
Abstract
Attention-deficit/hyperactivity disorder (ADHD) is a highly heritable neurodevelopmental disorder that often persists into adulthood. There is growing evidence that epigenetic dysregulation participates in ADHD. Given that only a limited number of epigenome-wide association studies (EWASs) of ADHD have been conducted so far and they have mainly focused on pediatric and population-based samples, we performed an EWAS in a clinical sample of adults with ADHD. We report one CpG site and four regions differentially methylated between patients and controls, which are located in or near genes previously involved in autoimmune diseases, cancer or neuroticism. Our sensitivity analyses indicate that smoking status is not responsible for these results and that polygenic risk burden for ADHD does not greatly impact the signatures identified. Additionally, we show an overlap of our EWAS findings with genetic signatures previously described for ADHD and with epigenetic signatures for smoking behavior and maternal smoking. These findings support a role of DNA methylation in ADHD and emphasize the need for additional efforts in larger samples to clarify the role of epigenetic mechanisms on ADHD across the lifespan.
Similar content being viewed by others

Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a common neurodevelopmental disorder characterized by age-inappropriate levels of inattention, impulsivity and hyperactivity1. ADHD is a disabling condition in childhood and adolescence which often persists into adulthood, interfering with the quality of social, academic, or occupational functioning2,3.
ADHD is a multifactorial disorder with an estimated heritability of 76%. Twenty-two percent of its phenotypic variance is explained by common genetic variants1,4 and the proportion of variance still to be explained might be, to some extent, accounted for by gene by environment interactions. In this context, epigenetic processes have emerged as a plausible mechanism by which environmental exposures can lead to long-lasting alterations, such as variation in brain structure or neuronal circuits, found in psychiatric disorders5,6,7. There is growing evidence that epigenetic dysregulation is a feature of ADHD6,8,9,10,11, depression12, autism13,14,15,16, schizophrenia17,18 and bipolar disorder19.
Studies of DNA methylation profiles in ADHD have been conducted using peripheral blood, cord blood, buccal samples or saliva6,9,10,11,20,21,22,23,24,25,26,27,28. Candidate gene studies have revealed differential methylation patterns in genes involved in the dopaminergic, serotoninergic and neurotrophic systems, including SLC6A4, DRD4, COMT, ANKK1, BDNF, or NGFR, associated with ADHD symptomatology and severity23,24,25,26,27,28. Seven epigenome-wide association studies (EWASs) on ADHD have been run to date, with sample sizes ranging from 54 subjects for clinical samples21 to 4,689 individuals in a meta-analysis considering ADHD symptomatology in general population9, yielding non-overlapping findings across them6,9,10,11,20,21,22. There is limited research on adults using this approach, given that most of the EWASs have focused on pediatric samples6,10,11,20,22. To the best of our knowledge, only two studies evaluated methylome-wide patterns on adults9,21. One identified methylation changes associated with ADHD symptomatology that did not remain significant when results were meta-analyzed across cohorts9. The second one found hypermethylated regions in genes involved in fatty acid metabolism and fatty acid oxidation pathways associated with ADHD persistence when compared to remittance21. In the childhood period, Wilmot et al. analyzed a population cohort of school-age boys and found lower methylation levels at the VIPR2 gene in ADHD subjects compared to their age- and sex- matched controls10, results that were recently replicated in the largest EWAS on ADHD in children conducted so far22. In a similar aged population cohort, Walton et al. investigated ADHD symptom trajectories from birth to adolescence and pointed to epigenetic marks in genes related to neural tube development and peroxisomal mechanisms as candidates to be involved in the different ADHD symptom trajectories across time6. In the most recent EWAS evaluating ADHD symptoms in population-based cohorts, aberrant methylation patterns at birth in different regions, lying in the ERC2 and CREB5 genes among others, were associated with later ADHD symptoms in childhood or adolescence11. And finally, the latest and largest EWAS conducted in a clinical sample of children with ADHD supported the association between ADHD polygenic risk and DNA methylation patterns at the GART and SON genes22.
Recent evidence supports a large genetic overlap between ADHD in children and adults29, but little is known about the co-occurrence between the epigenetic signatures characterizing both groups of age. In addition, although various studies report shared genetics between ADHD and several psychiatric and behavioral traits4,29, this overlap has not been assessed yet using epigenome-wide data.
Whereas most previous studies considered pediatric clinical samples or adult population-based cohorts with measures of ADHD symptoms, we report an EWAS on a clinical sample of adults with ADHD. With these data we (i) assessed DNA methylation signatures for ADHD in adults through an EWAS in peripheral blood mononuclear cells, (ii) tested whether either polygenic risk burden for ADHD or smoking status had an impact on those DNA methylation signatures, (iii) examined whether exposure to stressful life events had an effect on these methylation patterns in ADHD subjects and (iv) explored the overlap between these findings and results from previous meta-analyses of genome-wide association studies (GWAS-MA) on clinical ADHD or ADHD symptoms in population-based samples, and EWAS on ADHD symptoms or exposure to stressful life events.
Materials and methods
Participants and clinical assessment
The clinical sample consisted of 103 ADHD subjects that were referred to an ADHD program from primary care centers and adult community mental health services. All subjects were evaluated and recruited prospectively from a restricted geographic area of Catalonia (Spain) in a specialized out-patient program for Adult ADHD and by a single clinical group at Hospital Universitari Vall d’Hebron of Barcelona (Spain).
The clinical assessment consisted of structured interviews and self-reported questionnaires in two different steps: (i) assessment of ADHD diagnosis based on symptomatology using the Conner’s Adult ADHD Diagnostic Interview for DSM-IV (CAADID) by a psychiatrist and, (ii) assessment of the severity of ADHD symptoms, the levels of impairment and the presence of comorbid disorders by a psychologist to increase the diagnostic accuracy and reduce the likelihood of misdiagnosis with the Conners ADHD Rating Scale (CAARS), the ADHD Rating Scale (ADHD-RS), the Clinical Global Impression (CGI), the Wender Utah Rating Scale (WURS), the Sheehan Disability Inventory (SDS), and the Structured Clinical Interview for DSM-IV Axis I and II Disorders (SCID-I and SCID-II). Afterwards, the psychiatrist and psychologist integrate the clinical information and self-reports for the valid assessment of symptoms and impairments. In case of discordance between different raters of ADHD symptoms or inconsistencies between reporters in responses to items measuring similar symptoms, the clinician-identified symptoms on the CAADID prevailed. Exclusion criteria were IQ < 70; lifelong and current history of mood, psychotic, anxiety, substance abuse, and DSM-IV axis II disorders; pervasive developmental disorders; a history or the current presence of a condition or illness, including neurologic, metabolic, cardiac, liver, kidney, or respiratory disease; a chronic medication of any kind; birth weight≤1.5 kg; and other neurological or systemic disorders that might explain ADHD symptoms. For more detailed information on clinical assessment see Sánchez-Mora et al.30.
Data pertaining to exposure to 17 stressful life events (six gestational and 11 postnatal) were collected retrospectively with the CAADID Part I31 and were available from 98 subjects with ADHD. No information was available from controls. Specifically, this questionnaire includes: premature birth, illegal drug abuse during pregnancy, maternal smoking, prenatal exposure to drugs, maternal health problems during pregnancy, other problems during maternal pregnancy, exposure to heavy metals, malnutrition, financial stress and/or poverty, extreme familial stress, neglect, familiar violence, emotional and physical maltreatment, sexual abuse, death or separation from a loved one, and other trauma in childhood or adolescence.
The control sample consisted of 100 unrelated healthy blood donors matched by sex and ethnicity with the clinical group. Individuals with ADHD symptomatology were excluded retrospectively under the following criteria: (1) having been diagnosed with ADHD previously or (2) answering positively to the lifetime presence of the following ADHD symptoms: (a) often has trouble in keeping attention on tasks, (b) usually loses things needed for tasks, (c) often fidgets with hands or feet or squirms in seat, and (d) often gets up from seat when remaining in seat is expected.
All subjects reported European ancestry, which was confirmed through principal component analysis (PCA) using genetic data. The study was approved by the Clinical Research Ethics Committee (CREC) of Hospital Universitari Vall d’Hebron, all methods were performed in accordance to the relevant guidelines and regulations and written informed consent was obtained from all subjects before inclusion into the study.
DNA isolation, quantification, and genome-wide DNA methylation assays
Peripheral blood mononuclear cells (PBMCs) of patients with ADHD and controls were isolated using the Ficoll density gradient method, and DNA was extracted using the QIAamp DNA Mini Kit DNA Purification following manufacturer’s instructions (Qiagen, Hilden, Germany). The quality of the samples was checked by NanoDrop® ND-1000 (Thermo Fisher Scientific, MA) and by PicoGreen® (Thermo Fisher Scientific, MA). Genome-wide DNA methylation was assessed with the Illumina Infinium MethylationEPIC BeadChip Kit (EPIC array) (Illumina, San Diego, CA, USA) following sodium bisulfite treatment of genomic DNA.
DNA methylation analysis based on ADHD diagnosis
Data preprocessing and normalization
The 203 samples included in this study were assayed in three batches, which were preprocessed and normalized separately. Raw signal intensities of each probe were extracted using the Illumina Genome Studio software (https://support.illumina.com) and were imported into the R software (3.6.0 version; https://www.R-project.org) using the minfiData 0.2 package32. The bisulfite conversion control probes and the 59 single nucleotide polymorphism (SNP) probes of the EPIC array were used to calculate the bisulfite conversion reaction efficiency and to confirm the absence of sample contamination, respectively. Sex was confirmed for all samples using the getSex function of the minfi R package33. The Horvath Epigenetic Clock algorithm34 implemented by the agep function of the wateRmelon R package was used to calculate the estimated age of participants according to their DNA methylation data, which correlated with their reported age (ρ = 0.82, SE = 0.04, P < 2.00E−16). Poorly performing probes or samples were removed using the wateRmelon R package (version 0.9.9;35). The exclusion criteria for the probes included detection P-values >0.05 for >1% of the samples and a beadcount <3 for >5% of the samples. Probes that were cross-reactive, present in sexual chromosomes or that contained polymorphisms were also excluded from the study36,37. Samples with >1% of probes with a detection P-value >0.01 were also removed. Probes that passed the quality control filters were quantile normalized with the dasen function of the wateRmelon R package.
Bioinformatic and statistical analyses
PCA of methylation values was conducted using the prcomp function of the stats R package, first separately for each batch and then across all batches. Within batch, non-biological experimental variation (Sentrix Position and chip ID) of normalized methylation values was tested for association with the Principal Component loadings (PCs). Chip ID was associated with the first PC (PC1) in all three batches, which accounted for the 99% of the variation of samples. We therefore adjusted the beta values with the ComBat function of the SVA R package38 for this variable. The effect of batch and sex on adjusted methylation values of probes present in the three batches after quality control (n = 744,227) was tested for association with the PCs estimated in the overall sample. Evidence of clustering according to batch was visually detected and statistically confirmed with a significant association of PC1 with batch (P-value < 2.20E−16).
Given that detailed smoking information was not available for each individual, an individual smoking score (continuous measure) was generated based on DNA methylation sites known to be associated with current smoking using a method developed by Elliot and colleagues39. To account for methylation differences between cell types, we estimated the cell-type composition using the estimateCellCounts function of the FlowSorted.Blood.450k R package40.
Probe-wise differential methylation analysis was performed using the lmFit function of the limma R package41. Each CpG site was tested individually in a linear regression model with normalized, corrected beta values as the dependent variable and ADHD status as independent predictor, including covariates for sex, age, batch, smoking score and cell-type composition. Age was included as covariate in all the analysis, since it was significantly different between cases and controls. Multiple testing corrections were applied using false discovery rate (FDR) with a cut-off of 5%42. The qqman R package was used to generate the Manhattan plot.
The post-hoc power analysis in our sample calculated with the EPIC array online tool (https://epigenetics.essex.ac.uk/shiny/EPICDNAmPowerCalcs/)43 using the default significance threshold (P-value < 9.42E−08) showed that 6.12% of sites had > 90% power to detect a mean methylation difference of 1%.
At the differentially methylated CpG site, we tested the association between DNA methylation and the exposure to at least one stressful life event, and to each stressful life event separately using the lmFit function of the limma R package. As 17 stressful life events were tested, Bonferroni correction was set at P < 2.94E−03. We also tested the correlation between the number of stressful life events (sum of overall stressful life events and also separated in pre- and post-natal periods) and DNA methylation levels using Spearman’s correlation.
To identify differentially methylated regions (DMRs), we used the Python module comb-p44 to group spatially correlated CpG sites with a seed of P-value < 0.01 and 500 base pairs (bp) as the maximum distance. DMR P-values were corrected for multiple testing using the Šidák correction45 and significant regions were defined as those with at least two probes and an adjusted P-value < 0.05. DMRs were mapped to genes using the interface provided by the minfi R package or the UCSC Genome Browser to identify the closest gene when no genes were mapped to a region (https://genome.ucsc.edu/cgi-bin/hgGateway).
Sensitivity analyses were conducted with the same parameters described above for the probe-wise and regional analyses excluding smoking score as covariate in the model.
DNA methylation analysis based on ADHD diagnosis controlling for ADHD polygenic burden
Bioinformatic and statistical analyses
ADHD polygenic burden was inferred using a Polygenic Risk Score (PRS) built in a subset of 195 individuals with genotype data available, from three different genotyping waves (Illumina HumanOmni1-Quad BeadChip (n = 3), Illumina HumanOmni2.5-8 BeadChip (n = 29) and Infinium™ Global Screening Array-24 v2.0 (n = 163) (Illumina, San Diego, CA, USA), using summary statistics of the largest GWAS-MA performed to date on ADHD4, with different P-value thresholds ((PT) < 1e−04, 5e−04, 0.001, 0.005, 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 1). None of the samples used in this study were included in this GWAS-MA4, and thus did not contribute to defining the variants included in the PRS.
In this subset of 195 individuals, sensitivity analyses for the differentially methylated sites and regions were conducted with the same parameters used in the original EWAS but including the PRS explaining the most variance (Nagelkerke’s R2) as an additional covariate to control for ADHD polygenic risk burden.
ADHD PRSs for each individual were generated with PRSice2 (https://choishingwan.github.io/PRSice/) including sex and the first five PCs as covariates in the model. To set an empirical threshold for the best-fit PRS, 1,000 permutations were run. Information about the pre-imputation quality control at individual and SNP level for the 195 individuals in the target sample and about the phasing and imputation software used is described elsewhere29. The European ancestry panel of the 1000 Genomes Project was considered as reference for the imputation (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/) and best guess genotypes were filtered by excluding variants with MAF < 0.05, missing rate>0.01, Hardy-Weinberg Equilibrium (P < 1.00E−06). Ambiguous strand and multiallelic variants were removed and independent SNPs (obtained using the clumping parameters p1 = 1, p2 = 1, r2 = 0.2, kb=250 in PLINK1.946) present in all individuals were included (n = 37,527).
Enrichment analyses
We assessed whether probes in different categories: (i) showing a statistically significant proportion of methylation variance explained by additive genetic effects as reported by Zeng et al.47; (ii) probes identified in previous EWASs on exposure to adverse live events48,49,50; (iii) probes identified in previous EWASs on ADHD21,22 or ADHD symptoms6,9 or (iv) probes located in ADHD-associated loci identified through GWAS4,29,51 showed, on average, a stronger association with adult ADHD than other methylation sites by regressing our EWAS test statistics (Zscore) on each CpG category as described by van Dongen et al. 9:
where |Zscore| represents the absolute value of the Zscore from our EWAS on adult ADHD, category x represents whether a CpG belongs or not to a specific category and \(\beta\)category x represents the effect estimate for that category. A CpG was assigned to a category if it was associated to the phenotype of interest according to the P-value thresholds shown in Supplementary Table 1 [excel file]. For GWAS, we considered CpG sites within windows of 10 kb, 100 kb, and 1 Mb around significant variants (Supplementary Table 1 [excel file]). For each enrichment test, bootstrap standard errors were computed with 2,000 bootstraps using the “simpleboot” R package. Bonferroni correction was applied for multiple comparison correction (PBootstrap < 3.85E−03; accounting for the 13 analyses conducted).
We also tested for enrichment of regulatory domains, ontological categories and pathways, using CpG sites with P-value<1.00E−05 in our results. For the enrichment analysis of regulatory domains, ontological categories and pathways, probes were annotated with the Illumina Human EPIC array annotation R package (“IlluminaHumanMethylationEPICanno.ilm10b2.hg19”). The enrichment analyses for transcription factor binding sites (TFBS) and DNase I hypersensitive sites (DHS) from the ENCODE project52 were performed using a two-sided Fisher’s 2×2 exact test. The enrichment analyses for GO terms and KEGG, Reactome or Biocarta pathways were assessed using the gsameth function of the missMethyl R package53. Gene sets denoting canonical pathways were downloaded from MSigDB (http://www.broadinstitute.org/gsea/msigdb), which integrates Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/), BioCarta (http://www.biocarta.com/), Reactome (https://reactome.org/) and Gene Ontology (GO) (http://www.geneontology.org/) resources.
The datasets for this article are not publicly available because of limitations in ethical approvals and the summary data will be available upon request.
Results
Our sample consisted of 103 cases and 100 controls after quality control. The distribution of sexes was not significantly different between groups (χ2 = 2.60, P = 0.11), with 56% and 45% of cases and controls being male, respectively. Age of participants was significantly different between cases and controls (P = 3.61E−04), with a mean age of 31.90 (SD = 11.45) years in cases and of 37.25 (SD = 9.47) years in controls. In the case group, 35% of participants experienced no stressful life events, 35% were exposed to at least one prenatal stressful life event and 54% were exposed to at least one of them after birth (Supplementary Table 2; Supplementary Fig. 1).
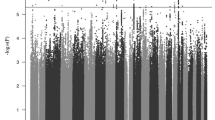
We identified one differentially methylated CpG site, cg07143296, in the EWAS (P.adj = 0.033; Fig. 1a, b; Table 1; EWAS inflation factor λ = 0.67). This CpG lies 77 bp upstream the PCNXL3 gene and was hypermethylated in patients, with a mean difference of 0.2% between groups (Table 1, Fig. 2). When evaluating the effect of prenatal and postnatal stressful life events on the methylation patterns of ADHD subjects at this CpG site, we found no significant differences in the methylation levels between individuals with ADHD exposed to stressful life events compared to those not exposed. The combined analysis of multiple correlated CpG sites showed evidence of association between ADHD and methylation levels in four genomic regions (P.adj < 0.02), with the most significant one spanning six CpG sites and located in the DENND2D gene (P.adj = 2.52E−07; Table 2). The smoking score was not significantly different between cases and controls (mean score in cases = −2.42, mean in controls = −3.34, P = 0.05). When we excluded it from the fitted model as a sensitivity analysis, cg07143296 (logFC = 0.0059, P = 1.19E−07, P.adj=0.07) and the region in chromosome 11 were no longer significant and the other regions remained significant (Table 2).

a Manhattan plot. Horizontal line indicates 5% FDR significance threshold (P-value=6.72E−08). b Quantile-quantile plot.

Boxplot showing the levels of DNA methylation in cases and controls at cg07143296.
We subsequently tested whether the polygenic risk burden for ADHD had an effect on the DNA methylation signatures. After constructing PRSs at different P-value thresholds from the largest GWAS-MA on ADHD in children and adults4, the PRS explaining the most variance in our sample was found for PT = 0.001 (NSNPs = 490, R2 = 0.052, Pperm = 0.029), and was significantly higher in ADHD patients than controls (P = 3.10E−03; Supplementary Fig. 2). After adding it as a covariate to the model fitted for the EWAS, we found that the cg07143296 CpG site (logFC = 0.066, P = 1.60E−08, P.adj=0.012) and three of the four genomic regions identified remained significant (Table 2).
We then tested whether CpG sites whose methylomic variation is mainly explained by additive genetic effects showed, on average, a stronger association with adult ADHD than other methylation sites included in the array, and found a significant enrichment of signal for adult ADHD among them (PBootstrap = 2.39E−04). In addition, when we assessed the overlap between genetic and epigenetic signatures of ADHD, we found suggestive evidence of overlap between our EWAS results and probes annotated to ADHD-associated loci in the largest GWAS meta-analyses on ADHD across the lifespan or GWAS-MA on ADHD symptoms in children (PBootstrap = 6.75E−03 and PBootstrap = 1.36E−02, respectively), but not with results of previous GWAS-MA on ADHD conducted separately in adults or children (Supplementary Table 1 [excel file]). We also considered CpG sites differentially methylated in previous EWAS on individuals exposed to adverse life events, on clinical ADHD or on ADHD symptoms and found that CpG sites previously associated with current vs never smoking and with maternal smoking showed a highly significant enrichment of signal for adult ADHD (PBootstrap = 9.03E−18 and PBootstrap = 4.62E−14, respectively) (Supplementary Table 1 [excel file]). However, no overlap was detected with findings of previous EWASs on ADHD, on ADHD symptoms and on physical/emotional neglect or abuse (Supplementary Table 1 [excel file]).
When we focused on the top 15 differentially methylated CpG sites (P < 1.00E−05) in our EWAS, we found no enrichment of regulatory domains (TFBS and DHS) from the ENCODE project52 nor ontological categories or pathways from GO terms, KEGG, Reactome or Biocarta (Supplementary Table 3 [excel file]).
Discussion
To the best of our knowledge, this is the first study evaluating DNA methylation signatures in a clinical sample of adults with ADHD and testing whether smoking status, polygenic risk burden for ADHD or exposure to stressful life events had an impact on the methylation signatures identified.
Methylation differences were found in regions that include genes related to cancer and pulmonary function (DENND2D)54,55, neuroticism and regulation of histone acetylation dynamics (PWWP2B)56,57 or regulation of immune signaling (UBASH3A)58. We also identified a CpG site (cg07143296) significantly hypermethylated in ADHD, located close to PCNXL3, a gene related to autoimmune diseases59. Although not achieving significance after multiple comparison correction, CpG sites in ADHD-related genes were found among the top ten signals of the EWAS, including CREM, which has been previously associated with impulsivity, hyperactivity, anxiety-like behavior, circadian rhythmicity and drug addiction60,61,62, ADK, whose deficiency may result in altered dopaminergic function, attentional impairment, and learning impairments63,64, or LAT, whose genetic variation has been associated with educational attainment65.
The lack of overlap between our EWAS results and those from previous EWASs on ADHD in childhood6,10,11,20,22 is in line with the fact that genome-wide DNA methylation is highly age dependent34. Contrary to some risk factors stably involved in ADHD throughout the lifespan, DNA methylation is developmental-stage specific and hence the patterns contributing to ADHD susceptibility may differ over time. The absence of overlap between our results and findings from previous EWASs on ADHD in the adulthood period9,21 could be ascribed to differences in the characteristics of the samples and on the array used (clinical vs population-based samples and EPIC vs Infinium Human Methylation 450K array9), to random variation and limited statistical power or, as previously suggested by Meijer et al.21, to the fact that the epigenetic effects identified may not be those with the strongest effect sizes on the phenotype21.
Results on the relationship between genetic and epigenetic signatures in ADHD were not conclusive. We found enrichment of signal for adult ADHD in CpGs whose methylation variance is mainly explained by additive genetic effects47 and suggestive evidence of enrichment in loci described in the largest GWAS-MA on ADHD4 and on ADHD symptoms51. However, no evidence was found for overlap between our EWAS results and loci from smaller GWAS-MAs on ADHD28 or for a substantial effect of the polygenic burden for ADHD on the methylation patterns identified. These inconsistent results should be interpreted in the context of the limited statistical power of the EWAS and warrant further investigation.
Our EWAS findings do not seem to be driven by an effect of current smoking since they were significant when we adjusted the model for it. When excluding smoking status from the model, we did not detect an effect of methylation on ADHD through smoking for cg07143296 or for the region in chromosome 11 but we cannot rule out a mediating effect for the remaining regions as their signal becomes more significant. Although bearing in mind that we used an estimated smoking score that might be a less accurate tool than clinical data, it has been postulated as a valid marker for current tobacco exposure13,39.
We also report preliminary data supporting overlap between epigenetic signatures of ADHD and smoking-related traits or behaviors. Enrichment of top-ranking CpGs from previous EWASs on smoking behavior49 or maternal smoking50 was obtained. In addition, methylation differences were identified in regions lying in or near genes (such as DENND2D or PWWP2B) related to phenotypes where tobacco exposure is a key risk factor66,67,68, and maternal smoking, which increases risk of ADHD in the offspring69,70,71, was the most frequently prenatal stressful life event reported by participants with ADHD.
To note, sixty-five percent of individuals with ADHD reported having been exposed to stressful life events, a circumstance that has been associated with the persistence of the disorder into adulthood72. Extreme familial stress was found among the most frequently reported postnatal exposures in individuals with ADHD, which is not surprising given that the presence of ADHD has been associated to varying degrees of disturbances in family and marital functioning73,74,75. However, no effect of stressful live events on DNA methylation patterns was found in ADHD subjects. Given that our study lacked data on exposure to stressful live events in controls, larger studies including cases and controls are needed to understand the impact of environmental factors on DNA methylation patterns associated with ADHD.
The results of the present study should be interpreted in the context of several limitations. First, the limited sample size of the present EWAS, which should be viewed as a pilot study whose findings await further replication. Second, our study design allowed the assessment of methylation patterns in a restricted clinical sample of medication-naïve subjects with no comorbid disorders. This design may have facilitated the identification of novel epigenetic signatures, which may not have been possible using a broader recruitment strategy. However, given that patients under medication and/or with lifetime comorbidities were excluded and this group accounts for a not negligible proportion of the overall ADHD group, further studies in larger samples including cases and controls meeting common inclusion criteria, more relaxed in terms of medication or comorbid disorders, will be required to clarify whether the results obtained could be generalized to a more realistic clinical situation. Third, the low inflation factor obtained indicates that the distribution of effect sizes in the present EWAS were not driven by systematic biases but also suggests that our study had limited statistical power and that the data may have been overcorrected, which may have prevented us from detecting methylation signatures with small effect sizes. And fourth, peripheral tissues used generally as proxies have limited utility for inferring variation in the brain76, although these novel signatures identified in blood might be used as biomarkers for the disorder.
In summary, we conducted the largest study assessing DNA methylation signatures in a clinical sample of adult patients with ADHD. Our results suggest that ADHD polygenic risk burden or current smoking status do not change substantially the methylomic variation between cases and controls, suggest an overlap between epigenetic signatures of ADHD and smoking-related traits, and point to an overlap between genetic and epigenetic signatures in ADHD. These results emphasize the need of additional efforts in larger samples and the inclusion of stressful life events in future studies to clarify the role of epigenetic mechanisms and environmental risk factors on ADHD across the lifespan.
References
Faraone, S. V. et al. Attention-deficit/hyperactivity disorder. Nat. Rev. Dis. Prim. 1, 15020 (2015).
Franke, B. et al. Live fast, die young? A review on the developmental trajectories of ADHD across the lifespan. Eur. Neuropsychopharmacol.: J. Eur. Coll. Neuropsychopharmacol. 28, 1059–1088 (2018).
Kupper, T. et al. The negative impact of attention-deficit/hyperactivity disorder on occupational health in adults and adolescents. Int. Arch. Occup. Environ. Health 85, 837–847 (2012).
Demontis, D. et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63–75 (2019).
Rutten, B. P. & Mill, J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophrenia Bull. 35, 1045–1056 (2009).
Walton, E. et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol. Psychiatry 22, 250–256 (2017).
Mill, J. & Petronis, A. Pre- and peri-natal environmental risks for attention-deficit hyperactivity disorder (ADHD): the potential role of epigenetic processes in mediating susceptibility. J. Child Psychol. Psychiatry Allied Discip. 49, 1020–1030 (2008).
Hamza, M. et al. Epigenetics and ADHD: toward an integrative approach of the disorder pathogenesis. J. Atten. Disord. 23, 655–664 (2019).
van Dongen, J. et al. Epigenome-wide association study of ADHD symptoms in adults. Biol. Psychiatry https://doi.org/10.1016/j.biopsych.2019.02.016 (2019).
Wilmot, B. et al. Methylomic analysis of salivary DNA in childhood ADHD identifies altered DNA methylation in VIPR2. J. Child Psychol. Psychiatry Allied Discip. 57, 152–160 (2016).
Neumann, A. et al. Association between DNA methylation and ADHD symptoms from birth to school age: a prospective meta-analysis. Preprint at https://www.biorxiv.org/content/10.1101/ (2019).
Li, M. et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 47(D1), D983–D988 (2019).
Hannon, E. et al. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 10, 19 (2018).
Loke, Y. J., Hannan, A. J. & Craig, J. M. The role of epigenetic change in autism spectrum disorders. Front. Neurol. 6, 107 (2015).
Sun, W. et al. Histone acetylome-wide association study of autism spectrum disorder. Cell 167, 1385–1397 e1311 (2016).
Tremblay, M. W. & Jiang, Y. H. DNA methylation and susceptibility to autism spectrum disorder. Annu. Rev. Med. 70, 151–166 (2019).
Pries, L. K., Guloksuz, S. & Kenis, G. DNA methylation in schizophrenia. Adv. Exp. Med. Biol. 978, 211–236 (2017).
Viana, J. et al. Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum. Mol. Genet. 26, 210–225 (2017).
Ludwig, B. & Dwivedi, Y. Dissecting bipolar disorder complexity through epigenomic approach. Mol. psychiatry 21, 1490–1498 (2016).
Gervin, K., Nordeng, H., Ystrom, E., Reichborn-Kjennerud, T. & Lyle, R. Long-term prenatal exposure to paracetamol is associated with DNA methylation differences in children diagnosed with ADHD. Clin. Epigenetics 9, 77 (2017).
Meijer et al. Genome-wide DNA methylation patterns in persistent attention-deficit/hyperactivity disorder and in association with impulsive and callous traits. Front. Genet 11, 16 (2020).
Mooney, M. A. et al. Large epigenome-wide association study of childhood ADHD identifies peripheral DNA methylation associated with disease and polygenic risk burden. Transl. Psychiatry 10, 8 (2020).
Heinrich, H. et al. Attention, cognitive control and motivation in ADHD: linking event-related brain potentials and DNA methylation patterns in boys at early school age. Sci. Rep. 7, 3823 (2017).
Sengupta, S. M., Smith, A. K., Grizenko, N. & Joober, R. Locus-specific DNA methylation changes and phenotypic variability in children with attention-deficit hyperactivity disorder. Psychiatry Res. 256, 298–304 (2017).
van Mil, N. H. et al. DNA methylation profiles at birth and child ADHD symptoms. J. Psychiatr. Res. 49, 51–59 (2014).
Xu, Y. et al. Multiple epigenetic factors predict the attention deficit/hyperactivity disorder among the Chinese Han children. J. Psychiatr. Res. 64, 40–50 (2015).
Dadds, M. R., Schollar-Root, O., Lenroot, R., Moul, C. & Hawes, D. J. Epigenetic regulation of the DRD4 gene and dimensions of attention-deficit/hyperactivity disorder in children. Eur. Child Adolesc. Psychiatry 25, 1081–1089 (2016).
Park, S. et al. Associations between serotonin transporter gene (SLC6A4) methylation and clinical characteristics and cortical thickness in children with ADHD. Psychological Med. 45, 3009–3017 (2015).
Rovira, P. et al. Shared genetic background between children and adults with attention deficit/hyperactivity disorder. Neuropsychopharmacology 1–10 (2020).
Sanchez-Mora, C. et al. Epigenetic signature for attention-deficit/hyperactivity disorder: identification of miR-26b-5p, miR-185-5p, and miR-191-5p as potential biomarkers in peripheral blood mononuclear cells. Neuropsychopharmacology 44, 890–897 (2019).
Epstein, J. N., Johnson, D. E. & Conners, C. K. Conners Adult ADHD Diagnostic Interview for DSM-IV. (Multi-Health Systems, North Tonawanda, NY, 1999).
Hansen, K. D., Aryee, M. & Timp, W. minfiData: example data for the Illumina Methylation 450k array. R package version 0.24.0. Bioconductor. https://doi.org/10.18129/B9.bioc.minfiData (2017).
Aryee, M. J. et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Pidsley, R. et al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC genomics 14, 293 (2013).
Chen, Y. A. et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209 (2013).
McCartney. et al. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genomics Data 9, 22–24 (2016).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Elliott, H. R. et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin. Epigenetics 6, 4 (2014).
Jaffe, A. E. FlowSorted.Blood.450k: Illumina HumanMethylation data on sorted blood cell populations. R package version 1.22.0. Bioconductor. https://doi.org/10.18129/B9.bioc.FlowSorted.Blood.450k (2019).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc.: Ser. B (Methodol.) 57, 289–300 (1995).
Mansell, G. et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC genomics 20, 366 (2019).
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 28, 2986–2988 (2012).
Šidák, Z. K. Rectangular confidence regions for the means of multivariate normal distributions. J. Am. Stat. Assoc. 62, 626–633 (1967).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Zeng, Y. et al. Parent of origin genetic effects on methylation in humans are common and influence complex trait variation. Nat. Commun. 10, 1383 (2019).
Cecil, C. A. et al. Epigenetic signatures of childhood abuse and neglect: implications for psychiatric vulnerability. J. Psychiatr. Res. 83, 184–194 (2016).
Joehanes, R. et al. Epigenetic signatures of cigarette smoking. Circulation Cardiovascular Genet. 9, 436–447 (2016).
Joubert, B. R. et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am. J. Hum. Genet. 98, 680–696 (2016).
Middeldorp, C. M. et al. A genome-wide association meta-analysis of attention-deficit/hyperactivity disorder symptoms in population-based pediatric cohorts. J. Am. Acad. Child Adolesc. Psychiatry 55, 896–905 e896 (2016).
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288 (2016).
Kanda, T., Ishii, K., Kawaguchi, H., Kitajima, K. & Takenaka, D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology 270, 834–841 (2014).
Shrine, N. et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat. Genet. 51, 481–493 (2019).
Okbay, A. et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 48, 624–633 (2016).
Zhang, T. et al. A variant NuRD complex containing PWWP2A/B excludes MBD2/3 to regulate transcription at active genes. Nat. Commun. 9, 3798 (2018).
Ge, Y., Paisie, T. K., Newman, J. R. B., McIntyre, L. M. & Concannon, P. UBASH3A mediates risk for type 1 diabetes through inhibition of T-cell receptor-induced NF-kappaB signaling. Diabetes 66, 2033–2043 (2017).
Sun, C. et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat. Genet. 48, 323–330 (2016).
Maldonado, R., Smadja, C., Mazucchelli, C. & Sassone-Corsi, P. Altered emotional and locomotor responses in mice deficient in the transcription factor CREM. Proc. Natl Acad. Sci. 96, 14094–14099 (1999).
Lahti, T. A. & Partonen, T. CREM mutations and ADHD symptoms. Med. Hypotheses 72, 544–545 (2009).
Miller, M. L. et al. Ventral striatal regulation of CREM mediates impulsive action and drug addiction vulnerability. Mol. Psychiatry 23, 1328–1335 (2018).
Boison, D. & Aronica, E. Comorbidities in neurology: is adenosine the common link? Neuropharmacology 97, 18–34 (2015).
Sandau, U. S. et al. Adenosine kinase deficiency in the brain results in maladaptive synaptic plasticity. J. Neurosci. 36, 12117–12128 (2016).
Lee, J. J. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Ling, B. et al. Suppression of non-small cell lung cancer proliferation and tumorigenicity by DENND2D. Lung Cancer 79, 104–110 (2013).
Zhang, T. et al. Downregulation of miR-522 suppresses proliferation and metastasis of non-small cell lung cancer cells by directly targeting DENN/MADD domain containing 2D. Sci. Rep. 6, 19346 (2016).
Park, S. L. et al. Mercapturic acids derived from the toxicants acrolein and crotonaldehyde in the urine of cigarette smokers from five ethnic groups with differing risks for lung cancer. PLoS ONE 10, e0124841 (2015).
Joelsson, P. et al. Prenatal smoking exposure and neuropsychiatric comorbidity of ADHD: a finnish nationwide population-based cohort study. BMC psychiatry 16, 306 (2016).
Huang, L. et al. Maternal smoking and attention-deficit/hyperactivity disorder in offspring: a meta-analysis. Pediatrics 141, e20172465 (2018).
He, C. W., Liao, C. P. & Pan, C. L. Wnt signalling in the development of axon, dendrites and synapses. Open Biol. 8, 180116 (2018).
Friedrichs, B., Igl, W., Larsson, H. & Larsson, J. O. Coexisting psychiatric problems and stressful life events in adults with symptoms of ADHD-a large Swedish population-based study of twins. J. Atten. Disord. 16, 13–22 (2012).
Eakin, L. et al. The marital and family functioning of adults with ADHD and their spouses. J. Atten. Disord. 8, 1–10 (2004).
Harpin, V. A. The effect of ADHD on the life of an individual, their family, and community from preschool to adult life. Arch. Dis. Child 90(Suppl 1), i2–i7 (2005).
Schermerhorn, A. C. et al. Offspring ADHD as a risk factor for parental marital problems: controls for genetic and environmental confounds. Twin Res. Hum. Genet. 15, 700–713 (2012).
Hannon, E., Lunnon, K., Schalkwyk, L. & Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 10, 1024–1032 (2015).
Acknowledgements
Over the course of this investigation, P.R. is a recipient of a predoctoral fellowship from the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR), Generalitat de Catalunya, Spain (2016FI_B 00899). C.S.M. is a recipient of a Sara Borrell contract from the Instituto de Salud Carlos III, Ministerio de Economía, Industria y Competitividad, Spain (CD15/00199). M.P has been a recipient of a predoctoral fellowship from the Vall d’Hebron Research Institute (PRED-VHIR-2013). L.V. is a recipient of a predoctoral fellowship from the Instituto de Salud Carlos III, Spain (FI18/00285). M.S.A. was a recipient of a contract from the Biomedical Network Research Center on Mental Health (CIBERSAM), Madrid, Spain, and now is a recipient of a Juan de la Cierva Incorporación contract from the Ministry of Science, Innovation and Universities, Spain (IJC2018-035346-I). M.R. is a recipient of a Miguel de Servet contract from the Instituto de Salud Carlos III, Spain (CP09/00119 and CPII15/00023). The research leading to these results has received funding from the European Union H2020 Programme (H2020/2014-2020) under grant agreements no. 667302 (CoCA) and no. 728018 (Eat2beNICE), from the Instituto de Salud Carlos III (PI15/01789, PI16/01505, PI17/00289, PI18/01788, PI19/00721, and P19/01224), from the Pla estratègic de recerca i innovació en salut (PERIS); Generalitat de Catalunya (MENTAL-Cat; SLT006/17/287) and from the Agència de Gestió d’Ajuts Universitaris i de Recerca-AGAUR, Generalitat de Catalunya (2017SGR1461) and cofinanced by the European Regional Development Fund (ERDF) and by “la Marató de TV3” (092330/31),. The work was also supported by the ECNP Network ‘ADHD across the Lifespan’ (https://www.ecnp.eu/research-innovation/ECNP-networks/List-ECNP-Networks/). This paper reflects only the authors’ views, and the European Union is not liable for any use that may be made of the information contained therein. The authors are grateful to patients and controls who kindly participated in this research. The Microarray-based DNA methylation profiling was performed by the Spanish “Centro Nacional de Genotipado” (CEGEN‐ISCIII) (www.cegen.org). We thank Dr. Walton for providing the list of 783 probes with P < 1.00E−03 in their EWAS on childhood ADHD symptoms. We thank Dr. Meijer for providing the lists of 723 and 658 probes with P < 1.00E−03 in their EWAS on persistent ADHD versus healthy controls and in their EWAS on persistent versus remittent ADHD, respectively. We thank Dr. Mooney for providing the list of 679 probes with P < 1.00E−03 in their EWAS on childhood ADHD. We thank Dr. Zeng and Dr. Haley for providing the list of probes of the Illumina EPIC array with a significant proportion of methylation variance explained by additive genetic effects.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Paula Rovira, Dr. Sánchez-Mora, Dr. Pagerols, Laura Vilar-Ribó, Lorena Arribas, Gemma Shireby, Dr. Hannon, Prof. Mill, Dr. Soler Artigas and Dr. Ribasés reported no biomedical financial interests or potential conflicts of interest. Vanesa Richarte has served on the speakers for Eli Lilly, Rubio and Shire in the last 5 years. She has received travel awards from Eli Lilly and Co. and Shire for participating in psychiatric meetings. The ADHD Program has received unrestricted educational and research support from Eli Lilly and Co., Janssen-Cilag, Shire, Rovi, Psious and Laboratorios Rubió in the past 2 years. Montserrat Corrales received travel awards for taking part in psychiatric meetings from Shire. Christian Fadeuilhe received fees to give talks for Shire, Ferrer, Italfarmaco and Otsuka in the last 5 years. He also received travel awards (air tickets +hotel) for taking part in psychiatric meetings from Janssen-Cilag, Rubió, Shire, Lundbeck, Otsuka and Ferrer. Prof. Casas has received travel grants and research support from Eli Lilly and Co., Janssen-Cilag, Shire and Lundbeck and served as consultant for Eli Lilly and Co., Janssen-Cilag, Shire and Lundbeck. Prof. Ramos-Quiroga was on the speakers’ bureau and/or acted as consultant for Eli-Lilly, Janssen-Cilag, Novartis, Shire, Lundbeck, Almirall, Braingaze, Sincrolab, Medice, and Rubió in the last 5 years. He also received travel awards (air tickets + hotel) for taking part in psychiatric meetings from Janssen-Cilag, Rubió, Shire, Medice and Eli- Lilly. The Department of Psychiatry chaired by him received unrestricted educational and research support from the following companies in the last 5 years: Eli-Lilly, Lundbeck, Janssen- Cilag, Actelion, Shire, Ferrer, Oryzon, Roche, Psious, and Rubió.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rovira, P., Sánchez-Mora, C., Pagerols, M. et al. Epigenome-wide association study of attention-deficit/hyperactivity disorder in adults. Transl Psychiatry 10, 199 (2020). https://doi.org/10.1038/s41398-020-0860-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-0860-4
This article is cited by
-
DNA methylation is associated with prenatal exposure to sulfur dioxide and childhood attention-deficit hyperactivity disorder symptoms
Scientific Reports (2023)
-
An overview on neurobiology and therapeutics of attention-deficit/hyperactivity disorder
Discover Mental Health (2023)
-
Comprehensive analysis of omics data identifies relevant gene networks for Attention-Deficit/Hyperactivity Disorder (ADHD)
Translational Psychiatry (2022)
-
Epigenetics and ADHD: Reflections on Current Knowledge, Research Priorities and Translational Potential
Molecular Diagnosis & Therapy (2022)
