
Abstract
Cutaneous squamous cell carcinoma (SCC) is a recurrent cancer that is prevalent in predisposed subjects such as immunosuppressed patients and patients being treated for other malignancies. Model systems to trial therapies at different stages of SCC development are lacking, therefore precluding efficient therapeutic interventions. Here, we have disrupted the expression of the tumor suppressor GRHL3 to induce loss of PTEN and activation of the PI3K/mTOR signaling pathway in mice and human skin, promoting aggressive SCC development. We then examined the potential for targeting PI3K/mTOR and an oncogenic driver miR-21, alone and in combination, for the prevention and treatment of SCC during the initiation, promotion/progression and establishment stages. Treatment with PI3K/mTOR inhibitors completely prevented tumor initiation, and these inhibitors significantly delayed the course of papilloma progression to malignancy. However, established SCC did not undergo any growth regression, indicating that this therapy is ineffective in established cancers. Mechanistically, the resistant SCCs displayed increased miR-21 expression in mice and humans where antagonists of miR-21 rescued expression levels of GRHL3/PTEN, but the combination of miR-21 antagonism with PI3K/mTOR inhibition resulted in acquired SCC resistance in part via c-MYC and OCT-4 upregulation. In conclusion, our data provide molecular evidence for the efficacy of targeting oncogenic drivers of SCC during the initiation and promotion stages and indicate that combination therapy may induce an aggressive phenotype when applied in the establishment stage.
Similar content being viewed by others


Introduction
The incidence of squamous cell carcinoma (SCC) of the skin is rising alarmingly [1] with a remarkable overrepresentation in immunosuppressed patients, especially those undergoing organ transplants [2], and patients being treated for other malignancies (e.g., melanoma therapy with B-Raf inhibitors) [3]. The success of current treatment modalities in aggressive SCC [4] is restricted not only because of tumor bulk, but also because poorly understood physiological and biological factors may contribute to the failure of targeting cancer drivers as well as resistance to therapies in some patients [5].
SCC is a multistep disease progressing over time with the accumulation of genetic defects disrupting tumor suppressor genes and inducing oncogenic proliferative advantage. Insights into SCC drivers have come mainly from chemically induced skin cancer in mice. The most efficient protocol for the genesis of mouse skin SCC is initiated by administration of the chemical carcinogen 7,12-Dimethyl-benz(a)anthracene (DMBA), inducing mutations in cancer-initiating cells (CICs), followed by repetitive applications of the tumor promoter 12-O-tetradecanoyl phorbol-13-acetate (TPA) promoting subsequent expansion of CICs [6]. Carcinogen-induced skin SCC in mice demonstrated that Ras mutation/activation is an important oncogenic event to initiate the development of papillomas and their progression to SCCs when combined with loss of tumor suppressor genes such as p53 [7]. Ras (N-, Ha-, Ki-) mutations are seen with high frequency in wild-type (WT) mouse skin SCC but only occasionally in primary human SCC [8]. Activating mutations in Ras leads particularly to engagement of extracellular signal–regulated kinase/mitogen-activated protein kinase (ERK/MAPK) and phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/mTOR) signaling pathways; [9] and it is evident that the engagement of the PI3K/mTOR pathway is most prominent in human skin SCC [6, 10]. Interestingly, PI3K/mTOR pathway activation occurs in the absence of oncogenic PI3KCA and AKT1 mutations [11] and, furthermore, human cutaneous SCCs lack somatic mutations in the PTEN gene [12].
We have developed a mouse model of Grhl3 deletion, which recapitulates PI3K/mTOR pathway activation and the multistage development of epidermal SCC [10]. Genetic deletion of a floxed allele of Grhl3 (Grhl3 fl/–) in epidermis using a keratin-14 (K14)-driven Cre (Grhl3∆/– /K14Cre +) leads to hyperproliferation of the skin, as well as head and neck tissues [10, 13]. These mice develop papillomas spontaneously, and aggressive SCC when challenged with DMBA/TPA, indicating that Grhl3 is a tumor suppressor gene. Mechanistically, downregulation of Pten, a direct transcriptional target of GRHL3, leads to activation of the PI3K/AKT/mTOR signaling and the initiation, promotion/progression and maintenance of skin SCC [10]. Interestingly, no activating hotspot mutations in Ras genes were found in Grhl3-deficient SCC, and p-ERK1/2 expression was barely detectable [10, 13]. The Grhl3 mouse model confirms studies showing that mutation/activation of Ha-Ras and complete loss of Pten are mutually exclusive in skin carcinoma [14], providing an ideal system to investigate antagonism of the PI3K/mTOR pathway.
Evidence that GRHL3 also functions as a major tumor suppressor in human skin SCC comes from studies showing that the proto-oncogenic microRNA, miR-21, inhibits expression of both GRHL3 and its target PTEN. This synchronous downregulation of two tumor suppressors by a solitary microRNA results in oncogenic hyperactivation of PI3K/mTOR signaling. Furthermore, simultaneous downregulation of GRHL3 and PTEN levels was shown to occur in over 95% of human SCC cases [10].
Based on these compelling data, two promising candidates for targeted therapy have been identified. One is the PI3K/mTOR dual-inhibitor NVP-BEZ235, a synthetic small molecular mass compound that potently inhibits both class-1 PI3K catalytic activity and mTOR catalytic activity [15]. The other is an antagonist to miR-21 (miRZip-21) [16]. The commonly used and readily accessible inhibitor BEZ235 is still in clinical trials for multiple solid cancers [17], demonstrating antitumor activity by reversing the hyperactivation of PI3K/mTOR [18]. In parallel, miR-21 is a crucial driver of SCC development in skin and head and neck tissues, and its inhibition provides an efficient therapeutic approach [10, 13].
Given the oncogenic role of both PI3K/mTOR and miR-21 in SCC of the skin, we examined the potential for targeting these drivers alone and in combination for the prevention of SCC development and treatment of established disease both in vivo, using the Grhl3/K14Cre mouse model, and in vitro, using the human skin SCC cell line SCC13.
Results
The initiation of skin SCC is prevented by BEZ235
A cohort (n = 20) of mice was administered a vehicle control or 35 mg/kg of BEZ235 by oral gavage, 3 times/week, at the tumor-initiation stage of the DMBA/TPA chemical-carcinogen protocol (Fig. 1a). DMBA was applied once at the start of the experiment followed by TPA “painting” that begun from the second week until the end of the experiment. The thrice-weekly BEZ235 administrations started in the first week alongside the solitary application of DMBA and continued for 8 weeks. Treatment of Grhl3/K14Cre mice with BEZ235 prevented tumor development in response to the carcinogens in contrast to mice receiving the vehicle control which developed papilloma and SCC as early as 7 weeks post DMBA initiation (Fig. 1b). The preventative result was further investigated in groups of Grhl3/K14Cre mice (n = 6) using BEZ235 treatment for durations of 2, 4, 8, 12 and 15 weeks (Fig. 1c) to determine the minimal treatment period that is necessary for prevention of SCC. Interestingly, treatments which started in the DMBA-initiation stage and continued for intervals as short as 4 weeks completely blocked the development of SCC for up to 22 weeks despite the concomitant ongoing exposure to TPA well beyond the 4-week BEZ235 treatment period (Fig. 1c).

BEZ235 prevented the initiation of skin SCC. a Schematic representation of the DMBA/TPA carcinogenesis protocol and BEZ235 treatment in Grhl3/K14Cre mice. A total of 25 μg DMBA dissolved in acetone was applied once topically to shaved area on the back of mice followed 1 week later by twice-weekly application of 7.6 nmol of TPA in acetone for up to 22 weeks. Red diamond represents the DMBA hit and each green diamond indicates 1 week of TPA treatment. Blue arrows correspond to BEZ235 administration. b Mice receiving carcinogens simultaneously with 35 mg/kg BEZ235 (8 weeks) by oral gavage were protected against SCC in comparison to mice receiving the vehicle control (n = 20 per group). c Administration of BEZ235 prevented SCC development from 4 weeks of treatment (n = 6 mice per group). d Genotyping of the Grhl3 floxed and deleted (∆) alleles and e mRNA expression of Grhl3 in skin from Grhl3/K14Cre mice treated with either vehicle or BEZ235. Error bars represent the SD (±SD). ***Corresponds to p-value of <0.001 using Student’s t-test
Normal epidermis from vehicle and BEZ235-treated cohorts were collected for analysis of Grhl3 expression. The ratio of allele deletion (∆ to flox) (Fig. 1d) and the mRNA level of Grhl3 (Fig. 1e) were similar in both epidermis. Multiple dorsal skin areas (~4 mm2) from vehicle and BEZ235-treated animals confirmed a consistent pattern of the deleted (∆) allele (Supplementary Fig. 1A). This indicates that the protection is due to PI3K/mTOR pathway inhibition independently of any remaining undeleted Grhl3 allele.
Furthermore, the effect of TPA on PI3K pathway activation was investigated in tumors treated with TPA only (no DMBA) in comparison to tumors that developed spontaneously (no DMBA/TPA) in Grhl3-deficient mice. Immunohistochemistry (IHC) staining was performed for p-AKT and p-S6 to assess PI3K/mTOR signaling and for p-PKCα [19] as a responsive marker to TPA treatment (Supplementary Fig. 2A). Protein kinase C-α (PKCα) is found active in tumors from both cohorts (TPA-treated and untreated) in correlation with PI3K pathway activation, which was sufficient to promote oncogene addiction to PI3K/mTOR and sensitivity to the dual-inhibitor BEZ235. IHC on papillomas and SCCs of –/+TPA groups were quantified for percentage (%) positivity (Supplementary Fig. 2B) and staining intensity of positive cells (Supplementary Fig. 2C). Together, these data indicate that baseline activation of PI3K/mTOR signaling is driving tumor development independently of TPA treatment.
BEZ235 delayed papilloma progression to malignancy
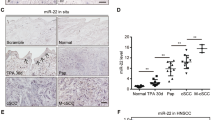
The therapeutic effect of BEZ235 on premalignant tumors was investigated during the progression stage using the DMBA/TPA model. A protocol was designed in which Grhl3-deficient mice developing more than 3 papillomas or at least 1 papilloma of >1 cm diameter were administered BEZ235 in the presence of ongoing TPA treatment, and assessed for progression to frank SCC (Fig. 2a). Interestingly, BEZ235 therapy significantly delayed the course of disease progression when compared to a group of mice receiving vehicle control (Fig. 2b). The inhibitory effect of BEZ235 on its PI3K/mTOR kinase targets was confirmed by p-S6 IHC on normal skin and papillomas from vehicle- and BEZ235-treated mice (Fig. 2c). Quantification of p-S6 positivity (Supplementary Fig. 3A) and staining intensity (Supplementary Fig. 3B) are shown as percentage of the p-S6/S6 ratio. To determine which resistant pathways evolved as an “escape” mechanism in remaining tumors, the level of miR-21 was analyzed in vehicle- and BEZ235-treated mice. Interestingly, most tumors from the BEZ235 cohort were papillomas and showed fivefold increase in miR-21 levels by real-time PCR (Fig. 2d). These findings coincide with those showing that Grhl3 loss induces upregulation of miR-21 in mice [20], and suggest that miR-21 induction following PI3K/mTOR inhibition could be involved in the maintenance of cell proliferation.

The progression of papilloma to SCC is delayed by BEZ235. a Schematic representation of the DMBA/TPA carcinogenesis protocol and BEZ235 treatment of papillomas. b BEZ235 administration (for up to 20 weeks) significantly delayed the progression of papillomas to SCC in Grhl3/K14Cre mice (n = 10) when compared to a group of mice receiving vehicle control (n = 10) (p < 0.05). The green arrow points to the start of BEZ235 administration. c IHC for p-S6 relative to total S6 shows inhibition of the PI3K/mTOR signaling in normal skin and papilloma samples from BEZ235-treated Grhl3/K14Cre mice. Scale bars correspond to 50 μm. d Real-time PCR showing miR-21 relative expression in tumors that developed on Grhl3/K14Cre mice treated with BEZ235 vs. vehicle control (N, normal skin). Mean of 3 independent experiments ± SD. *P-value < 0.05
Established SCCs are resistant to BEZ235 therapy
To trial therapies in established cancers, a cohort (n = 26) of mice was painted with DMBA/TPA until they developed frank SCC (Fig. 3a). After SCC establishment, Grhl3/K14Cre mice were separated into two groups receiving either vehicle or BEZ235 while being continuously painted with TPA. Despite displaying hyperactivation of PI3K/mTOR signaling and ERK inhibition [10], the total number of SCCs was not reduced in response to BEZ235 treatment (Fig. 3b), and these SCCs did not show any significant difference in their average size (Supplementary Fig. 3C). The inhibitory effect of BEZ235 was confirmed by IHC (Fig. 3c) and immunoblots (IBs) (Fig. 3d) for p-S6 relative to total S6 for mTOR activity and by IBs for p110α relative to p85 for PI3K activity. Quantification of p-S6 positivity (Supplementary Fig. 3D) and staining intensity (Supplementary Fig. 3E) are shown as percentage of the p-S6/S6 ratio. These data indicate that inhibitors of PI3K/mTOR, while effective in preventing SCC initiation and progression, are ineffective in treating established SCC.

Resistance of established SCC to BEZ235. a Schematic representation of DMBA/TPA carcinogenesis protocol and BEZ235 treatment of established SCC in Grhl3/K14Cre mice. These SCCs were scored macroscopically based on their thickened appearance, exophytic rounded shape and firm to hard texture with a keratinized center. b BEZ235 administration did not reduce SCC growth when compared to SCC that developed on vehicle-treated Grhl3/K14Cre mice. The green arrow points to the start of BEZ235 administration. c IHC for p-S6 shows inhibition of the PI3K/mTOR signaling in SCC samples from BEZ235-treated Grhl3/K14Cre mice. Scale bars correspond to 50 μm. d IBs using antibodies against p100α, p85, p-S6 and total S6 on SCC samples from Grhl3/K14Cre mice treated with vehicle and BEZ235 after SCC establishment. The ratio of p110α/p85 and p-S6/S6 from the SCC with the strongest signal was set to 1 as a reference and the ratio of p110α/p85 and p-S6/S6 from other SCCs was calculated relative to this reference. e Analysis of Ras (Ha- and Ki-) mutations on Exon I (Codons 12 and 13) and Exon II (Codon 61) in 6 papillomas and 6 SCC induced with DMBA/TPA in Grhl3/K14Cre mice and treated with either vehicle or BEZ235 as compared to Ras mutations in DMBA/TPA-induced tumors (papillomas) from WT mice. Mutations of Ha-Ras in WT mice are shown as positive controls
Analysis of resistance pathways in BEZ235-treated tumors
The crosstalk between Ras/ERK and PI3K/mTOR signaling is well described in human cancers [21]. Since Ras mutations are rare events in human skin SCC, primary patient samples were assessed for (active) p-ERK1/2 by IBs in comparison to their normal adjacent skin (Supplementary Fig. 3F). Although some SCC samples express p-ERK1/2 at levels similar to normal skin, most cancers display lower expression. These data are consistent with previous findings showing low percentage of mutant Ras and inactive-ERK in tumors derived from Grhl3/K14Cre mice [10].
Papillomas and SCCs treated with vehicle or BEZ235 after establishment (Fig. 3a) were sequenced for Ras mutations to determine whether the resistance to BEZ235 is occurring by a positive selection for specific mutations. No Ras mutations were found in either tumor series (Fig. 3e). These tumors were then analyzed for mechanisms involved in the evolution of skin SCC (Fig. 4a). The tumor pathology was histologically confirmed using hematoxylin and eosin (H&E) staining. p-ERK was absent in all Grhl3-deficient tumors suggesting that ERK inactivation could be due to a transcriptional control preventing a feedback response. The expression of hypoxia-inducible factor-1α (HIF-1α), which is associated with tumor growth and metastasis in cutaneous SCC [22], was unchanged, and the lack of active-Caspase-3 indicated no increased apoptosis. Since skin SCC is believed to originate from a cancer stem cell (CSC) [23], we examined whether resistance to BEZ235 could be caused by inducing CSC factors. IHCs were performed against the stem cell factors c-Myc, Sox-2, Oct-4 and Nanog [24]. c-Myc was upregulated in BEZ235-treated SCC supporting previous data where c-Myc induction conferred resistance to BEZ235 [25]. The staining intensity of the CSC-initiating factor Sox-2 [26] was weak in BEZ235-resistant papillomas and SCCs in comparison to Oct-4. However, Nanog was not detectable in those tumors. Metamorph quantification is shown for c-Myc and Oct-4 by the percentage of positive cells and staining intensity (Fig. 4b) indicating an increased expression in BEZ235-treated tumors. The IHC quantification of p-ERK1/2, Sox-2, HIF-1α, active-Caspase-3 and Nanog (Supplementary Fig. 4A and B) show absent/low expression for p-ERK, active-Caspase-3 and Nanog in all groups. However, the expression of HIF-1α was not affected in the BEZ235 cohort and only BEZ235-treated papillomas showed significant increase of Sox-2. These preliminary observations, particularly for c-Myc and Oct-4 expression, suggest that mechanisms allowing CSCs to resist treatment may play important roles in the evolution of skin SCC [27].

Analyses of pathway activation in BEZ235-resistant tumors. a H&E staining (×20 magnification) and IHC (×40) using antibodies against c-Myc, p-ERK, HIF-1α, active-Caspase-3, Sox-2, Oct-4 and Nanog on papillomas and SCCs from vehicle- and BEZ235-treated Grhl3/K14Cre mice. Scale bars, 200 μm (white) and 50 μm (black). b Metamorph quantification of c-Myc and Oct-4 IHC represented by percentage positive cells and staining intensity showing increased expression in papillomas and SCC treated with BEZ235. *P < 0.05 and **p < 0.01; n.s. statistically not significant
Targeting the miR-21 oncogenic driver in human SCC13 cells
Human SCC13 cells originating from a skin SCC and containing a homozygous point mutation in p53 alleles [28] were used to address the resistance mechanism. No Ras mutation has been reported in this line. We have previously shown that SCC13 illustrates GRHL3-dependent SCC with high levels of miR-21 and reduced GRHL3 and PTEN expression, and hyperactivation of PI3K/mTOR signaling [10]. Here, a miR-21-inhibiting lentiviral vector (miRZip-21) was trialed as a therapeutic approach [10]. We found that miRZip-21 significantly reduced miR-21 expression (Fig. 5a), leading to rescued expression of GRHL3 and PTEN and inhibition of PI3K/mTOR signaling (Fig. 5b). In parallel, the inhibition of miR-21 significantly decreased cellular proliferation in two-dimensional (2D) cell cultures (Fig. 5c). We have also analyzed the enrichment for ALDH+ cells as a preliminary read-out of CSC number [29] and found that miR-21 inhibition reduced ALDH+SCC13 with stem cell properties by fourfold (Fig. 5d), correlating with the antiproliferative effect.

miR-21 inhibition decreases PI3K/mTOR-dependent SCC proliferation. a Quantitative PCR (Q-PCR) shows miRZip-21 inhibition of miR-21 expression in human skin SCC13. b IBs using antibodies against protein of the PI3K and ERK pathways in SCC13 transduced with either Scr or miRZip-21. The expression level, relative to loading controls (HSP70 and Actin), in SCC13 was set to 1 as a reference and the expression in SCC13+miRZip-21 was calculated relative to this reference. c miRZip-21 reduced SCC13 proliferation in vitro over a period of 8 days. Error bars represent ± SD. d Percentage of ALDH+ cells in SCC13 transduced with miRZip-21. Error bars represent the SEM ( ± SEM). *p < 0.05 and **p < 0.01
The combination of BEZ235 with miRZip-21 does not confer benefit against human SCC proliferation
SCC13 were firstly treated with either BEZ235 or NVP-BKM120 (a pan-class-1 PI3K inhibitor), at 1 μM each. Both inhibitors were able to significantly reduce cellular proliferation (Supplementary Fig. 5A). Following this, we minimized the concentrations of BEZ235 to reduce toxicity and to establish a dose-dependent response. We found that BEZ235 induced growth arrest at low concentration (10 nM) (Fig. 6a), which was associated with a complete inhibition of p-S6 and a feedback loop inducing p-AKT (Fig. 6b). Similar results were obtained with BKM120 (Supplementary Fig.5B and C). This suggests that BEZ235 treatment inhibits SCC13 proliferation by specific blockage of PI3K/mTOR signaling that is unlikely due to any off-target effects.

Combination of BEZ235 and miRZip-21 in human skin SCC13. a The dose-dependent response of SCC13 treated with BEZ235 (10, 100, 1000 nM) shows significant reduction in cell number in comparison to vehicle-treated cells. Error bars represent ± SD. **P < 0.01 at day 8. b The therapeutic result shown at day 4 in (a green arrow) correlates with p-S6 inhibition using BEZ235 at the stated concentrations, with induction of p-AKT at 10 nM as a feedback resistance mechanism. c Organotypic 3D cultures of SCC13 treated with either miRZip-21, BEZ235 or both. IHC for Oct-4, c-Myc, Sox-2 and Nanog showed that miR-21 inhibition reverses the multilayer growth of SCC13 in a similar fashion to BEZ235 treatment. Expression of Oct-4 was specifically maintained in the combination therapy, which induced cell-intrinsic advantage growth of SCC cells on mouse embryonic fibroblasts in vitro. Scale bars correspond to 100 μm
We next examined whether using anti-miR-21 and BEZ235 would be more effective in combination than either treatment alone. miRZip-21-transduced SCC13 cells were treated with BEZ235 (at 10 nM) in 2D cultures. While the combination therapy led to complete blockade of PI3K/mTOR signaling (Supplementary Fig. 5D), it was no more effective at inhibiting cellular proliferation than BEZ235 alone (Supplementary Fig. 5E).
To determine whether these effects are cell intrinsic, we grew organotypic three-dimensional (3D) cultures on a matrix layer of fibroblasts in the presence of either miRZip-21, BEZ235 or both. The monotherapies each reduced SCC13 proliferation in 3D cultures. However, combined treatment (BEZ235+miRZip-21) did not show any synergistic effect. The lack of synergy was supported by H&E staining showing high number of resistant cells (Supplementary Fig. 5F). IHCs show that expression of Oct-4 was specifically maintained in the combination therapy (Fig. 6c).
Resistance of skin SCC to anti-PI3K/mTOR and anti-miR-21 therapy in vivo
SCC13 cells transduced with either a Scr control or with miRZip-21 were injected subcutaneously into the flank of immunocompromised NSG mice. After 4 weeks, when tumors of >1 cm diameter had developed, both Scr and miRZip-21 cohorts of mice were gavaged with BEZ235 (35 mg/kg), 3 times/week for 4 weeks. We found that Scr-containing xenografts treated with BEZ235 alone and miRZip-21-containing xenografts grew slower in comparison to untreated tumors (Fig. 7a). Surprisingly, the miRZip-21-containing xenografts treated with BEZ235 not only increased tumor weight (Fig. 7a) but also tumor volume (Fig. 7b), and these xenografts showed increased growth and invasion into the mice dermis. These data confirm the resistance to therapies in vivo and the acquisition of an aggressive behavior. Tumors from different treatment groups were further analyzed for markers of invasive growth such as Podoplanin, which was highly expressed in the combination therapy (Fig. 7c) as confirmed by the quantification of positive cells (Fig. 7d) and staining intensity (Fig. 7e). Our results indicate that each inhibitor reduces cancer growth but when used in combination they lead to activation of an alternative molecular program that accelerates SCC development.

SCC13 xenografts are resistant to the combination of BEZ235 and miRZip-21 therapy. a Tumor weights and b volumes of SCC13 xenografts treated with either miRZip-21, BEZ235 or the combination of both over a period of 4 weeks in NSG mice. c IHC with anti-Podoplanin on xenograft samples from different mice. Scale bars correspond to 50 μm. Metamorph quantification of Podoplanin IHC represented by d percentage positive cells and e staining intensity shows significant increase of Podoplanin expression in xenografts of SCC13 treated with the combination of miRZip-21 and BEZ235 but not with single inhibitor. *P < 0.05 and **p < 0.01
Furthermore, the xenografts maintained the parental signature with low miR-21 and increased GRHL3 and PTEN mRNA in miRZip-21-transduced cells (Supplementary Fig. 6A). SCC13 treated with BEZ235 alone showed significant increase of miR-21 similar to what is observed in SCCs from BEZ235-treated Grhl3/K14Cre mice. Also, the expression of GRHL3 and PTEN is similar in cells treated with either miRZip-21 or with both inhibitors. When assessed by IHC, these tumors did not show differences in p-AKT, p-S6 or p-ERK expression regardless of treatment (Supplementary Fig. 7A). Their number of positive cells and staining intensity was not significantly different in combination treatment (Supplementary Fig. 7B) as was also the case for HIF-1α, Sox-2 and Nanog (Supplementary Fig. 8A). Interestingly, Oct-4 was found to be upregulated in SCC13 treated with both miRZip-21 and BEZ235 (Supplementary Fig. 8A), suggesting that this factor could be driving resistance. The IHC quantification (Supplementary Fig. 9A and B) showed significant increase in Oct-4 expression. Moreover, IBs (Supplementary Fig. 9C) confirmed our mouse in vivo observations that SCCs treated with either miRZip-21 or BEZ235 induced high levels of c-Myc, which was further increased in BEZ235+miRZip-21 combination-treated cells. We also analyzed the expression profile of epithelial (E-cadherin)-to-mesenchymal (Vimentin) transition markers as well as markers for differentiation (K1, K10 and Involucrin) and activation (K6) of keratinocytes (Supplementary Fig. 10A), which demonstrated increased miRZip-21-dependent cellular differentiation as reflected in the IHC quantification (Supplementary Fig. 11A and B). Combined, our data suggest that miRZip-21 induced differentiation and slowed proliferation of SCC13 cells in vivo, but when treated in combination with BEZ235 the cells upregulate c-Myc and Oct-4 to become resistant and continue to grow.
Discussion
The present study focuses on two oncogenic drivers: (1) PI3K/AKT and (2) miR-21, which are two of the most potent oncogenes involved in the pathogenesis of a wide range of human cancers [30, 31]. Both oncogenes are known to converge on the mTOR complex [32]; however, despite novel and more specific inhibitors of mTOR being evaluated in preclinical trials, making the translation to the clinic has thus far been challenging due to the lack of animal models that accurately mimic the human setting [33]. Studies investigating inhibitors of PI3K/mTOR signaling at specific stages of SCC evolution are emerging. For example, the mTOR inhibitor rapamycin was recently shown to prevent ultraviolet (UV)-induced cutaneous SCC when applied following exposure to solar-stimulated light; in contrast, the application of rapamycin during UV exposure actually increased tumor yields [34]. This suggests that care needs to be given to the timing of drug administration for stage-dependent therapeutic efficacy against SCC. In line with this concept, using the Grhl3/K14Cre mouse model, we found that the dual PI3K/mTOR inhibitor BEZ235 exerts a continued robust preventative effect against SCC development when administered in conjunction with chemical carcinogens. These data confirmed oncogenic addiction to PI3K/mTOR signaling and suggests that this preventative therapy might be acting directly on DMBA-initiated cells [35] and/or indirectly on the surrounding microenvironment to repress the propagation of CICs [36]. It is also consistent with the timing of epidermal stem cell activation and tissue renewal of around 4 weeks [37] considering that BEZ235 treatment for 4 weeks was sufficient to abort tumor initiation. These data recapitulate re-expression of PTEN that was shown to reproduce the same effect by inhibiting PI3K/mTOR signaling in Grhl3-deficient keratinocytes [10]. Interestingly, despite having a distinct molecular signature from human cancers, chemically induced DNA insult in mice was shown to result in the selection of different activating variants of the same oncogenes as seen in human tumors [7]. Therefore, extended investigations into the role of next-generation PI3K/mTOR inhibitors in preventing SCC emergence could provide preventative approaches in predisposed subjects, and against the recurrence of SCC in patients with primary cancers displaying the Grhl3 signature.
When BEZ235 was administered during the premalignant stage, a significant delay in the course of cancer evolution was achieved. This suggests that BEZ235 is able to restrain the malignant potential of specific CIC populations. However, clone(s) of resistant cells with alternative drivers might have emerged eventually progressing into malignancy [38,39,40,41].
Despite efficient inhibition of the PI3K/mTOR pathway, BEZ235 did not affect the growth of established SCC in Grhl3/K14Cre mice. These studies clearly underscore the notion that blocking and delaying tumorigenesis are not equivalent to treating a cancer that is already established. Further analyses will determine whether combined inhibitors modulate specific mutant genes, gene copy-number alterations and the acquisition of chromosomal rearrangements that are associated with malignant progression [7] and whether using BEZ235 in combination with inhibitors that act downstream of c-Myc and Oct-4 such as the RNA Polymerase I inhibitor (CX-5461) [42, 43] would have better therapeutic potentials.
The established Grhl3-deficient tumors that were resistant to BEZ235 showed increased miR-21 levels suggesting that miR-21 could be promoting drug resistance. Interestingly, blocking miR-21 in SCC13 reduced the number of ALDH+ cells. This was predicted as miR-21 inhibition was shown to reduce CSC numbers and to enhance chemosensitivity in tongue SCC [44], lung adenocarcinoma [45], glioblastoma [46], leukemia [47] and other cancers [48]. Combination of both inhibitors was then hypothesized to provide a cumulative benefit similar to the effective combination of PI3K/mTOR inhibitors in reducing T-cell leukemia growth [49]. Surprisingly, combined inhibition of miR-21 and PI3K/mTOR was not beneficial against human skin SCC despite demonstrated PI3K/mTOR pathway inhibition, mirroring other interventions that had controversial effects such as hypoxia-induced invasion [50] and anti-angiogenic therapy-induced progression of tumors to distant metastasis [51].
The combination therapy induced a cell-intrinsic resistance phenotype in organotypic 3D -cultures of SCC13. Podoplanin, a key factor in tissue remodeling [52] and controlling the initial steps of SCC invasion [53], was expressed in 3D cultures of dual inhibitor-resistant SCC13 and also in vivo when xenografted into NSG mice. A recent study using neurospheres found a similar mechanism of evasive resistance in which PI3K inhibitor therapy reprograms mitochondrial trafficking to fuel tumor cell invasion [54], supporting our findings. The resistance to PI3K/mTOR inhibition was shown through c-MYC upregulation in human cancer cells treated with BEZ235 [55]. In our Grhl3 model, while c-Myc overexpression is not required for skin SCC development, it is induced in response to the inhibition of either miR-21 or PI3K/mTOR. In addition, Oct-4 was also overexpressed in resistant SCC mimicking its induction in acquired resistance to cisplatin, warranting further investigations on the role of these stem cell factors in SCC therapies [56].
Together, our data provide molecular evidence for the efficacy of preventing SCC initiation using PI3K/mTOR inhibitors. These inhibitors can also significantly delay the progression of benign tumors to malignant SCC. However, contrary to the majority of combination therapies, inhibitors that target the PI3K/mTOR pathway in combination with anti-miR-21 may induce an aggressive phenotype in established SCC. Further insights into rewiring mechanisms of the adaptive resistance could ultimately be used to test potential combination therapies together with next-generation PI3K/mTOR inhibitors in a stage-specific manner.
Materials and methods
Mouse models
Age-matched (6–10 weeks old) Grhl3∆/– /K14Cre + mice (C57Bl/6 background) were treated for tumor development using the skin carcinogenesis protocol of DMBA/TPA, as previously described [10]. The concentration of NVP-BEZ235 from Novartis (Basel, Switzerland) was determined based on the maximum tolerance dose and treatments were performed by oral gavage at 35 mg/kg at intervals of 3 times/week. When tumors reached a critical ethical size (1000 mm3), the animals were killed and tumors excised and sampled for gene expression analyses, histology, IHC and IB. Mutational analysis of Exon I (Codons 12 and 13) and Exon II (Codon 61) of Ha-Ras and Ki-Ras genes was performed on 6 papillomas and 6 SCC induced with DMBA/TPA and treated with either vehicle or BEZ235 in Grhl3-deficient mice (cKO) together with tumors from WT mice as previously described [10]. Animal housing, handling and all procedures involving mice were approved by Monash University, The Peter MacCallum Cancer Centre and The University of Melbourne animal Ethics Committees, in compliance with the Australian national regulations for experimental animal studies.
Human skin SCC samples
Primary samples of human skin SCC were supplied by the Victorian Tissue Biobank. The protocol of sample collection was approved by the Human Ethics Committees of the Alfred Hospital and the Royal Melbourne Hospital.
Reverse transcription and quantitative PCR
Normal and tumor samples were homogenized in TRIzol (Invitrogen) and RNA extracted according to the manufacturer’s instructions. Quantitative real-time PCR was carried out as detailed previously [10].
Histology, immunohistochemistry and immunoblotting assays
For histology, normal and tumor skin samples were fixed in 4% paraformaldehyde overnight at 4 °C, embedded in paraffin and stained with H&E. For IHC, paraffin-embedded samples were sectioned at 6 μm onto Superfrost-Plus slides and processed as per standard protocols using DAB staining. Primary antibodies were purchased from Covance (CA, USA) and were used at the following dilutions: for K1, K6, K10, Oct-4 and Podoplanin (1:1000); from Cell Signaling (MA, USA) for Nanog, E-cadherin, Vimentin, Involucrin, S6 and p-S6, PTEN, HIF-1α, Sox-2, active-Caspase-3, p85, p110-α, p27 and ERK1/2, p-ERK1/2 (1:1000), 4EBP1, p-4EBP1, AKT, p-AKT (S473) and p-AKT (T308) (1:500); from Santa Cruz (CA, USA) for HSP70 (1:3000), Actin (1:1000), c-Myc (1:500); and from Aviva Systems Biology (CA, USA) for Grhl3 (1:2000). IHC quantification was determined using the Metamorph (Molecular Devices) image analysis software on representative tumor sections (from at least 3 different tumors). Five to eight images from each tumor microscopic field at ×40 magnification were blindly loaded into Metamorph where positive (brown-staining) cells for p-S6, p-AKT, p-PKCα, p-ERK, c-Myc and Sox-2 were counted relative to nuclei and the number of cells staining with the corresponding total unphosphorylated antibodies. From all treatment groups, the brightest staining was acquired to find the fixed exposure time, and the rest of the pictures were taken at the same exposure time to determine the intensity of staining. The Y-axes show the percentage positive cells or the percentage maximum staining intensity. IBs and quantification of protein expression were performed as previously described [57].
2D and 3D cell cultures and treatments
The skin-derived SCC13 cells were grown in K-SFM media (Gibco, ThermoFisher Scientific, USA) without any serum, and supplemented with growth factors (epidermal growth factor and bovine pituitary extract) and 1% of penicillin/streptomycin solution from Gibco (ThermoFisher Scientific,USA). All cultures were in a humidified atmosphere of 95% O2 and 5% CO2 at 37 °C. Cells grown in 2D were divided into four groups: transduced with either Scr control or miRZip-21, and untreated and treated with BEZ235 (or BKM120) concentrations as indicated in the figures. Protocols for Scr and miRZip-21 viral transductions as well as determining cell proliferation were performed as previously described [10, 13]. For organotypic 3D cultures, SCC13 cells transduced with either Scr or miRZip-21 were cultured on a fibroblast matrix layer [58]. The fibroblasts were first seeded in 6-well plates (Transwell Carrier, Organogenesis, TS01-001) containing 6 inserts (Corning Costar Transwell 3414). Following that, 0.5 × 106 SCC cells were cultured on top of the fibroblast layer for 5 days. The medium was changed every 2 days and the treatment was performed using 10 nM BEZ235 for a period of 6 days to allow growth of resistant SCC13 cells. ALDH labeling experiments were measured using the AldeRed live cell-permeable dye reagent (Life Sicence Research). According to the manufacturer’s instructions, CSCs in SCC13 were identified and isolated by flow cytometry using the AldeRed fluo, which is a red-shifted fluorescent substrate for ALDH.
SCC cell grafting and in vivo treatments
3 × 106 SCC13 cells, transduced with Scr or miRZip-21, were injected subcutaneously into the flank of non-obese diabetic/severe combined immunodeficient interleukin-2 receptor-gamma chain null (NSG) mice. Tumor growth was monitored for 8–12 weeks following grafting. After establishment of SCCs, treatments with BEZ235 (35 mg/kg) were performed 3 times/week for 4 weeks by oral gavage. The volume of tumors was measured at each treatment point using digital caliper. At the end of the experiment, mice were killed and their tumors were excised, weighed and processed for downstream analyses.
Statistical data analysis
Experiments were performed on n = 6–26 mice per group of treatment and repeated 3 times with biological triplicates in cells. Staining was performed on at least 3 sections per sample from 3 mice per cohort. Two-way analysis of variance or Student’s t-test were used to determine statistical differences in treated vs. control groups and in expression levels, respectively, and results were analyzed using the GraphPad Prism and Microsoft Excel Data Analysis. Values were expressed as mean ± SD and differences with p-value < 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001).
References
Lomas A, Leonardi-Bee J, Bath-Hextall F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br J Dermatol. 2012;166:1069–80.
Stasko T, Brown MD, Carucci JA, Euvrard S, Johnson TM, Sengelmann RD, et al. Guidelines for the management of squamous cell carcinoma in organ transplant recipients. Dermatol Surg. 2004;30:642–50.
Sufficool KE, Hepper DM, Linette GP, Hurst EA, Lu D, Lind AC, et al. Histopathologic characteristics of therapy-associated cutaneous neoplasms with vemurafenib, a selective BRAF kinase inhibitor, used in the treatment of melanoma. J Cutan Pathol. 2014;41:568–75.
Neville JA, Welch E, Leffell DJ. Management of nonmelanoma skin cancer in 2007. Nat Clin Pract Oncol. 2007;4:462–9.
Massacesi C, di Tomaso E, Fretault N, Hirawat S. Challenges in the clinical development of PI3K inhibitors. Ann N Y Acad Sci. 2013;1280:19–23.
Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009;4:1350–62.
Nassar D, Latil M, Boeckx B, Lambrechts D, Blanpain C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med. 2015;21:946–54.
Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26:1657–67.
Chow HY, Jubb AM, Koch JN, Jaffer ZM, Stepanova D, Campbell DA, et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–75.
Darido C, Georgy SR, Wilanowski T, Dworkin S, Auden A, Zhao Q, et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell. 2011;20:635–48.
Hafner C, Landthaler M, Vogt T. Activation of the PI3K/AKT signalling pathway in non-melanoma skin cancer is not mediated by oncogenic PIK3CA and AKT1 hotspot mutations. Exp Dermatol. 2010;19:e222–7.
Kubo Y, Urano Y, Hida Y, Arase S. Lack of somatic mutation in the PTEN gene in squamous cell carcinomas of human skin. J Dermatol Sci. 1999;19:199–201.
Georgy SR, Cangkrama M, Srivastava S, Partridge D, Auden A, Dworkin S, et al. Identification of a novel proto-oncogenic network in head and neck squamous cell carcinoma. J Natl Cancer Inst. 2015;107:pii: djv152
Mao JH, To MD, Perez-Losada, Wu J, Del Rosario D, Balmain R. A. mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev. 2004;18:1800–5.
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63.
Huang S, Li XQ, Chen X, Che SM, Chen W, Zhang XZ. Inhibition of microRNA-21 increases radiosensitivity of esophageal cancer cells through phosphatase and tensin homolog deleted on chromosome 10 activation. Dis Esophagus.2013;26:823–31.
Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14:34–43.
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–30.
Palazzo E, Kellett MD, Cataisson C, Bible PW, Bhattacharya S, Sun HW, et al. A novel DLX3-PKC integrated signaling network drives keratinocyte differentiation. Cell Death Differ. 2017;(24):717–730.
Bhandari A, Gordon W, Dizon D, Hopkin AS, Gordon E, Yu Z et al. The Grainyhead transcription factor Grhl3/Get1 suppresses miR-21 expression and tumorigenesis in skin: modulation of the miR-21 target MSH2 by RNA-binding protein DND1. Oncogene 2013; 32: 1497–1507.
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;(118):3065–3074.
An X, Xu G, Yang L, Wang Y, Li Y, McHepange UO, et al. Expression of hypoxia-inducible factor-1alpha, vascular endothelial growth factor and prolyl hydroxylase domain protein 2 in cutaneous squamous cell carcinoma and precursor lesions and their relationship with histological stages and clinical features. J Dermatol. 2014;41:76–83.
Cangkrama M, Ting SB, Darido C. Stem cells behind the barrier. Int J Mol Sci. 2013;14:13670–86.
Qi H, Pei D. The magic of four: induction of pluripotent stem cells from somatic cells by Oct4, Sox2, Myc and Klf4. Cell Res. 2007;17:578–80.
Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA. 2011;108:E699–708.
Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;(511):246–250.
da Silva-Diz V, Simon-Extremera P, Bernat-Peguera A, de Sostoa J, Urpi M, Penin RM et al. Cancer stem-like cells act at via distinct signaling pathways in promoting late stages of malignant progression. Cancer Res. 2016;76:1245-59.
Burns JE, Baird MC, Clark LJ, Burns PA, Edington K, Chapman C, et al. Gene mutations and increased levels of p53 protein in human squamous cell carcinomas and their cell lines. Br J Cancer. 1993;(67):1274–1284.
Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011;7:292–306.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501.
Fu X, Han Y, Wu Y, Zhu X, Lu X, Mao F, et al. Prognostic role of microRNA-21 in various carcinomas: a systematic review and meta-analysis. Eur J Clin Invest. 2011;41:1245–53.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84.
Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–53.
Dickinson SE, Janda J, Criswell J, Blohm-Mangone K, Olson ER, Liu Z, et al. Inhibition of Akt enhances the chemopreventive effects of topical rapamycin in mouse skin. Cancer Prev Res. 2016;9:215–24.
Dao V, Pandeswara S, Liu Y, Hurez V, Dodds S, Callaway D, et al. Prevention of carcinogen and inflammation-induced dermal cancer by oral rapamycin includes reducing genetic damage. Cancer Prev Res. 2015;8:400–9.
Devaud C, John LB, Westwood JA, Darcy PK, Kershaw MH. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology. 2013;2:e25961.
Blanpain C, Horsley V, Fuchs E. Epithelial stem cells: turning over new leaves. Cell. 2007;128:445–58.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Einspahr JG, Calvert V, Alberts DS, Curiel-Lewandrowski C, Warneke J, Krouse R, et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev Res. 2012;(5):403–413.
Darido C, Georgy SR, Jane SM. The role of barrier genes in epidermal malignancy. Oncogene. 2016;35:5705–12.
Devlin JR, Hannan KM, Hein N, Cullinane C, Kusnadi E, Ng PY, et al. Combination therapy targeting ribosome biogenesis and mRNA translation synergistically extends survival in MYC-driven lymphoma. Cancer Discov. 2016;6:59–70.
Rebello RJ, Kusnadi E, Cameron DP, Pearson HB, Lesmana A, Devlin JR et al. The dual inhibition of RNA Pol I transcription and PIM kinase as a new therapeutic approach to treat advanced prostate cancer. Clin Cancer Res. 2016;22:5539-5552.
Ren W, Wang X, Gao L, Li S, Yan X, Zhang J, et al. MiR-21 modulates chemosensitivity of tongue squamous cell carcinoma cells to cisplatin by targeting PDCD4. Mol Cell Biochem. 2014;390:253–62.
Jeon YJ, Middleton J, Kim T, Lagana A, Piovan C, Secchiero P, et al. A set of NF-kappaB-regulated microRNAs induces acquired TRAIL resistance in lung cancer. Proc Natl Acad Sci USA. 2015;112:E3355–64.
Wong ST, Zhang XQ, Zhuang JT, Chan HL, Li CH, Leung GK. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012;32:2835–41.
Wang WZ, Pu QH, Lin XH, Liu MY, Wu LR, Wu QQ, et al. Silencing of miR-21 sensitizes CML CD34+stem/progenitor cells to imatinib-induced apoptosis by blocking PI3K/AKT pathway. Leuk Res. 2015;39:1117–24.
Hong L, Han Y, Zhang Y, Zhang H, Zhao Q, Wu K, et al. MicroRNA-21: a therapeutic target for reversing drug resistance in cancer. Expert Opin Ther Targets. 2013;17:1073–80.
Ikezoe T, Nishioka C, Bandobashi K, Yang Y, Kuwayama Y, Adachi Y, et al. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk Res. 2007;31:673–82.
Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–61.
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31.
Martin-Villar E, Borda-d’Agua B, Carrasco-Ramirez P, Renart J, Parsons M, Quintanilla M, et al. Podoplanin mediates ECM degradation by squamous carcinoma cells through control of invadopodia stability. Oncogene. 2015;34:4531–44.
Wicki A, Christofori G. The potential role of podoplanin in tumour invasion. Br J Cancer. 2007;96:1–5.
Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci USA. 2015;112:8638–43.
Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–93.
Nor C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, et al. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. 2014;16:137–46.
Darido C, Buchert M, Pannequin J, Bastide P, Zalzali H, Mantamadiotis T, et al. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res. 2008;68:4258–68.
Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, et al. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc. 2012;7:235–46.
Acknowledgements
This study was supported by the Australian National Health and Medical Research (NHMRC, APP1049870, APP1106697). CD was supported by a fellowship from the Victorian Cancer Agency (Clare Oliver Memorial, COF11_04) and by a grant (11-0060) from The Association for International Cancer Research (AICR). RBP is supported by an NHMRC Research fellowship and CD is currently holding a Victorian Cancer Agency Mid-Career Fellowship (CR_16/5985). NVP-BEZ235 and NVP-BKM120 were obtained from Novartis (Basel, Switzerland). We thank Susan Jackson and Kerry Ardley for animal technical support and Dr Andrew Cuddihy for his critical reading and correction of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Darido, C., Georgy, S.R., Cullinane, C. et al. Stage-dependent therapeutic efficacy in PI3K/mTOR-driven squamous cell carcinoma of the skin. Cell Death Differ 25, 1146–1159 (2018). https://doi.org/10.1038/s41418-017-0032-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0032-0
This article is cited by
-
YBX1 integration of oncogenic PI3K/mTOR signalling regulates the fitness of malignant epithelial cells
Nature Communications (2023)
-
New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury
Angiogenesis (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}