196 A biochemical analysis of RPS19 variants revealed that a subset of mutant RPS19 proteins failed to localize to the nucleolus and exhibited a dramatically reduced level of protein expression. Proteasome inhibitors effectively restored mutant RPS19 protein expression levels and nucleolar localization.196 Reduced stability of mutant RPs via proteasomal degradation was also observed in the cells expressing mutant RPS15 proteins.197 These findings thus underscore a proteasome-based mechanism of quality control of mutant RPs.
Conversely, RP mutations have been demonstrated to alter proteasomal activity. Cells with an RPL10-R98S mutation exhibit aberrant expression of several proteasomal proteins including upregulation of Psmb10 and Psmb9, the catalytic subunits for the immunoproteasome, along with reduced chymotrypsin-like and caspase-like activities of the proteasome.198 The altered expression and activities of proteasomes in RPL10- R98S expressing cells led to an elevated sensitivity to proteasome inhibitors. Targeting the proteasome is also proposed to be a novel therapeutic approach for treatment of TCS. Degradation of the cellular nucleic acid-binding protein (Cnbp), a protein involved in craniofacial development, was identified to antagonize the TCS phenotype in a zebrafish model of TCS. Proteasome inhibitors ameliorate cranial skeleton anomalies exhibited in TCS-like embryos by decreasing degradation of Cnbp, strengthening a critical role of the functional ribosomes in cell proteostasis.199
Cancer
Cancer susceptibility in ribosomopathies
Patients with ribosomopathies have an elevated risk of developing cancer throughout their life, despite the hypo-proliferative phenotypes associated with early symptoms, and for particular cancer types the risk can be up to 200-fold higher23 (Table 2). The cancer susceptibility of DBA has been recapitulated in the mouse model with heterozygous deletion of Rpl11.200 The paradoxical transition from an early hypo-proliferative cellular response to the hyper-proliferation oncogenic phenotype later in life, was first reported by Dameshek in 1967 and referred to as Dameshek’s riddle.24 The recent discoveries of somatic mutations in RP genes in hematological cancers and solid tumors reinforce the link between defects in ribosome biogenesis and oncogenic transformation.17,19,20 Here, we describe a range of common cancer-associated mutations mainly reported in RPL5, RPL11, RPL10, RPL22, RPS15 and RPS15A occurring in overlapping and distinct cancers (Table 1) and we discuss the current understanding of the mechanisms of oncogenesis of RP mutations including impaired nucleolar stress response and altered mRNA translation.
Somatic RP mutations in cancer
RPL5 and RPL11
As mentioned above, RPL5 and RPL11 mediate activation of the nucleolar stress response by inhibiting Mdm2 leading to p53 induction. Heterozygous RPL5 and RPL11 mutations or deletions have been identified in spontaneous human cancer. A comprehensive analysis of TCGA/ICGC pan-cancer dataset including 19,000 cancer samples across 49 cancer types detected 139 RPL5 and 74 RPL11 cancer-associated mutations, of which the majority are missense mutations (66% and 73% in RPL5 and RPL11 mutations, respectively).201 Various frequencies of RPL5 mutations/deletions have been reported in breast cancer (34%), melanoma (28%), multiple myeloma (up to 30%) and T-ALL (2%).202 In multiple myeloma, patients with low RPL5 expression level have a worse survival outcome. However, analysis of clinical trial data has revealed that multiple myeloma patients with low RPL5 mRNA expression levels respond better to the proteasome inhibitor bortezomib by comparison with patients with high RPL5 expression,203 highlighting the link described above between RP mutations and altered proteasomal activity.
RPL10
Somatic mutations in RPL10 at residues R98 and Q123, including R98S, R98C, and Q123P are thought to play an active role in tumorigenesis. Particularly, R98S has been described as a mutation hotspot with >90% of RPL10 mutations at this residue, accounting for ~8% of pediatric T-ALL.204,205 The mutants including I33V, E66G, I70M and I70L in RPL10 have been reported in multiple myeloma with low frequency (2%) and cluster in a region that is distinct from the mutation hotspot identified in T-ALL.206 An increase in RPL10 expression has been observed in ovarian and pancreatic cancers and linked to enhanced cell proliferation, invasion, survival, and resistance to oxidative stress.207,208
RPL22
Inactivation of RPL22 due to heterozygous deletion has been observed in 10% T-ALL patient samples. Rpl22 haploinsufficiency accelerated the development of thymic lymphoma in a mouse model of T-cell lymphoma driven by hyperactivation of AKT2 in T-cell progenitors, supporting a tumor-suppressive role for RPL22 in T-ALL.209 Homozygous deletion of Rpl22 resulted in distinct phenotypic characteristics with expanded thymic tumors at the primary site in the absence of dissemination and migration to peripheral organs, suggesting a genetic dosage-dependent model in which loss of a single allele of Rpl22 promotes lymphomagenesis and cancer dissemination, while loss of both copies impairs migration capacity and restricts malignant cells to the thymus.210 RPL22 mutations have also been described in ∼10% of gastric, endometrial, and colorectal and adrenocortical solid cancer samples.211,212,213,214
RPS15 ad RPS15A
RPS15 mutations have been reported in 19.5% of aggressive CLL and in 12% in CLL with 17p deletion that is typically refractory to most conventional therapies.215 The RPS15 heterozygous S138F mutation drove CLL-like disease development in mice and combination with heterozygous Trp53 deletion shortened the latency of CLL and generated a more aggressive disease course.216 In contrast to the tumor-suppressive role of RPS15, RPS15A has been reported to promote oncogenic transformation and progression in lung,217 glioblastoma,218,219 gastric,220 liver221 and colorectal cancer.222 A decrease in RPS15A expression inhibited proliferation of human glioblastoma cells and gastric cancer cells via downregulation of AKT pathway acitvity,218,220 and colorectal cancer cells via activation of the p53 pathway,222 and impaired angiogenesis in hepatocellular carcinoma by increasing FGF18 expression via the Wnt/Beta-Catenin pathway.221
Mechanisms of oncogenic potential RPs
Extensive research of cancer susceptibility in patients with ribosomopathies and somatic RP mutations in cancer has reveal an oncogenic potential of ribosomes. Three mechanisms for formation of “onco-ribosomes” have been proposed, including: (i) a shift in translational profiles towards synthesis of growth-promoting and pro-oncogenic proteins; (ii) extra-ribosomal functions of RPs beyond ribosome synthesis contributing to oncogenic transformation; and (iii) ribosomal defects-associated metabolic alterations promoting secondary mutations and genomic instability. These mechanistic insights not only assist in resolving Dameshek’s riddle, but also facilitate identifying therapeutic vulnerabilities and novel treatments for patients with ribosomopathies and cancer.
Translational reprogramming
While the DBA-associated germline mutations in RP genes are usually different from the cancer-associated somatic mutations, with only several variants in common,223 it is proposed that mutations in RPs can lead to common effects in deregulating ribosome synthesis and function by either reducing the number of competent ribosomes or forming heterogenous ribosomes (Fig. 3). These ribosomal defects lead to altered translational fidelity and capacity. The evidence for ribosomal defects-induced proteomic changes in the pathogenesis of ribosomopathies has been discussed above. Similarly, structural analysis of somatic cancer-associated RP mutations, including the mutation hotspots in RPL10 and RPS15 revealed that these RP mutations are localized in the regions associated with the catalytic core of ribosome, consistent with their influence on mRNA translation and thus global protein synthesis.197,224
An altered translation pattern in cancer cells with RP mutations may promote oncogenic protein synthesis. For instance, the T-ALL-associated RPL10-R98S mutation protects leukemia cell survival via specific upregulation of IRES-dependent translation of the antiapoptotic molecule BCL2.198 Studies of cancer susceptibility in ribosomopathies also provide the evidence for oncogenic transformation through translation reprogramming. In SDS, SBDS mutations specifically affect translation of the C/EBPα and β proteins, which are important regulators of hematopoietic granulocyte differentiation. The inability to translate C/EBP α and β is associated with impaired hematopoiesis in SBDS, and more importantly, the 200-fold elevated risk of developing AML in SDS patients,225 as loss-of-function mutations in C/EBPα have a known role in AML pathogenesis.226 The current findings support the hypothesis that altered translational capacity and fidelity resulting from defective ribosomes contribute to cancer development by facilitating the production of oncoproteins and/or activation of oncogenic pathways.
Impaired nucleolar stress response and pro-oncogenic extra-ribosomal functions of RPs
The well-characterized nucleolar stress response whereby the 5S-RNP complex formed by RPL5, RPL11 and 5S rRNA mediates regulation of p53 activation is considered to be an important barrier to cancer development upon defects in ribosome biogenesis. RPL5 or RPL11 loss-of-function disrupts p53 activation and consequently contributes to the propensity of DBA patients to develop cancer. Studies in Rpl11 heterozygous mice showed increased susceptibility to radiation-induced lymphomagenesis. In this regard, total or partial deletion of Rpl11 compromised p53 activation upon ribosome stress or DNA damage in fibroblasts.200 RPL5 mutations in human cancer cell lines impair p53 activation, providing supporting data for RPL5/RPL11 haploinsufficiency in promoting malignant transformation via an impaired nucleolar stress response.200,201 In agreement with this, the nucleolar proteins SPIN1 and PICT1, which sequester RPL5 and RPL11, respectively in the nucleolus impair the Mdm2-p53 pathway and promote tumor growth by preventing RPL5 and RPL11 from interacting with Mdm2 in the nucleoplasm.227,228
Moreover, the extra-ribosomal functions of RPs may confer pro-oncogenic capacity. As noted above, RPL11,229 RPL5131 and RPS14132 can repress MYC expression by either binding directly to the promoter region and reducing transcription or accelerating mRNA degradation. Thus, the loss of these RPs may promote transformation by oncogenic c-MYC overexpression. Indeed, c-MYC upregulation has been described in lymphoma models of heterozygous Rpl11 or Rpl22.200,209
Metabolic alterations and oxidative stress
The metabolic alterations-associated with ribosome defects also contribute to the oncogenic potential of RPs. The T-ALL-associated RPL10-R98S mutation has been linked to oxidative stress,189 which potentially increases genomic instability and places selective pressure towards acquire rescuing mutations, ultimately leading to oncogenic transformation. Metabolic changes can occur though transcriptional, translational and post-translational modulation. The upregulation of phosphoserine phosphatase (PSPH) transcription and translation detected in the lymphoid cells with the RPL10-R98S mutation was thought to contribute to ribosomal mutation-driven serine/glycine synthesis in T-ALL.230 In addition, altered proteasome activity in RP mutant cells induces proteomic changes and promotes pro-oncogenic pathway activation. A decrease in Jak1 degradation was found in RPL10-R98S mutant cells and led to activation of the JAK-STAT signaling pathway, an established oncogenic driver in T-ALL.198 These findings thus highlight the critical role of metabolic reprogramming in RP mutation-driven oncogenesis.
Dysregulation of Pol I transcription of 47S rRNA genes in cancer
In contrast to cancers associated with RP mutations and deletions, oncogene-driven cancers are linked to hyperactive Pol I transcription.21 Moreover, the rDNA loci are inherently unstable and susceptible to DNA damage and chromosomal recombination events resulting in large copy number variations.75,76,231 However, increased rRNA synthesis can be achieved even if rDNA copy number is reduced,232 which can occur by increasing the rate of Pol I transcription per rDNA repeat and/or the number of active rDNA repeats. Furthermore, variation in rDNA copy number independent of Pol I transcription rate has been associated with cancer.233 Deregulation of rDNA silencing and/or increased rDNA instability have been proposed to promote global genomic instability and tumorigenesis.231,232,234,235,236
For many decades, an increase in nucleolar size and number, indicative of high rates of Pol I transcription and ribosome biogenesis, has been used as a biomarker of poor cancer prognosis.32,237 Indeed, recent studies have confirmed that hyperactivation of Pol I transcription of rRNA genes is a key step in malignant transformation.39,41,238 The upregulation of Pol I transcription rates observed in cancer is largely mediated through the deregulation of upstream oncogenic and tumor-suppressive signaling pathways known to modulate ribosome biogenesis, including RAS/RAF/ERK, PI3K/AKT/mTOR, c-MYC, p53, pRb, and PTEN.31,45,133,239,240,241,242,243 These oncogenic growth-promoting pathways converge to directly control key players of Pol I transcription initiation such as RRN3 and UBF, leading to enhanced Pol I transcription and cellular transformation.15,244 For example, c‐MYC, a potent transcriptional driver of ribosome biogenesis, including the synthesis of rRNAs and RPs,240,245,246,247,248,249,250 is dysregulated or amplified in greater than 70% of all cancers.251 RAS and RAF are also mutated in 30% and 6–7% of human cancers, respectively.252,253 Activating mutations in the PIK3CA gene or inactivating mutations in the negative regulator PTEN are drivers of ~30% of human sporadic tumors.254,255 These oncogenic networks cooperate to enhance rRNA synthesis, ribosome biogenesis and protein synthesis at multiple levels and promote “translation addiction” in cancer cells. Thus, targeting ribosome biogenesis has emerged as a potential therapeutic approach to combat cancer.41,256
Therapies targeting ribosomal defects
Therapies for ribosomopathies
The current standard of care for ribosomopathies encompasses management of the symptoms associated with tissue-specific phenotypes such as bone marrow failure and anemia. This includes chronic blood transfusions followed by steroids to allow patients to survive this disease phase (Table 2), but with many undesirable and even fatal side effects after long-term of steroids, including hypertension, diabetes mellitus, and growth retardation.257 There is clearly an unmet need in developing new therapeutic strategies for ribosomopathies.
Here, we summarize the recent research of prospective therapeutics for DBA and del(5q) MDS, the two ribosomopathies related to RP mutations. DBA patients are treated with transfusions followed by corticosteroids. Patients who stop responding to steroid treatment but are transfusion-dependent are then treated with hematopoietic stem cell transplantation (HSCT), which requires an HLA-matched donor and a lifelong immunosuppressive therapy. Thus, the implementation of alternative therapies for the cure of DBA warrants investigation. A range of new compounds have been tested for DBA treatment. The amino acid Leucine improves anemia and developmental defects in DBA zebrafish and mouse models and patients.258,259 l-leucine stimulates mRNA translation through activation of the mTOR (the mammalian target of rapamycin) pathway, which regulates cap-dependent translation of TOP mRNAs to synthesize proteins involved in the translational apparatus.259,260 The efficacy of l-leucine for the treatment of transfusion-dependent DBA is being evaluated in Phase I/II clinical trials (NCT01362595, NCT02386267, https://clinicaltrials.gov/ct2/home). A recent finding showing that p53 induces the expression of the translation inhibitor 4E-BP1, a negative regulator of eIF4E-mediated cap-dependent translation upon RP haploinsufficiency in a murine model of DBA261 also strongly supports targeting cap-dependent translation as a promising approach to treating ribosomopathies. Furthermore, the loss of p53 and thus the ability to repress eIF4E-mediated translation could be a mechanism of the increased cancer risk associated with DBA.261 Indeed, increased eIF4E activity has been shown to promote cellular transformation.262,263,264 Moreover, RP genes are routinely deleted across human cancers, particularly in concert with TP53 mutations.25 Therefore, inhibitors of cap-dependent translation such as rapamycin and other inhibitors of mTOR may also serve as potential candidates to treat cancers characterized by RP gene deletions and loss of p53.
In addition to l-leucine, there are three drugs currently being evaluated in clinical trials for treatment of bone marrow failures in DBA: Sotatercept (NCT01464164), TFP (NCT03966053) and EPAG (NCT04269889). Sotatercept is a recombinant human fusion protein containing the extracellular domain of activin receptor type IIA, which inhibits TGF-β signaling. Its murine analog RAP-011 improved erythropoiesis in a DBA zebrafish model.265 Trifluoperazine (TFP), a calmodulin inhibitor, rescued the anemia phenotype in different DBA zebrafish and murine models by decreasing p53 translation and accumulation.266 Eltrombopag (EPAG), a thrombopoietin (TPO) receptor agonist, increased hemoglobin and platelet numbers in one case,267 and rescued erythroid maturation defects in a cellular model of DBA.268 In addition, recent studies utilizing gene therapy gene to deliver wild-type RPS19 in lymphoblastoid cell lines (LCLs) established from RPS19-deficient DBA patients rescued impaired ribosome synthesis and reduced p53 levels in LCLs.269 A similar approach also rescued the anemia and bone marrow failure phenotype in RPS19-deficient DBA murine model, demonstrating the in vivo efficacy of gene therapy and its potential for curing genetic disorders in ribosomopathies.174,270
MDS with del(5q) is managed with red blood cell transfusion (RBC) or recombinant erythropoietin, thalidomide, and retinoid injections to treat the primary symptoms of chronic anemia and fatigue. The majority of patients become RBC-dependent and long-term blood transfusion have multiple adverse effects, including iron accumulation, myocardial infarction, renal failure, infection and malignancy, resulting in increased morbidity, and mortality. Lenalidomide, an immunomodulatory agent has been approved for the treatment of transfusion-dependent anemia in del(5q) MDS with prognostic implication defined as low-risk according to the International Prognostic Score System (IPSS) and the revised IPSS (IPSS-R).271 The cytogenetic responses were assessed in a phase II study (MDS-003) involving 148 transfusion-dependent del(5q) MDS patients272 with 73% responding to lenalidomide treatment at 10 mg daily and 45% achieving a complete cytogenetic remission, and a phase III study (MDS-004) involving 204 patients with 50% responding in the 10mg- cohorts.273 Current clinical data indicates that lenalidomide treatment significantly improved overall survival in patients with low-risk MDS with and without del(5q).274 Mechanistically, lenalidomide selectively suppresses MDS clones in patients with del(5q) by targeting the haploinsufficient genes in the CDR of chromosome 5q and the associated pathways. The Csnk1a1 gene that resides within the CDR for del(5q) MDS encodes for Casein kinase 1a1 (CK1a). In a hematopoietic-specific conditional knockout murine model, heterozygous loss of Csnk1a1 causes β-catenin accumulation and stem cell expansion while homozygous loss of Csnk1a1 cause cell apoptosis via of p53 activation.151 Lenalidomide induced ubiquitination and degradation of CK1a in heterozygous Csnk1a1 cells, leading to p53 induction and apoptosis by binding to Cereblon (CRBN), the substrate adaptor for a CUL4A-RBX1-DDB1 E3 ubiquitin ligase complex and modulating the substrate specificity of the enzyme.275 Therefore, the synthetic lethality between lenalidomide and Csnk1a1 haploinsufficiency in chromosome 5q deletion may underpin the clinical efficacy of lenalidomide in del(5q) MDS. In line with this, TP53 mutations have been linked to the lenalidomide resistance in del(5q) MDS,276,277 confirming the primary mechanism of action of lenalidomide is via p53 activation. Indeed, almost 40% of patients progress to acute leukemia by 5 years after starting lenalidomide treatment, most commonly due to clonal evolution associated with acquiring mutations in in TP53, RUNX1, and TET2.278
Targeting ribosome biogenesis as a therapeutic approach in oncogene-driven cancer
Inhibiting oncogene-driven upregulation of ribosome biogenesis in cancer provides a therapeutic specificity for selectively targeting cancer cells over normal cells.13,41,256,279 Classical chemotherapeutics (e.g., oxaliplatin, cisplatin, actinomycin D and 5-FU) and poly-ADP ribose polymerase (PARP) inhibitors, recently FDA approved for ovarian cancer treatment, have been discovered to act through distinct mechanisms of action that include inhibition of rRNA synthesis, rRNA processing or ribosome biogenesis34,35,37,38,280,281 (Fig. 1b). Indeed, many cancer therapies originally intended to cause DNA damage and kill cancer cells actually impair ribosome biogenesis via multiple mechanisms subsequently leading to cell cycle arrest and senescence.36,280
A drug screen by Burger et al., has shown that 21 of 36 chemotherapeutic agents affect ribosome biogenesis at the level of rDNA transcription or rRNA processing.34 For example, actinomycin D, which intercalates GC-rich regions of DNA, selectively targets rDNA at concentrations as low as 5 nM and prevents Pol I transcription elongation.282 The anti-metabolite 5-FU, an inhibitor of nucleotide synthesis via depleting the intracellular deoxynucleotide pool,283 also disrupts rRNA processing.35 In addition, the platinum-containing compound cisplatin known to act as a DNA cross-linker inhibits Pol I transcription with a high degree of specificity284 through its ability to cross-link DNA at HMG-protein affinity sites, thus preventing the transcription factor UBF from associating with rDNA promoters.38,285 Moreover, oxaliplatin, another platinum compound used in the treatment of colorectal cancer, was also shown to kill cells by inhibiting Pol I transcription and inducing ribosome biogenesis stress but not by activating the DNA damage response.280 These observations provide the rationale for selectively targeting the Pol I transcription apparatus as a new class of anticancer therapies with reduced toxicity and improve efficacy compared to conventional genotoxic chemotherapeutic agents. Indeed, this has spurred the development of agents such as BMH-21, which can non-covalently bind GC-rich DNA, degrade Pol I and does not induce DNA damage.42,286 In this review, we focus on CX-5461, a novel anticancer agent that is showing increasing promise in clinical investigations.287
CX-5461: A first in class selective inhibitor of Pol I transcription
CX-5461 selectively inhibits Pol I transcription relative to Pol II and Pol III transcription279,288 and has demonstrated single-agent therapeutic efficacy in multiple preclinical cancer models, including lymphoma, AML, breast, prostate, and ovarian cancer.39,43,289,290,291,292,293,294,295
Using genetically engineered models of Eμ-Myc lymphoma, Bywater et al. demonstrated that CX-5461 induces nucleolar disruption, resulting in the binding of unassembled RPL5 and RPL11 to Mdm2 and subsequently rapidly activating p53-mediated cell cycle arrest and apoptosis39 (Fig. 4). Furthermore, this response was triggered in the absence of changes in the total levels of functional ribosomes or the rates of protein synthesis, demonstrating that certain tumor cells are highly dependent on accelerated Pol I transcription levels.28 The in vivo doses of CX-5461 that activate p53 and induce p53-mediated apoptotic cell death in MYC-driven B-cell lymphoma cells did not cause deleterious genotoxic effects to normal B-cell populations of the same lineage.39 Furthermore, a strong correlation between the mutational status of p53 and the level of drug sensitivity was also found, whereby p53-wildtype lymphoma and AML cell lines were significantly more sensitive towards CX-5461 than p53-null or -mutant cell lines.39,291 Inhibition of Pol I transcription with low-dose actinomycin D treatment and subsequent activation of the nucleolar stress response was also shown to delay the in vivo growth of p53-wildtype Eμ-Myc lymphomas but had no impact on the expansion of p53-null Eμ-Myc lymphomas.296 Nevertheless, CX-5461 demonstrated significant efficacy in p53-null AML in vivo. The significant survival advantage in both p53-wildtype and p53-null leukemic mice treated with CX-5461 was associated with activation of the G2/M cell cycle checkpoint, induction of myeloid differentiation and targeting of the leukemia-initiating cell population.291

Inhibition of RNA polymerase I transcription by CX-5461 induces both p53-dependent and -independent responses. A schematic representation of CX-5461’s mode of action and its downstream stress response pathways. CX-5461 inhibits the initiation of Pol I-mediated transcription by disrupting the association between SL-1 and Pol I, thus preventing Pol I recruitment to the rDNA promoter. This displacement leads to “exposed” rDNA repeats devoid of Pol I and the presence of defects associated with an open chromatin structure and the recruitment and phosphorylation of RPA to single-stranded rDNA, a marker for replication stress. CX-5461-mediated alterations in rRNA synthesis and rDNA chromatin and topology in turn trigger the downstream activation of two major signaling pathways: (i) a canonical p53-dependent nucleolar stress response leading to accumulation of p53 and/or (ii) a p53-independent DNA damage response (DDR) involving the activation of ATM/ATR kinase signaling. Each pathway induces various cellular responses including G1/S and G2/M cell cycle defects, apoptosis and senescence. Pol I, RNA polymerase I; SL-1, selectivity factor 1; rDNA, ribosomal RNA gene; TBP, TATAbinding protein; UBF, upstream binding factor; UCE, upstream control element; RRN3, RNA polymerase I-specific transcription initiation factor; Mdm2, mouse double minute 2; CHK, checkpoint kinases; CDK, cyclin-dependent kinases; ATM indicates ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related
P53-independent responses to CX-5461 in acute lymphoblastic leukemia (ALL), human ovarian cancer models and human immortalized fibroblasts were shown to involve a G2/M checkpoint arrest and cell death via the ATM/ ATR kinase pathways.43,292,297,298 In addition to inhibiting Pol I transcription, CX-5461 was shown to induce perturbations in rDNA chromatin and replication stress, triggering non-canonical activation of ATM/ATR kinase signaling within the nucleoli. Strikingly, these occurred in the absence of detectable γH2AX foci, a marker of double-stranded DNA breaks (DSBs)298 (Fig. 4). The net results of the unique CX-5461-mediated unique DNA damage response is replication stress, leading to replication-dependent DNA damage and cell cycle arrest and cell death.43,294 The combination of CX-5461 with dual inhibition of CHK1/2 has been reported to significantly enhance the therapeutic outcome against p53-null MYC-driven lymphoma in vivo.36
The first-in-human phase I dose escalation study with CX-5461 was recently completed in 16 patients with advanced relapsed or refractory hematological malignancies at the Peter MacCallum Cancer Centre, Australia (Australia and New Zealand Clinical Trials Registry, #12613001061729). CX-5461 was found to be both safe and tolerable at doses associated with a clinical benefit, with overall manageable dermatologic side effects.287 Clinical efficacy was also identified in ~30% (6/16) of the patients. In particular, one patient with anaplastic large-cell lymphoma had a prolonged partial response for >12 months, and 5 patients with either diffuse large B-cell lymphoma or multiple myeloma achieved periods of disease stabilization for varying lengths of time.
In addition to hematological cancers, a phase I/II clinical trial is also currently being conducted by the Canadian Cancer Trials Group (CCTG) in patients with advanced solid tumors characterized by DNA-repair deficiencies (NCT02719977, opened May 2016). CX-5461 thus far has been found to be well tolerated with 4 patients achieving a partial response and an additional 6 patients maintaining stable disease.299 Preliminary activity for CX-5461 has been observed in patients with homologous recombination (HR)-deficient tumors. An expansion cohort for patients with metastatic breast cancer with confirmed HR deficiency is currently open.
Sensitivity and resistance to ribosome-targeting therapy
While CX-5461 represents an exciting therapeutic option for multiple cancer types, the identification of predictive biomarkers of response to identify patients who will benefit from this therapy is essential for further clinical development. In a study utilizing a panel of ovarian cancer cell lines, sensitivity to CX-5461 was shown to be associated with a high baseline rate of Pol I transcription and higher proportion of active to inactive rDNA repeats, and independent of p53 mutation status.43,44 This is consistent with CX-5461’s mode of action in inhibiting Pol I transcription and triggering defects associated with open chromatin and replication stress at the rDNA leading to activation of p53-independent DDR.295,298 Therefore, cancer cells with a higher proportion of active rDNA are more sensitive to CX-5461-mediated nucleolar DDR and activation of cell cycle checkpoints.
Moreover, biomarkers of sensitivity to CX-5461 in ovarian cancer models include BRCA-mutated and MYC targets gene expression signatures that were found to be enriched in a subset of primary and relapsed ovarian cancer.43 As MYC is a master regulator of ribosome biogenesis, MYC-driven Pol I transcription and/or MYC-driven global transcription and replication stress may underlie sensitivity to CX-5461. In a recent genome-wide siRNA screen in ovarian cancer cells treated without or with CX-5461, a number of genes known to be important in HR DNA repair and other DNA repair pathways were identified to be synthetic lethal with CX-5461.295 The loss of DNA topoisomerase I (TOP1) was shown to cooperate with CX-5461 in inhibiting cell proliferation. TOP1 plays an important role in resolving topological stress at the rDNA loci,300,301 and TOP1 inhibitors such as topotecan have been used as a salvage therapy for relapsed ovarian cancer patients302 though their clinical use has been limited due to hematological toxicity. The combination of CX-5461 and low-dose topotecan markedly enhanced nucleolar replication stress and the DDR without enhancing DNA damage levels compared to single-agent treatment, leading to cell cycle arrest and decreased clonogenic survival of cancer cells.295 When CX-5461 and low-dose topotecan were combined to treat HGSOC tumors in vivo, significant inhibition of tumor growth occurred without observing side effects.295 This suggests that the toxicity often associated with using standard topotecan doses could potentially be lessened by using lower doses when combined with CX-5461.
CX-5461 has shown efficacy in combination with other therapies targeting ribosome biogenesis in a number of MYC-driven cancer models.290,293 PIM kinase is co-elevated in MYC-driven prostate cancer303 and is responsible for increasing MYC transcriptional activity and stability, and increasing translational activity via stimulation of phosphorylation of 4E-BP1. Elevation of PIM kinase has also been found to correlate with therapeutic resistance in prostate cancer. CX-5461 in combination with a PIM kinase inhibitor demonstrated marked therapeutic benefit compared to single-agent treatment, highlighting the promising therapeutic benefit in effective targeting of ribosome biogenesis in prostate cancer.293
It is well accepted that combinatorial targeting of multiple growth signaling pathways and/or processes required for cell growth and proliferation can prove effective in cancer treatment. Targeting of the PI3K/mTOR pathway upstream of ribosome biogenesis is not sufficient for robust clinical responses in many tumor types, due to feedback loops that maintain pathway activity and compensatory activation of RAS signaling,45 which also regulates ribosome biogenesis. Therefore, targeting the signaling networks that controls ribosome synthesis and function at multiple steps can improve therapeutic efficacy downstream of these oncogenic networks and delay the onset of acquired resistance.45 Indeed, CX-5461 in combination with everolimus, another inhibitor of mRNA translation that targets mTORC1 has shown remarkable therapeutic efficacy in the Eμ-Myc lymphoma murine model,290 with the combination therapy being well tolerated and demonstrating a prolonged survival benefit compared to single-agent therapy, with no negative effects on the wild-type B-cell population. Thus, multi-pronged targeting of ribosome synthesis and function at different steps or in combination of therapeutic targeting of upstream signaling pathways can provide substantial improvement in the efficacy of ribosome-targeting therapy. Together, the effective targeting of ribosome biogenesis and protein synthesis may prove effective in overcoming growth signaling pathway redundancies and tumor heterogeneity and can potentially be used for the treatment of a large subset of human cancer with RP gene deletions.25
In addition to activation of the nucleolar stress response and pro-apoptotic responses, inhibition of ribosome biogenesis by CX-5461 and everolimus combination therapy impairs mRNA translation capacity.304 Global translation profiling and metabolomic analysis have shown that the marked improvement in the in vivo efficacy of CX-5461 and everolimus combination therapies is associated with specific suppression of translation of mRNAs encoding regulators of cellular energetic metabolism.304 Consistent with the critical role of suppressed translation in therapeutic efficacy, acquired resistance to this co-treatment is driven by translational rewiring that results in deregulated mitochondrial respiration. Inhibition of mitochondrial function and energy biosynthesis with metformin, a widely used anti-diabetic agent, re-sensitized the resistant B-cell lymphoma cells to CX-5461 and everolimus treatment. These discoveries reveal that translational plasticity drives resistance to ribosome-directed therapy, expanding our understanding of ribosome addiction in cancer cells and reprogramming of mRNA translation and protein synthesis that drives drug resistance.
These findings have direct relevance to understanding cancer predisposition in patients with ribosomopathies and provide an understanding of the adaptive molecular alterations that enable the switch from a hypo-proliferative to hyper-proliferative state. The data substantiate an evolutionary model of ribosomopathies whereby chronically compromised ribosome biogenesis results in the evolution of subclones with altered translation of a select subset of transcripts and/or acquired mutations that promote pro-survival mechanisms including translationally driven elevated metabolism and escape from cell cycle arrest/senescence (Fig. 5).

A model of the transition from cellular hypo-proliferation to hyper-proliferation in ribosomopathies. Defects in ribosome biogenesis and ribosome function induce p53 activation via the nucleolar stress response, but also activate the p53-independent DNA damage response (DDR). Defects in ribosome biogenesis also result in the selective translation of subsets of mRNAs involved in the regulation of cellular metabolism. In turn, deregulation of metabolism leads to oxidative stress that further impairs ribosome biogenesis and ribosome function. These nucleolar and metabolic stresses result in hypo-proliferative responses including cell cycle arrest, senescence or apoptosis that parallel the hypo-proliferative phenotypes associated with ribosomopathies. Chronic deregulation of ribosome biogenesis and cellular metabolism promotes genomic instability and secondary mutations, leading to the outgrowth of clones harboring translationally driven elevated metabolism and pro-survival mechanisms that underpin the transition from hypo-proliferation to hyper-proliferation phenotypes and cancer predisposition in ribosomopathies
Conclusions and perspectives
Genetic defects such as mutations in RPs as well as therapy-induced defects in ribosome biogenesis result in hypo-proliferative phenotypes, including cell cycle arrest, senescence, or apoptosis. The impairment of ribosome biogenesis, at any step from rRNA synthesis to ribosome assembly induces p53 activation via the nucleolar stress response and activates the DDR and other p53-independent stress pathways. Thus, combination therapeutic approaches that target signaling networks at multiple steps upstream of ribosome synthesis and function can provide potent and effective treatment options for oncogene-driven cancers.
Defects in ribosome biogenesis also result in an altered pattern of mRNA translation due to impaired translation capacity and fidelity, which may also contribute to the hypo-proliferative phenotypes associated with ribosomopathies. Oxidative stress and other metabolic changes exacerbate ribosome stress and induce DNA damage, promoting secondary mutations and genomic instability. The outgrowth of clones with enhanced survival capacity due to genetic mutations, translatomic and metabolic changes under the pressure of chronically compromised ribosome biogenesis, may underpin the transition from hypo-proliferative to hyper-proliferative phenotypes. Such selection may lead to malignant transformation in ribosomopathy patients and development of resistance to ribosome-targeting therapy. Thus, systematic and comprehensive analyses of the changes in mRNA translation, genetic mutations and metabolic alterations in response to RP mutations and compromised ribosome biogenesis will enable the identification of new vulnerabilities for RP mutation-associated diseases.
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Thomson, E., Ferreira-Cerca, S. & Hurt, E. Eukaryotic ribosome biogenesis at a glance. J. Cell Sci. 126, 4815–4821 (2013).
Fromont-Racine, M., Senger, B., Saveanu, C. & Fasiolo, F. Ribosome assembly in eukaryotes. Gene 313, 17–42 (2003).
Kressler, D., Hurt, E. & Baßler, J. Driving ribosome assembly. Biochim. Biophys. Acta 6, 673–683 (2010).
Moss, T. At the crossroads of growth control; making ribosomal RNA. Curr. Opin. Genet. Dev. 14, 210–217 (2004).
Warner, J. R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24, 437–440 (1999).
Cmarko, D., Smigova, J., Minichova, L. & Popov, A. Nucleolus: the ribosome factory. Histol. Histopathol. 23, 1291–1298 (2008).
Derenzini, M., Thiry, M. & Goessens, G. Ultrastructural cytochemistry of the mammalian cell nucleolus. J. Histochem. Cytochem. 38, 1237–1256 (1990).
Fatica, A. & Tollervey, D. Making ribosomes. Curr. Opin. Cell Biol. 14, 313–318 (2002).
Grummt, I. In Progress in Nucleic Acid Research and Molecular Biology 1st Edn, Vol 62 (ed Kivie Moldave) p109-154 (Elsevier, 1999).
Henras, A. K. et al. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 65, 2334–2359 (2008).
Nazar, R. N. Ribosomal RNA processing and ribosome biogenesis in eukaryotes. IUBMB Life 56, 457–465 (2004).
Drygin, D., Rice, W. G. & Grummt, I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 50, 131–156 (2010).
Salifou, K. et al. The histone demethylase JMJD2A/KDM4A links ribosomal RNA transcription to nutrients and growth factors availability. Nat. Commun. 7, 10174 (2016).
Stefanovsky, V. Y. et al. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell 8, 1063–1073 (2001).
Kang, J. et al. Amino acid-dependent signaling via S6K1 and MYC is essential for regulation of rDNA transcription. Oncotarget 7, 48887–48904 (2016).
Sulima, S. O., Kampen, K. R. & De Keersmaecker, K. Cancer biogenesis in ribosomopathies. Cells 8, 229 (2019).
Narla, A. & Ebert, B. L. Ribosomopathies: human disorders of ribosome dysfunction. Blood 115, 3196–3205 (2010).
Farley-Barnes, K. I., Ogawa, L. M. & Baserga, S. J. Ribosomopathies: old concepts, new controversies. Trends Genet. 35, 754–767 (2019).
Aspesi, A. & Ellis, S. R. Rare ribosomopathies: insights into mechanisms of cancer. Nat. Rev. Cancer 19, 228–238 (2019).
Hannan, K. M., Sanij, E., Rothblum, L. I., Hannan, R. D. & Pearson, R. B. Dysregulation of RNA polymerase I transcription during disease. Biochim. Biophys. Acta 1829, 342–360 (2013).
Mills, E. W. & Green, R. Ribosomopathies: There’s strength in numbers. Science 358, eaan2755 (2017).
Kampen, K. R., Sulima, S. O., Vereecke, S. & De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 48, 1013–1028 (2020).
Dameshek, W. Riddle: what do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and” hypoplastic” leukemia have in common? Blood 30, 251–254 (1967).
Ajore, R. et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP53 mutations. EMBO Mol. Med. 9, 498–507 (2017).
Bywater, M. J., Pearson, R. B., McArthur, G. A. & Hannan, R. D. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. Cancer 13, 299–314 (2013).
Orsolic, I. et al. The relationship between the nucleolus and cancer: Current evidence and emerging paradigms. Semin. Cancer Biol. 37-38, 36–50 (2016).
Pelletier, J., Thomas, G. & Volarevi, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 18, 51–63 (2017).
Penzo, M., Montanaro, L., Trere, D. & Derenzini, M. The ribosome biogenesis-cancer connection. Cells 8, 55 (2019).
Turi, Z., Lacey, M., Mistrik, M. & Moudry, P. Impaired ribosome biogenesis: mechanisms and relevance to cancer and aging. Aging (Albany NY) 11, 2512–2540 (2019).
Barna, M. et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–975 (2008).
Derenzini, M. et al. Nucleolar function and size in cancer cells. Am. J. Pathol. 152, 1291–1297 (1998).
Montanaro, O., Treré, D. & Derenzini, M. The emerging role of RNA polymerase I transcription machinery in human malignancy: a clinical perspective. OncoTargets Ther. 6, 909–916 (2013).
Burger, K. et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 285, 12416–12425 (2010).
Ghoshal, K. & Jacob, S. T. Specific inhibition of pre-ribosomal RNA processing in extracts from the lymphosarcoma cells treated with 5-fluorouracil. Cancer Res. 54, 632–636 (1994).
Quin, J. E. et al. Targeting the nucleolus for cancer intervention. Biochim. et. Biophys. Acta (BBA)-Mol. Basis Dis. 1842, 802–816 (2014).
Trask, D. K. & Muller, M. T. Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D. Proc. Natl Acad. Sci. USA 85, 1417–1421 (1988).
Treiber, D. K., Zhai, X., Jantzen, H. M. & Essigmann, J. M. Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc. Natl Acad. Sci. USA 91, 5672–5676 (1994).
Bywater, M. J. et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 22, 51–65 (2012).
Catez, F. et al. Ribosome biogenesis: an emerging druggable pathway for cancer therapeutics. Biochemical Pharmacol. 159, 74–81 (2019).
Hein, N., Hannan, K. M., George, A. J., Sanij, E. & Hannan, R. D. The nucleolus: an emerging target for cancer therapy. Trends Mol. Med. 19, 643–654 (2013).
Peltonen, K. et al. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 25, 77–90 (2014).
Sanij, E. et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 11, 2641 (2020).
Son, J. et al. rDNA Chromatin Activity Status as a Biomarker of Sensitivity to the RNA Polymerase I Transcription Inhibitor CX-5461. Front. Cell Dev. Biol. 8, 568 (2020).
Yan, S. et al. The potential of targeting ribosome biogenesis in high-grade serous ovarian cancer. Int. J. Mol. Sci. 18, 210 (2017).
Moore, P. B. & Steitz, T. A. The structural basis of large ribosomal subunit function. Annu. Rev. Biochem. 72, 813–850 (2003).
Shen, P. S. et al. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 347, 75–78 (2015).
Wilson, D. N. & Doudna Cate, J. H. The structure and function of the eukaryotic ribosome. Cold Spring Harb. Perspect. Biol. 4, a011536 (2012).
Nakao, A., Yoshihama, M. & Kenmochi, N. RPG: the ribosomal protein gene database. Nucleic Acids Res. 32, D168–D170 (2004).
Trumtel, S., Leger-Silvestre, I., Gleizes, P. E., Teulieres, F. & Gas, N. Assembly and functional organization of the nucleolus: ultrastructural analysis of Saccharomyces cerevisiae mutants. Mol. Biol. Cell 11, 2175–2189 (2000).
Tschochner, H. & Hurt, E. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 13, 255–263 (2003).
Puvion-Dutilleul, F., Puvion, E. & Bachellerie, J. P. Early stages of pre-rRNA formation within the nucleolar ultrastructure of mouse cells studied by in situ hybridization with a 5’ETS leader probe. Chromosoma 105, 496–505 (1997).
Szymański, M., Barciszewska, M. Z., Erdmann, V. A. & Barciszewski, J. 5 S rRNA: structure and interactions. Biochem. J. 371, 641–651 (2003).
Anger, A. M. et al. Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85 (2013).
Panse, V. G. & Johnson, A. W. Maturation of eukaryotic ribosomes: acquisition of functionality. Trends Biochem. Sci. 35, 260–266 (2010).
Schmickel, R. D. Quantitation of human ribosomal DNA: Hybridization of human DNA with ribosomal RNA for quantitation and fractionation. Pediatr. Res. 7, 5–12 (1973).
Worton, R. G. et al. Human ribosomal RNA genes: Orientation of the tandem array and conservation of the 5′ end. Science 239, 64–68 (1988).
Henderson, A. S., Warburton, D. & Atwood, K. C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl Acad. Sci. USA 69, 3394–3398 (1972).
McStay, B. Nucleolar organizer regions: genomic ‘dark matter’ requiring illumination. Genes Dev. 30, 1598–1610 (2016).
Nemeth, A. & Langst, G. Genome organization in and around the nucleolus. Trends Genet. 27, 149–156 (2011).
McStay, B. & Grummt, I. The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 24, 131–157 (2008).
Sanij, E. et al. UBF levels determine the number of active ribosomal RNA genes in mammals. J. Cell Biol. 183, 1259–1274 (2008).
Sanij, E. & Hannan, R. D. The role of UBF in regulating the structure and dynamics of transcriptionally active rDNA chromatin. Epigenetics 4, 374–382 (2009).
Conconi, A., Widmer, R. M., Koller, T. & Sogo, J. M. Two different chromatin structures coexist in ribosomal RNA genes throughout the cell cycle. Cell 57, 753–761 (1989).
Hamperl, S. et al. Chromatin states at ribosomal DNA loci. Biochim. Biophys. Acta 1829, 405–417 (2013).
Hamdane, N. et al. Conditional inactivation of Upstream Binding Factor reveals its epigenetic functions and the existence of a somatic nucleolar precursor body. PLoS Genet. 10, e1004505 (2014).
Hung, S. S. et al. Cell cycle and growth stimuli regulate different steps of RNA polymerase I transcription. Gene 612, 36–48 (2017).
Schneider, D. A. RNA polymerase I activity is regulated at multiple steps in the transcription cycle: recent insights into factors that influence transcription elongation. Gene 493, 176–184 (2012).
Xie, W. et al. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl Acad. Sci. USA 109, 8161–8166 (2012).
Goodfellow, S. J. & Zomerdijk, J. C. B. M. Basic mechanisms in RNA polymerase I transcription of the ribosomal RNA genes. Sub-Cell. Biochem. 61, 211–236 (2013).
Diesch, J. et al. Changes in long-range rDNA-genomic interactions associate with altered RNA polymerase II gene programs during malignant transformation. Commun. Biol. 2, 39 (2019).
Ide, S., Miyazaki, T., Maki, H. & Kobayashi, T. Abundance of ribosomal RNA gene copies maintains genome integrity. Science 327, 693-696 (2010).
Li, J., Langst, G. & Grummt, I. NoRC-dependent nucleosome positioning silences rRNA genes. EMBO J. 25, 5735–5741 (2006).
Murayama, A. et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133, 627-639 (2008).
Kobayashi, T. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell Mol. Life Sci. 68, 1395–1403 (2011).
Salim, D. & Gerton, J. L. Ribosomal DNA instability and genome adaptability. Chromosome Res. 27, 73–87 (2019).
Gibbons, J. G., Branco, A. T., Yu, S. & Lemos, B. Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat. Commun. 5, 4850 (2014).
Killen, M. W., Stults, D. M., Adachi, N., Hanakahi, L. & Pierce, A. J. Loss of Bloom syndrome protein destabilizes human gene cluster architecture. Hum. Mol. Genet. 18, 3417–3428 (2009).
O’Donohue, M. F., Choesmel, V., Faubladier, M., Fichant, G. & Gleizes, P. E. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J. Cell Biol. 190, 853–866 (2010).
Kim, H. J., Maiti, P. & Barrientos, A. Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 47, 67–81 (2017).
Sylvester, J. E., Fischel-Ghodsian, N., Mougey, E. B. & O’Brien, T. W. Mitochondrial ribosomal proteins: candidate genes for mitochondrial disease. Genet. Med. 6, 73–80 (2004).
Wang, W. et al. Ribosomal proteins and human diseases: pathogenesis, molecular mechanisms, and therapeutic implications. Med. Res. Rev. 35, 225–285 (2015).
Warner, J. R. & McIntosh, K. B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell 34, 3–11 (2009).
Boulon, S., Westman, B. J., Hutten, S., Boisvert, F. M. & Lamond, A. I. The nucleolus under stress. Mol. Cell 40, 216–227 (2010).
de Las Heras-Rubio, A., Perucho, L., Paciucci, R., Vilardell, J. & LLeonart, M. E. Ribosomal proteins as novel players in tumorigenesis. Cancer Metastasis Rev. 33, 115–141 (2014).
Zhang, Y. & Lu, H. Signaling to p53: ribosomal proteins find their way. Cancer Cell 16, 369–377 (2009).
Rubbi, C. P. & Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 22, 6068–6077 (2003).
Liu, Y., Deisenroth, C. & Zhang, Y. RP-MDM2-p53 pathway: linking ribosomal biogenesis and tumor surveillance. Trends Cancer 2, 191–204 (2016).
Pestov, D. G., Strezoska, Ž. & Lau, L. F. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: Effects of Nucleolar Protein Bop1 on G1/S transition. Mol. Cell. Biol. 21, 4246-4255 (2001).
Pan, W., Issaq, S. & Zhang, Y. The in vivo role of the RP-Mdm2-p53 pathway in signaling oncogenic stress induced by pRb inactivation and Ras overexpression. PLoS ONE 6, e21625 (2011).
Haupt, Y., Maya, R., Kazaz, A. & Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 (1997).
Kubbutat, M. H., Jones, S. N. & Vousden, K. H. Regulation of p53 stability by Mdm2. Nature 387, 299–303 (1997).
Momand, J., Zambetti, G. P., Olson, D. C., George, D. & Levine, A. J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245 (1992).
Oliner, J. D. et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362, 857–860 (1993).
Macias, E. et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 18, 231–243 (2010).
Dai, M. S. & Lu, H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 279, 44475–44482 (2004).
Bai, D., Zhang, J., Xiao, W. & Zheng, X. Regulation of the HDM2-p53 pathway by ribosomal protein L6 in response to ribosomal stress. Nucleic Acids Res. 42, 1799–1811 (2014).
Lohrum, M. A., Ludwig, R. L., Kubbutat, M. H., Hanlon, M. & Vousden, K. H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3, 577–587 (2003).
Zhang, Y. et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell Biol. 23, 8902–8912 (2003).
Fahl, S. P., Harris, B., Coffey, F. & Wiest, D. L. Rpl22 loss impairs the development of B lymphocytes by activating a p53-dependent checkpoint. J. Immunol. 194, 200–209 (2015).
Dai, M. S. et al. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell Biol. 24, 7654–7668 (2004).
Jin, A., Itahana, K., O’Keefe, K. & Zhang, Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell Biol. 24, 7669–7680 (2004).
Zhang, Y. et al. Negative regulation of HDM2 to attenuate p53 degradation by ribosomal protein L26. Nucleic Acids Res. 38, 6544–6554 (2010).
Daftuar, L., Zhu, Y., Jacq, X. & Prives, C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS ONE 8, e68667 (2013).
Yadavilli, S. et al. Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair (Amst.) 8, 1215–1224 (2009).
Chen, D. et al. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene 26, 5029–5037 (2007).
Zhu, Y. et al. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol. Cell 35, 316–326 (2009).
Zhou, X., Hao, Q., Liao, J., Zhang, Q. & Lu, H. Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress. Oncogene 32, 388–396 (2013).
Singh, S. A. et al. p53-Independent cell cycle and erythroid differentiation defects in murine embryonic stem cells haploinsufficient for Diamond Blackfan anemia-proteins: RPS19 versus RPL5. PLoS ONE 9, e89098 (2014).
Zhang, X. et al. Identification of ribosomal protein S25 (RPS25)-MDM2-p53 regulatory feedback loop. Oncogene 32, 2782–2791 (2013).
Cui, D. et al. The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 33, 2225–2235 (2014).
Xiong, X., Zhao, Y., He, H. & Sun, Y. Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene 30, 1798–1811 (2011).
Sun, X. X., DeVine, T., Challagundla, K. B. & Dai, M. S. Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J. Biol. Chem. 286, 22730–22741 (2011).
Xiong, X. et al. Ribosomal protein S27-like is a physiological regulator of p53 that suppresses genomic instability and tumorigenesis. Elife 3, e02236 (2014).
Deisenroth, C. & Zhang, Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 29, 4253–4260 (2010).
Lindstrom, M. S. & Nister, M. Silencing of ribosomal protein S9 elicits a multitude of cellular responses inhibiting the growth of cancer cells subsequent to p53 activation. PLoS ONE 5, e9578 (2010).
Morgado-Palacin, L. et al. Non-genotoxic activation of p53 through the RPL11-dependent ribosomal stress pathway. Carcinogenesis 35, 2822–2830 (2014).
Donati, G., Peddigari, S., Mercer, C. A. & Thomas, G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 4, 87–98 (2013).
Horn, H. F. & Vousden, K. H. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 27, 5774–5784 (2008).
Teng, T., Mercer, C. A., Hexley, P., Thomas, G. & Fumagalli, S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Mol. Cell Biol. 33, 4660–4671 (2013).
Lindstrom, M. S., Jin, A., Deisenroth, C., White Wolf, G. & Zhang, Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol. Cell Biol. 27, 1056–1068 (2007).
Mitrea, D. M. & Kriwacki, R. W. On the relationship status for Arf and NPM1 - it’s complicated. FEBS J. 285, 828–831 (2018).
Zhang, Q. et al. The role of IMP dehydrogenase 2 in Inauhzin-induced ribosomal stress. Elife 3, e03077 (2014).
Ofir-Rosenfeld, Y., Boggs, K., Michael, D., Kastan, M. B. & Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 32, 180–189 (2008).
Fumagalli, S. et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 11, 501–508 (2009).
Golomb, L., Volarevic, S. & Oren, M. p53 and ribosome biogenesis stress: the essentials. FEBS Lett. 588, 2571–2579 (2014).
Raiser, D. M., Narla, A. & Ebert, B. L. The emerging importance of ribosomal dysfunction in the pathogenesis of hematologic disorders. Leuk. Lymphoma 55, 491–500 (2014).
Zhou, X. et al. Ribosomal proteins L11 and L5 activate TAp73 by overcoming MDM2 inhibition. Cell Death Differ. 22, 755–766 (2015).
Dai, M. S., Arnold, H., Sun, X. X., Sears, R. & Lu, H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 26, 3332–3345 (2007).
Challagundla, K. B. et al. Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol. Cell Biol. 31, 4007–4021 (2011).
Liao, J. M., Zhou, X., Gatignol, A. & Lu, H. Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene 33, 4916–4923 (2014).
Zhou, X., Hao, Q., Liao, J. M., Liao, P. & Lu, H. Ribosomal protein S14 negatively regulates c-Myc activity. J. Biol. Chem. 288, 21793–21801 (2013).
Van Riggelen, J., Yetil, A. & Felsher, D. W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301–309 (2010).
Wang, A. et al. Ribosomal protein RPL41 induces rapid degradation of ATF4, a transcription factor critical for tumour cell survival in stress. J. Pathol. 225, 285–292 (2011).
Lee, S. B. et al. Ribosomal protein S3, a new substrate of Akt, serves as a signal mediator between neuronal apoptosis and DNA repair. J. Biol. Chem. 285, 29457–29468 (2010).
Yang, H. J. et al. Phosphorylation of ribosomal protein S3 and antiapoptotic TRAF2 protein mediates radioresistance in non-small cell lung cancer cells. J. Biol. Chem. 288, 2965–2975 (2013).
Simsek, D. & Barna, M. An emerging role for the ribosome as a nexus for post-translational modifications. Curr. Opin. Cell Biol. 45, 92–101 (2017).
Dougherty, S. E., Maduka, A. O., Inada, T. & Silva, G. M. Expanding role of ubiquitin in translational control. Int. J. Mol. Sci. 21, 1151 (2020).
Draptchinskaia, N. et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 21, 169–175 (1999).
Ulirsch, J. C. et al. The genetic landscape of diamond-Blackfan anemia. Am. J. Hum. Genet. 103, 930–947 (2018).
Da Costa, L., Narla, A. & Mohandas, N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Res 7, F1000 (2018).
Engidaye, G., Melku, M. & Enawgaw, B. Diamond Blackfan anemia: genetics, pathogenesis, diagnosis and treatment. EJIFCC 30, 67–81 (2019).
Ludwig, L. S. et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 20, 748–753 (2014).
Gastou, M. et al. The severe phenotype of Diamond-Blackfan anemia is modulated by heat shock protein 70. Blood Adv. 1, 1959–1976 (2017).
Gripp, K. W. et al. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. A 164A, 2240–2249 (2014).
Arber, D. A. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Boultwood, J. et al. Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood 99, 4638–4641 (2002).
Schneider, R. K. et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat. Med. 22, 288–297 (2016).
Ebert, B. L. et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451, 335–339 (2008).
Chen, T. H. et al. Knockdown of Hspa9, a del(5q31.2) gene, results in a decrease in hematopoietic progenitors in mice. Blood 117, 1530–1539 (2011).
Schneider, R. K. et al. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 26, 509–520 (2014).
Starczynowski, D. T. et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat. Med. 16, 49–58 (2010).
Warren, A. J. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv. Biol. Regul. 67, 109–127 (2018).
Boocock, G. R. et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 33, 97–101 (2003).
Weis, F. et al. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 22, 914–919 (2015).
Menne, T. F. et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 39, 486–495 (2007).
Finch, A. J. et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 25, 917–929 (2011).
Tan, S. et al. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood 134, 277–290 (2019).
Dhanraj, S. et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood 129, 1557–1562 (2017).
Heiss, N. S. et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 19, 32–38 (1998).
Mason, P. J. & Bessler, M. The genetics of dyskeratosis congenita. Cancer Genet. 204, 635–645 (2011).
Goldfarb, K. C. & Cech, T. R. Targeted CRISPR disruption reveals a role for RNase MRP RNA in human preribosomal RNA processing. Genes Dev. 31, 59–71 (2017).
Wise, C. A. et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc. Natl Acad. Sci. USA 94, 3110 (1997).
Valdez, B. C., Henning, D., So, R. B., Dixon, J. & Dixon, M. J. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl Acad. Sci. USA 101, 10709–10714 (2004).
Gonzales, B. et al. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum. Mol. Genet. 14, 2035–2043 (2005).
Calo, E. et al. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature 554, 112–117 (2018).
Sakai, D., Dixon, J., Achilleos, A., Dixon, M. & Trainor, P. A. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat. Commun. 7, 1–13 (2016).
Vincent, M. et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet. Med. 18, 49–56 (2016).
Dauwerse, J. G. et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 43, 20–22 (2011).
Noack Watt, K. E., Achilleos, A., Neben, C. L., Merrill, A. E. & Trainor, P. A. The roles of RNA polymerase I and III subunits Polr1c and Polr1d in craniofacial development and in zebrafish models of Treacher Collins syndrome. PLoS Genet. 12, e1006187 (2016).
Lau, M. C. C. et al. Pathogenesis of POLR1C-dependent Type 3 Treacher Collins Syndrome revealed by a zebrafish model. Biochim. et. Biophys. Acta (BBA)-Mol. Basis Dis. 1862, 1147–1158 (2016).
Yelick, P. C. & Trainor, P. A. Ribosomopathies: Global process, tissue specific defects. Rare Dis. 3, e1025185 (2015).
Chao, H. & Liu, J. M. Premature Senescence in Hematopoietic Stem and Progenitor Cells in Ribosomopathy-Associated Bone Marrow Failure Syndromes. Blood 126, 298 (2015).
Jaako, P. et al. Gene therapy cures the anemia and lethal bone marrow failure in a mouse model of RPS19-deficient Diamond-Blackfan anemia. Haematologica 99, 1792–1798 (2014).
Dutt, S. et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 117, 2567–2576 (2011).
Danilova, N., Sakamoto, K. M. & Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 112, 5228–5237 (2008).
Jones, N. C. et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 14, 125–133 (2008).
Fok, W. C. et al. p53 mediates failure of human definitive hematopoiesis in dyskeratosis congenita. Stem Cell Rep. 9, 409–418 (2017).
Choesmel, V. et al. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood 109, 1275–1283 (2007).
Nicolas, E. et al. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 7, 11390 (2016).
Khajuria, R. K. et al. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 173, 90–103 e119 (2018).
Boussaid, I. et al. Integrated analyses of translatome and proteome identify the rules of translation selectivity in RPS14-deficient cells. Haematologica 106, 746–758 (2021).
Horos, R. et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 119, 262–272 (2012).
Yoon, A. et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312, 902–906 (2006).
Dinman, J. D. Pathways to specialized ribosomes: the Brussels lecture. J. Mol. Biol. 428, 2186–2194 (2016).
Tiu, G. C. et al. A p53-dependent translational program directs tissue-selective phenotypes in a model of ribosomopathies. Dev. Cell 56, 2089-2102 (2021).
Kapralova, K. et al. Inflammatory signature, oxidative stress, and DNA damage response in DBA pathogenesis. Blood 130, 2452–2452 (2017).
Pereboeva, L. et al. DNA damage responses and oxidative stress in dyskeratosis congenita. PLoS ONE 8, e76473 (2013).
Sulima, S. O. et al. Ribosomal lesions promote oncogenic mutagenesis. Cancer Res. 79, 320–327 (2019).
Willi, J. et al. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 46, 1945–1957 (2018).
Shcherbik, N. & Pestov, D. G. The impact of oxidative stress on ribosomes: from injury to regulation. Cells 8, 1379 (2019).
Sakai, D. & Trainor, P. A. Face off against ROS: Tcof1/Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development. Dev. Growth Differ. 58, 577–585 (2016).
Ravera, S. et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci. Rep. 6, 25441 (2016).
Rio, S. et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 133, 1358–1370 (2019).
Zambetti, N. A. et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell 19, 613–627 (2016).
Cretien, A. et al. Study of the effects of proteasome inhibitors on ribosomal protein S19 (RPS19) mutants, identified in patients with Diamond-Blackfan anemia. Haematologica 93, 1627–1634 (2008).
Bretones, G. et al. Altered patterns of global protein synthesis and translational fidelity in RPS15-mutated chronic lymphocytic leukemia. Blood 132, 2375–2388 (2018).
Girardi, T. et al. The T-cell leukemia-associated ribosomal RPL10 R98S mutation enhances JAK-STAT signaling. Leukemia 32, 809–819 (2018).
Rosas, M. G., Lorenzatti, A., Porcel de Peralta, M. S., Calcaterra, N. B. & Coux, G. Proteasomal inhibition attenuates craniofacial malformations in a zebrafish model of Treacher Collins Syndrome. Biochem Pharm. 163, 362–370 (2019).
Morgado-Palacin, L. et al. Partial loss of Rpl11 in adult mice recapitulates diamond-blackfan anemia and promotes lymphomagenesis. Cell Rep. 13, 712–722 (2015).
Oršolić, I. et al. Cancer-associated mutations in the ribosomal protein L5 gene dysregulate the HDM2/p53-mediated ribosome biogenesis checkpoint. Oncogene 39, 3443–3457 (2020).
Fancello, L., Kampen, K. R., Hofman, I. J., Verbeeck, J. & De Keersmaecker, K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget 8, 14462–14478 (2017).
Hofman, I. J. F. et al. RPL5 on 1p22.1 is recurrently deleted in multiple myeloma and its expression is linked to bortezomib response. Leukemia 31, 1706–1714 (2017).
Liu, Y. et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211–1218 (2017).
De Keersmaecker, K. et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 45, 186–190 (2013).
Hofman, I. J. et al. Low frequency mutations in ribosomal proteins RPL10 and RPL5 in multiple myeloma. Haematologica 102, e317 (2017).
Yang, J., Chen, Z., Liu, N. & Chen, Y. Ribosomal protein L10 in mitochondria serves as a regulator for ROS level in pancreatic cancer cells. Redox Biol. 19, 158–165 (2018).
Shi, J. et al. Biological function of ribosomal protein L10 on cell behavior in human epithelial ovarian cancer. J. Cancer 9, 745–756 (2018).
Rao, S. et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood 120, 3764–3773 (2012).
Rao, S. et al. Ribosomal protein Rpl22 controls the dissemination of T-cell lymphoma. Cancer Res. 76, 3387–3396 (2016).
Novetsky, A. P. et al. Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol. Oncol. 128, 470–474 (2013).
Nagarajan, N. et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 13, R115 (2012).
Ferreira, A. M. et al. High frequency of RPL22 mutations in microsatellite-unstable colorectal and endometrial tumors. Hum. Mutat. 35, 1442–1445 (2014).
Zheng, S. et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 29, 723–736 (2016).
Yu, L. et al. Survival of Del17p CLL depends on genomic complexity and somatic mutation. Clin. Cancer Res. 23, 735–745 (2017).
Gutierrez, C. et al. RPS15 and TP53 co-mutation drives B cell malignancy through altered translation and MYC activation in a murine model. Blood 136, 28–29 (2020).
Zhao, X. et al. Decreased expression of RPS15A suppresses proliferation of lung cancer cells. Tumor Biol. 36, 6733–6740 (2015).
Yao, Y. et al. Down-regulation of ribosomal protein S15A inhibits proliferation of human glioblastoma cells in vivo and in vitro via AKT pathway. Tumor Biol. 37, 4979–4990 (2016).
Zhang, C. et al. Knockdown of ribosomal protein S15A induces human glioblastoma cell apoptosis. World J. Surg. Oncol. 14, 1–7 (2016).
Liu, C. et al. RPS15A promotes gastric cancer progression via activation of the Akt/IKK‐β/NF‐κB signalling pathway. J. Cell Mol. Med. 23, 2207–2218 (2019).
Guo, P. et al. Ribosomal protein S15a promotes tumor angiogenesis via enhancing Wnt/beta-catenin-induced FGF18 expression in hepatocellular carcinoma. Oncogene 37, 1220–1236 (2018).
Chen, J. et al. Ribosomal protein S15A promotes malignant transformation and predicts poor outcome in colorectal cancer through misregulation of p53 signaling pathway. Int. J. Oncol. 48, 1628–1638 (2016).
Rahman, N. Realizing the promise of cancer predisposition genes. Nature 505, 302–308 (2014).
Pollutri, D. & Penzo, M. Ribosomal protein L10: from function to dysfunction. Cells 9, 2503 (2020).
Alter, B. P., Giri, N., Savage, S. A. & Rosenberg, P. S. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30 (2018).
Pabst, T. & Mueller, B. U. Transcriptional dysregulation during myeloid transformation in AML. Oncogene 26, 6829–6837 (2007).
Sasaki, M. et al. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 17, 944–951 (2011).
Fang, Z. et al. SPIN1 promotes tumorigenesis by blocking the uL18 (universal large ribosomal subunit protein 18)-MDM2-p53 pathway in human cancer. Elife 7, e31275 (2018).
Dai, M.-S., Sears, R. & Lu, H. Feedback regulation of c-Myc by ribosomal protein L11. Cell Cycle 6, 2735–2741 (2007).
Kampen, K. R. et al. Translatome analysis reveals altered serine and glycine metabolism in T-cell acute lymphoblastic leukemia cells. Nat. Commun. 10, 2542 (2019).
Valori, V. et al. Human rDNA copy number is unstable in metastatic breast cancers. Epigenetics 15, 85–106 (2020).
Wang, M. & Lemos, B. Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLoS Genet. 13, e1006994 (2017).
Stults, D. M. et al. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104 (2009).
Diesch, J., Hannan, R. D. & Sanij, E. Perturbations at the ribosomal genes loci are at the centre of cellular dysfunction and human disease. Cell Biosci. 4, 43 (2014).
Udugama, M. et al. Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc. Natl Acad. Sci. USA 115, 4737–4742 (2018).
Xu, B. et al. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 13, e1006771 (2017).
Derenzini, M. et al. Distribution of silver-stained interphase nucleolar organizer regions as a parameter to distinguish neoplastic from nonneoplastic reactive cells in human effusions. Acta Cytologica 33, 491–498 (1989).
Chan, J. C. et al. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci. Signal. 4, ra56 (2011).
Hannan, K. M., Sanij, E., Hein, N., Hannan, R. D. & Pearson, R. B. Signaling to the ribosome in cancer–It is more than just mTORC1. IUBMB Life 63, 79–85 (2011).
Jiang, B. H. & Liu, L. Z. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 102, 19–65 (2009).
Kim, E. K. & Choi, E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 (2010).
Poortinga, G. et al. MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J. 23, 3325–3335 (2004).
Hannan, R. D., Drygin, D. & Pearson, R. B. Targeting RNA polymerase i transcription and the nucleolus for cancer therapy. Expert Opin. Therap. Targets 17, 873–878 (2013).
Zhao, J., Yuan, X., Frodin, M. & Grummt, I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol. Cell 11, 405–413 (2003).
Dang, C. V. MYC on the path to cancer. Cell 149, 22–35 (2012).
Dang, C. V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 3, a014217 (2013).
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G. & Thompson, C. B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008).
Ji, H. et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 6, e26057 (2011).
Poortinga, G., Quinn, L. M. & Hannan, R. D. Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene 34, 403–412 (2015).
Poortinga, G. et al. c-MYC coordinately regulates ribosomal gene chromatin remodeling and Pol I availability during granulocyte differentiation. Nucleic Acids Res. 39, 3267–3281 (2011).
Murphy, D. J. et al. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14, 447–457 (2008).
Osborne, J. K., Zaganjor, E. & Cobb, M. H. Signal control through Raf: in sickness and in health. Cell Res. 22, 14–22 (2012).
Pylayeva-Gupta, Y., Grabocka, E. & Bar-Sagi, D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774 (2011).
Yuan, T. L. & Cantley, L. C. PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510 (2008).
Zhang, S. & Yu, D. PI(3)king apart PTEN’s role in cancer. Clin. Cancer Res. 16, 4325–4330 (2010).
Ruggero, D. & Pandolfi, P. P. Does the ribosome translate cancer? Nat. Rev. Cancer 3, 179–192 (2003).
Orgebin, E. et al. Ribosomopathies: new therapeutic perspectives. Cells 9, 2080 (2020).
Jaako, P. et al. Dietary L-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood 120, 2225–2228 (2012).
Payne, E. M. et al. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 120, 2214–2224 (2012).
Pospisilova, D., Cmejlova, J., Hak, J., Adam, T. & Cmejla, R. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica 92, e66–e67 (2007).
Sonenberg, N. & Hinnebusch, A. G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 (2009).
Furic, L. et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl Acad. Sci. USA 107, 14134–14139 (2010).
Mamane, Y. et al. eIF4E–from translation to transformation. Oncogene 23, 3172–3179 (2004).
Ruggero, D. et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat. Med. 10, 484–486 (2004).
Ear, J. et al. RAP-011 improves erythropoiesis in zebrafish model of Diamond-Blackfan anemia through antagonizing lefty1. Blood 126, 880–890 (2015).
Taylor, A. M. et al. Calmodulin inhibitors improve erythropoiesis in Diamond-Blackfan anemia. Sci. Transl. Med. 12, 5831 (2020).
Winkler, T. et al. Successful treatment of a Diamond-Blackfan anemia (DBA) patient with eltrombopag. Blood 128, 2682–2682 (2016).
Qanash, H. et al. Eltrombopag improves erythroid differentiation in a human iPSC model of Diamond Blackfan anemia. Blood 134, 1214 (2019).
Aspesi, A. et al. Lymphoblastoid cell lines from Diamond Blackfan anaemia patients exhibit a full ribosomal stress phenotype that is rescued by gene therapy. Sci. Rep. 7, 12010 (2017).
Debnath, S. et al. Lentiviral vectors with cellular promoters correct anemia and lethal bone marrow failure in a mouse model for Diamond-Blackfan anemia. Mol. Ther. 25, 1805–1814 (2017).
Greenberg, P. et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89, 2079–2088 (1997).
List, A. et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N. Engl. J. Med. 355, 1456–1465 (2006).
Fenaux, P. et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 118, 3765–3776 (2011).
Lian, X. Y. et al. Efficacy and safety of lenalidomide for treatment of low-/intermediate-1-risk myelodysplastic syndromes with or without 5q deletion: a systematic review and meta-analysis. PLoS ONE 11, e0165948 (2016).
Kronke, J. et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 523, 183–188 (2015).
Jadersten, M. et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 29, 1971–1979 (2011).
Bally, C. et al. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk. Res. 38, 751–755 (2014).
Scharenberg, C. et al. Progression in patients with low- and intermediate-1-risk del(5q) myelodysplastic syndromes is predicted by a limited subset of mutations. Haematologica 102, 498–508 (2017).
Drygin, D. et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 71, 1418–1430 (2011).
Bruno, P. M. et al. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 23, 461–471 (2017).
Kim, D. S. et al. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol. Cell 75, 1270–1285 e1214 (2019).
Fetherston, J., Werner, E. & Patterson, R. Processing of the external transcribed spacer of murine rRNA and site of action of actinomycin D. Nucleic Acids Res. 12, 7187–7198 (1984).
Santi, D. V., McHenry, C. S. & Sommer, H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 13, 471–481 (1974).
Jordan, P. & Carmo-Fonseca, M. Cisplatin inhibits synthesis of ribosomal RNA in vivo. Nucleic Acids Res. 26, 2831–2836 (1998).
Hamdane, N. et al. Depletion of the cisplatin targeted HMGB-box factor UBF selectively induces p53-independent apoptotic death in transformed cells. Oncotarget 6, 27519–27536 (2015).
Wei, T. et al. Small-molecule targeting of RNA polymerase I activates a conserved transcription elongation checkpoint. Cell Rep. 23, 404–414 (2018).
Khot, A. et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: Results of a phase I dose-escalation study. Cancer Discov. 9, 1036–1049 (2019).
Haddach, M. et al. Discovery of CX-5461, the first direct and selective inhibitor of RNA polymerase I, for cancer therapeutics. ACS Medicinal Chem. Lett. 3, 602–606 (2012).
Cornelison, R. et al. Targeting RNA-polymerase I in both chemosensitive and chemoresistant populations in epithelial ovarian cancer. Clin. Cancer Res. 23, 6529–6540 (2017).
Devlin, J. R. et al. Combination therapy targeting ribosome biogenesis and mRNA translation synergistically extends survival in MYC-driven lymphoma. Cancer Discov. 6, 59–70 (2016).
Hein, N. et al. Inhibition of Pol I transcription treats murine and human AML by targeting the leukemia-initiating cell population. Blood 129, 2882–2895 (2017).
Negi, S. S. & Brown, P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 6, 18094–18104 (2015).
Rebello, R. J. et al. The dual inhibition of RNA Pol I transcription and PIM kinase as a new therapeutic approach to treat advanced prostate cancer. Clin. Cancer Res. 22, 5539–5552 (2016).
Xu, H. et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 8, 14432 (2017).
Yan, S. et al. The RNA polymerase I transcription inhibitor CX-5461 cooperates with topoisomerase 1 inhibition by enhancing the DNA damage response in homologous recombination-proficient high-grade serous ovarian cancer. Br. J. Cancer 124, 616–627 (2020).
Domostegui, A. et al. Impaired ribosome biogenesis checkpoint activation induces p53-dependent MCL-1 degradation and MYC-driven lymphoma death. Blood 137, 3351–3364 (2021).
Negi, S. S. & Brown, P. Transient rRNA synthesis inhibition with CX-5461 is sufficient to elicit growth arrest and cell death in acute lymphoblastic leukemia cells. Oncotarget 6, 34846–34858 (2015).
Quin, J. et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget 7, 49800–49818 (2016).
Hilton, J. et al. Abstract PD4-02: Canadian cancer trials group trial IND.231: A phase 1 trial evaluating CX-5461, a novel first-in-class G-quadruplex stabilizer in patients with advanced solid tumors enriched for DNA-repair deficiencies. Cancer Res. 80, PD4-02–PD04-02 (2020).
Manzo, S. G. et al. DNA topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 19, 100 (2018).
Muller, M. T., Pfund, W. P., Mehta, V. B. & Trask, D. K. Eukaryotic type I topoisomerase is enriched in the nucleolus and catalytically active on ribosomal DNA. EMBO J. 4, 1237–1243 (1985).
McGuire, W. P., Blessing, J. A., Bookman, M. A., Lentz, S. S. & Dunton, C. J. Topotecan has substantial antitumor activity as first-line salvage therapy in platinum-sensitive epithelial ovarian carcinoma: a Gynecologic Oncology Group Study. J. Clin. Oncol. 18, 1062–1067 (2000).
Wang, J. et al. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 29, 2477–2487 (2010).
Kusnadi, E. P. et al. Reprogrammed mRNA translation drives resistance to therapeutic targeting of ribosome biogenesis. Embo J. 39, e105111 (2020).
Gazda, H. T. et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 83, 769–780 (2008).
Wen, Y., An, Z., Qiao, B., Zhang, C. & Zhang, Z. RPS7 promotes cell migration through targeting epithelial-mesenchymal transition in prostate cancer. Urologic Oncol. 37, 297.e1–297. e297 (2019).
Zhang, C. et al. Kinase PIM1 promotes prostate cancer cell growth via c-Myc-RPS7-driven ribosomal stress. Carcinogenesis 40, 52–60 (2019).
Zhang, W. et al. RPS7 inhibits colorectal cancer growth via decreasing HIF-1α-mediated glycolysis. Oncotarget 7, 5800 (2016).
Wang, Z. et al. Small ribosomal protein subunit S7 suppresses ovarian tumorigenesis through regulation of the PI3K/AKT and MAPK pathways. PLoS ONE 8, e79117 (2013).
Doherty, L. et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 86, 222–228 (2010).
Ljungström, V. et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations. Blood. J. Am. Soc. Hematol. 127, 1007–1016 (2016).
Ikeda, F. et al. Exome sequencing identified RPS15A as a novel causative gene for Diamond-Blackfan anemia. Haematologica 102, e93–e96 (2017).
Cmejla, R., Cmejlova, J., Handrkova, H., Petrak, J. & Pospisilova, D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum. Mutat. 28, 1178–1182 (2007).
Yu, C. et al. Identification of key genes and pathways involved in microsatellite instability in colorectal cancer. Mol. Med. Rep. 19, 2065–2076 (2019).
Willig, T. N. et al. Mutations in ribosomal protein S19 gene and diamond blackfan anemia: wide variations in phenotypic expression. Blood 94, 4294–4306 (1999).
Chen, K.-C. et al. Flavonoids Luteolin and Quercetin Inhibit RPS19 and contributes to metastasis of cancer cells through c-Myc reduction. J. Food Drug Anal. 26, 1180–1191 (2018).
Nieminen, T. T. et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 147, 595–598.e595 (2014).
Krishnan, R., Boddapati, N. & Mahalingam, S. Interplay between human nucleolar GNL1 and RPS20 is critical to modulate cell proliferation. Sci. Rep. 8, 1–16 (2018).
Paolini, N. A. et al. A ribosomopathy reveals decoding defective ribosomes driving human dysmorphism. Am. J. Hum. Genet. 100, 506–522 (2017).
Choesmel, V. et al. Mutation of ribosomal protein RPS24 in Diamond-Blackfan anemia results in a ribosome biogenesis disorder. Hum. Mol. Genet. 17, 1253–1263 (2008).
Wang, Y. et al. RPS24c isoform facilitates tumor angiogenesis via promoting the stability of MVIH in colorectal cancer. Curr. Mol. Med. 20, 388–395 (2020).
Wang, R. et al. Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond-Blackfan anaemia. Br. J. Haematol. 168, 854–864 (2015).
Dutton-Regester, K. et al. A highly recurrent RPS27 5’UTR mutation in melanoma. Oncotarget 5, 2912–2917 (2014).
Mirabello, L. et al. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood 124, 24–32 (2014).
Bolze, A. et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science 340, 976–978 (2013).
Wu, Y. et al. ITGA6 and RPSA synergistically promote pancreatic cancer invasion and metastasis via PI3K and MAPK signaling pathways. Exp. Cell Res. 379, 30–47 (2019).
Van Raay, T. J., Connors, T. D., Klinger, K. W., Landes, G. M. & Burn, T. C. A novel ribosomal protein L3-like gene (RPL3L) maps to the autosomal dominant polycystic kidney disease gene region. Genomics 37, 172–176 (1996).
Lezzerini, M. et al. Ribosomal protein gene RPL9 variants can differentially impair ribosome function and cellular metabolism. Nucleic Acids Res. 48, 770–787 (2020).
Baik, I. H. et al. Knockdown of RPL9 expression inhibits colorectal carcinoma growth via the inactivation of Id-1/NF-κB signaling axis. Int. J. Oncol. 49, 1953–1962 (2016).
Klauck, S. M. et al. Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol. Psychiatry 11, 1073–1084 (2006).
Brooks, S. S. et al. A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X-linked microcephaly in humans. Genetics 198, 723–733 (2014).
Uchi, R. et al. PICT1 regulates TP53 via RPL11 and is involved in gastric cancer progression. Br. J. Cancer 109, 2199–2206 (2013).
Le Caignec, C. et al. RPL13 variants cause spondyloepimetaphyseal dysplasia with severe short stature. Am. J. Hum. Genet. 105, 1040–1047 (2019).
Landowski, M. et al. Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond-Blackfan anemia. Hum. Genet. 132, 1265–1274 (2013).
Ebright, R. Y. et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science 367, 1468–1473 (2020).
Yan, T.-T. et al. Downregulation of RPL15 may predict poor survival and associate with tumor progression in pancreatic ductal adenocarcinoma. Oncotarget 6, 37028–37042 (2015).
Dong, Z. et al. Ribosomal protein L15 is involved in colon carcinogenesis. Int. J. Med. Sci. 16, 1132–1141 (2019).
Wang, H. et al. Overexpression of ribosomal protein L15 is associated with cell proliferation in gastric cancer. BMC Cancer 6, 1–8 (2006).
Mirabello, L. et al. Novel and known ribosomal causes of Diamond-Blackfan anaemia identified through comprehensive genomic characterisation. J. Med. Genet. 54, 417–425 (2017).
Zhou, C. et al. Mutation in ribosomal protein L21 underlies hereditary hypotrichosis simplex. Hum. Mutat. 32, 710–714 (2011).
Li, C. et al. RPL21 siRNA blocks proliferation in pancreatic cancer cells by inhibiting DNA replication and inducing G1 arrest and apoptosis. Front. Oncol. 10, 1730 (2020).
Cao, B. et al. Cancer-mutated ribosome protein L22 (RPL22/eL22) suppresses cancer cell survival by blocking p53-MDM2 circuit. Oncotarget 8, 90651–90661 (2017).
Gazda, H. T. et al. Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in diamond-blackfan anemia. Hum. Mutat. 33, 1037–1044 (2012).
Sim, E. U., Ang, C. H., Ng, C. C., Lee, C. W. & Narayanan, K. Differential expression of a subset of ribosomal protein genes in cell lines derived from human nasopharyngeal epithelium. J. Hum. Genet. 55, 118–120 (2010).
Maruyama, Y. et al. Short hairpin RNA library-based functional screening identified ribosomal protein L31 that modulates prostate cancer cell growth via p53 pathway. PLoS ONE 9, e108743 (2014).
Fan, H. et al. Silencing of ribosomal protein L34 (RPL34) inhibits the proliferation and invasion of esophageal cancer cells. Oncol. Res. 25, 1061–1068 (2017).
Liu, H. et al. RNAi-mediated RPL34 knockdown suppresses the growth of human gastric cancer cells. Oncol. Rep. 34, 2267–2272 (2015).
Yang, S. et al. Over-expressed RPL34 promotes malignant proliferation of non-small cell lung cancer cells. Gene 576, 421–428 (2016).
Wei, F. et al. Ribosomal protein L34 promotes the proliferation, invasion and metastasis of pancreatic cancer cells. Oncotarget 7, 85259–85272 (2016).
Ji, P. et al. Knockdown of RPL34 inhibits the proliferation and migration of glioma cells through the inactivation of JAK/STAT3 signaling pathway. J. Cell. Biochem. 120, 3259–3267 (2019).
Luo, S. et al. Highly expressed ribosomal protein L34 indicates poor prognosis in osteosarcoma and its knockdown suppresses osteosarcoma proliferation probably through translational control. Sci. Rep. 6, 1–14 (2016).
Zhu, Y. et al. RPL34‐AS1–induced RPL34 inhibits cervical cancer cell tumorigenesis via the MDM2‐P53 pathway. Cancer Sci. 112, 1811–1821 (2021).
Liu, P. Y. et al. The long noncoding RNA lncNB1 promotes tumorigenesis by interacting with ribosomal protein RPL35. Nat. Commun. 10, 1–17 (2019).
Farrar, J. E. et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood 112, 1582–1592 (2008).
Hu, Y.-W. et al. LncRNA PLAC2 down-regulates RPL36 expression and blocks cell cycle progression in glioma through a mechanism involving STAT1. J. Cell Mol. Med. 22, 497–510 (2018).
Song, M. J. et al. RPL36 as a prognostic marker in hepatocellular carcinoma. Pathol. Int. 61, 638–644 (2011).
Tian, X., Sun, D., Zhao, S., Xiong, H. & Fang, J. Screening of potential diagnostic markers and therapeutic targets against colorectal cancer. OncoTargets Ther. 8, 1691–1699 (2015).
Acknowledgements
This work was supported by the National Health and Medical Research Council (NHMRC) of Australia (Project grants #1053792 and #1162052) and Cancer Council Victoria Project Grant #1184873. R.B.P was supported by Senior Research NHMRC Fellowship #1058586. E.S. is supported by a Victorian Cancer Agency Mid‐Career Research Fellowship (MCRF19007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, J., Brajanovski, N., Chan, K.T. et al. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Sig Transduct Target Ther 6, 323 (2021). https://doi.org/10.1038/s41392-021-00728-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00728-8
This article is cited by
-
PELI1: key players in the oncogenic characteristics of pancreatic Cancer
Journal of Experimental & Clinical Cancer Research (2024)
-
Characterizing influence of rCHOP treatment on diffuse large B-cell lymphoma microenvironment through in vitro microfluidic spheroid model
Cell Death & Disease (2024)
-
Pien Tze Huang Inhibits Proliferation of Colorectal Cancer Cells through Suppressing PNO1 Expression and Activating p53/p21 Signaling Pathway
Chinese Journal of Integrative Medicine (2024)
-
Domain definition and preliminary functional exploration of the endonuclease NOBP-1 in Strongyloides stercoralis
Parasites & Vectors (2023)
-
Combinatorial interventions in aging
Nature Aging (2023)
