
Abstract
The loss of p53 function is a central event in the genesis of osteosarcoma (OS). How mutation of p53 enables OS development from osteoblastic lineage cells is poorly understood. We and others have reported a key role for elevated and persistent activation of the cAMP/PKA/Creb1 pathway in maintenance of OS. In view of the osteoblast lineage being the cell of origin of OS, we sought to determine how these pathways interact within the context of the normal osteoblast. Normal osteoblasts (p53 WT) rapidly underwent apoptosis in response to acute elevation of cAMP levels or activity, whereas p53-deficient osteoblasts tolerated this aberrant cAMP/Creb level and activity. Using the p53 activating small-molecule Nutlin-3a and cAMP/Creb1 activator forskolin, we addressed the question of how p53 responds to the activation of cAMP. We observed that p53 acts dominantly to protect cells from excessive cAMP accumulation. We identify a Creb1-Cbp complex that functions together with and interacts with p53. Finally, translating these results we find that a selective small-molecule inhibitor of the Creb1-Cbp interaction demonstrates selective toxicity to OS cells where this pathway is constitutively active. This highlights the cAMP/Creb axis as a potentially actionable therapeutic vulnerability in p53-deficient tumors such as OS. These results define a mechanism through which p53 protects normal osteoblasts from excessive or abnormal cAMP accumulation, which becomes fundamentally compromised in OS.
Similar content being viewed by others


Introduction
Osteosarcoma (OS) is the most common cancer of bone and primarily affects teenagers and young adults. Whilst our understanding of the genetics of OS have rapidly advanced, clinical outcomes have stagnated for several decades. OS is a cancer with many complex genetic abnormalities, but few genetic drivers. Conventional human OS has very high, to near universal, rates of TRP53 mutation with recurrent mutations of RB1, ATRX, and DLG2 in 29–53% of cases1,2,3. Genome-wide association studies (GWAS) and sequencing studies have identified mutations in key components of the cAMP pathway within the mutational spectrum of human OS4,5. Several recent studies in murine models have provided further evidence for involvement of the cAMP-PKA pathway in OS6,7,8,9, but how these pathways interact in the normal osteoblasts has not been resolved.
During normal bone development, osteoblastic lineage cells communicate, regulate, and activate each other through the secretion of specific molecules including parathyroid hormone-related protein (PTHrP). PTHrP acts through its cell surface receptor PTHR1, with evidence also for an intracrine action10,11. Osteoblast-specific ablation of Pthrp in mice resulted in impaired bone formation both in vivo and ex vivo12,13. These findings identified a central role for osteoblast lineage generated PTHrP in the physiological regulation of bone formation. This paracrine role was later extended when PTHrP production by osteocytes was found to be necessary for normal bone formation and strength14. As osteoblastic cells commit to form mature osteoblasts and ultimately osteocytes, PTHR1 expression increases and so does signaling via PTHrP14. PTHR1/PTHrP acts primarily to activate adenylyl cyclase and stimulate cAMP production15. Primary tumor cell cultures from mouse models of OS demonstrate both elevated and persistently active cAMP signaling, significantly contributed to by an autocrine PTHR1-PTHrP loop6,8,16.
Inactivating TRP53 mutations are one of the most common mutations in human cancer17. The most frequent mutation type is point mutation resulting in P53 proteins with altered function18. Unlike most cancers, in OS unique genomic rearrangements and other mutation types often result in null alleles of TRP531,3, and TRP53 is the most recurrently mutated gene in OS1. TRP53 mutations are also hallmark of the hereditary cancer predisposition disorder Li-Fraumeni syndrome3,7,19,20, and Tp53 knockout mice develop OS at high penetrance amongst other tumors21,22. P53 is activated upon genotoxic or oncogenic stress and regulates cell cycle, survival, and apoptosis23,24,25,26,27,28,29. P53 also regulates “non-canonical” programs such as differentiation, autophagy, metabolism, cellular pluripotency, and plasticity30. P53 can mediate its non-canonical action via its interaction with a large number of transcription factors and coactivators31. Of relevance to OS, P53 regulates osteoblastic differentiation and Tp53−/− mice have altered osteoblast differentiation and a high bone mass phenotype32.
In osteoblastic cells, activation of the cAMP pathway ultimately leads to activation of the transcription factor Creb1. Creb1 activates transcription of many cellular genes in response to cAMP. Numerous reports describe the role of CREB1 in malignancies such as AML, prostate cancer and other solid tumors including OS8,33,34,35. When phosphorylated on serine 133, CREB1 can facilitate the interaction between CBP and P53 on P53-responsive promoters36. We recently described the interaction of the cAMP/Creb1 pathway with p53 deletion in the maintenance of OS8. Herein, we have explored the interaction between these pathways in normal osteoblasts. We find that p53 normally acts to protect osteoblasts from aberrantly elevated cAMP/Creb activity, and that activation of this pathway can be tolerated once p53 is lost. Moreover, this dependence reveals a new therapeutic vulnerability in OS cells.
Materials and methods
Study approval
All animal experiments were approved by the AEC (AEC#030/14 and AEC#031/15; St. Vincent’s Hospital, Melbourne).
Animals and cell culture
Long-bone primary osteoblastic cells and mouse OS cell cultures were cultured in αMEM (Lonza), 10% non-heat inactivated FBS (SAFC Biosciences) and 1% Penicillin/Streptomycin/Glutamine (Life Technologies). OS cell cultures were derived by mincing tumor tissue with a scalpel. The resulting tumor cells were transferred to a 6 well plate and allowed to establish in standard culture conditions. Primary cultures were passaged at 60–80% confluence with media changes every 2–3 days. The primary osteoblastic cells were derived from long-bone tissue, cleaned, and crushed lightly with a mortar-pestle. The suspension was rinsed with PBS, filtered to remove the majority of the hematopoietic (solution was clear). The bone fragments were placed in 15 ml of collagenase I (3 mg/ml) (Worthington), and incubated in a shaking 37 °C water bath for 45 min. A volume of 35 ml of PBS/2% FBS was then added and the cell suspension was sieved. The resulting population of long-bone derived cells was centrifuged at 400× g for 5 min, the cells were resuspended in culture media and plated onto a 6-well plate. On the next day, the 6-well plate was washed with PBS before adding fresh culture media to remove floating debris. At 48 h post-derivation, the cells were used for experiments.
Generation of isogenic p53WT/WT and p53KO/KO normal osteoblastic cells
For experiments involving R26-CreERT2ki/+p53fl/fl, primary osteoblasts were isolated from long bones using collagenase digestion protocol as above. After 48 h the cells were split into two separate T25cm2 flask containing α-MEM (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO, USA; non-heat inactivated), 2 mM Glutamax (Life Technologies, Carlsbad, CA, USA). The cells were treated with and without 500 nM 4-hydroxy-tamoxifen (Merck Millipore) to induce CRE activity and deletion of p53 over 21 days. Deletion of p53 was confirmed by genomic DNA PCR and western blotting in the KO cultures treated with tamoxifen, compared to non-tamoxifen treated isogenic R26-CreERT2ki/+p53fl/fl cultures. Three independently derived R26-CreERT2ki/+p53fl/fl cultures were generated and used for experiments.
OS cells
Primary mouse OS cell cultures were derived from primary tumors from murine models of fibroblastic OS (Osx-Cre p53fl/flpRbfl/fl) or osteoblastic OS (Osx-Cre Tgshp53.1224pRbfl/fl) and were maintained and studied for less than 15 passages16,22. All cells used were obtained directly from mouse models of OS, no further authentication performed by the authors, mycoplasma negative.
In vitro differentiation of p53WT/WT and p53KO/KO cells
Three independently derived p53WT/WT and p53KO/KO cells were seeded at 3000 cells/cm2 on 6-well plates in α-MEM with 10% FBS three days prior to differentiation induction. When cells had reached 100% confluence (Day 0), control cells were harvested, and all other cells were replenished three times per week with osteoblastic differentiation media: α-MEM (Lonza), 10% (v/v) FBS, 25 mM HEPES, 1% (v/v) (Gibco), Penicillin-Streptomycin (Gibco), 2 mM GlutaMAX™ (Gibco), 50 μg/ml ascorbate (Sigma), 0.01 M β-glycerophosphate (Sigma).
IBMX treatment and cAMP assays
p53WT/WT and p53KO/KO cells were seeded at 1.0 × 105 cells on 6-well plates in α-MEM with 10% FBS 1 day prior to IBMX treatment. 1 mM IBMX was added to the medium and the cells were incubated for 1 h before taking images. The cells were further incubated for 16–18 h before extracting lysates for western blotting. cAMP assay was performed as described previously6,8.
Radioimmunoassay (RIA) for PTHrP
After the cells became confluent, fresh α-MEM (Lonza), 10% (v/v) FBS, 25 mM HEPES, 1% Penicillin-Streptomycin (Gibco), 2 mM GlutaMAX™ (Gibco). The medium was aspirated and the cells were washed once with PBS. RIA was carried out using polyclonal goat antiserum raised in goats against PTHrP(1–40), using recombinant PTHrP(1–84) as standard6. The sensitivity of this assay is 2pmol/l.
PI staining
Adherent cells were washed in PBS, trypsinised, and pooled with the dead population of the same well. The cells were counted, resuspended in PBS and approximately 500,000 cells were transferred to a V-bottom plate. The plate was centrifuged, the cell pellet was dispersed and 100–150 μl of −20 °C chilled Ethanol was added to the cells. After brief incubation on ice, cells were pelleted, the ethanol was removed, and the cells were washed in PBS. Finally, the cells were resuspended in 100 μl PBS containing 40 μg/ml PI (Sigma-Aldrich, MO, USA) and 40 μg/ml RNAseA (Qiagen). Following PI addition, the cells were incubated in the dark for 30–60 min at 37 °C and analysed using a Fortessa FACS analyser (BD). The data were further analysed using flow cytometry analysis software FlowJo and the DNA cycles were determined using software Modfit LtTM.
Plasmids and constructs
Two independent p53WT/WT cell lines were used for overexpressing pLenti6/V5-p53_wt p53 construct, a gift from Bernard Futscher (Addgene plasmid # 2294537). Lentiviral packaging vector psPax2 (plasmid #12260) was obtained from Addgene (Cambridge, MA, USA), the pCMV-Eco Envelope (Cat No. RV112) vector was purchased from Cell Biolabs (San Diego, CA, USA). Two independent isogenic p53WT/WT cell lines were infected with lentivirus overexpressing human V5 tagged-P53. After 48 h cells were selected with 10 µg/ml Blasticidine S. The cells were confirmed for overexpression of WT V5-p53 by western blot analysis and used for experiments. For the construction of plasmid encoding a mouse p53 transcript, a cDNA encoding the mouse p53 transcript (IDT DNA) was cloned in pLVX vector (Clontech). Three independent Trp53WT/WT cell lines were used for overexpressing pLVX-p53_wt p53 construct. The cells were confirmed for overexpression of p53 by western blot analysis and qPCR and used for experiments.
Flow cytometry
Three independent isogenic p53WT/WT and p53KO/KO cell lines were prepared by trypsinization. Antibodies against murine CD45, Mac1, Gr1, F4/80, B220, IgM, CD2, CD3, CD4, CD8, Ter119, Sca1, CD51, PDGFRα (CD140a), CD31, either biotinylated or conjugated with eF450, PE, PerCP-Cy5.5, or APC were obtained from eBioscience (San Diego, CA) or BD Pharmingen. Biotinylated antibodies were detected with Streptavidin-Qdot605 (Invitrogen). Flow cytometry was performed on an LSRII Fortessa (BD Bioscience) interfaced with Cell Quest software, data were analysed on FlowJo (TreeStar).
Annexin V and 7AAD staining
Three independent isogenic p53WT/WT and p53KO/KO cell lines were treated with 1 mM IBMX and 10 µM Forskolin for 1 h and 24 h, respectively. Cells were washed and collected after trypsinization and then stained in 1× Annexin Binding buffer (eBioscience) diluted 1:20 with Annexin V-APC (1 mg/ml) (eBioscience) and 7-Aminoactinomycin D (7AAD) (100 μg/ml) (Life Technologies) for 15 min. Following the addition of 4 volumes of 1× Annexin Binding buffer, apoptotic cells were detected and quantified using FACS (LSRFortessa). Live cells (Annexin V negative, 7AAD low) and cells in early and late stages of apoptosis (Annexin V positive, 7AAD low/high) were quantified.
RNA extraction, cDNA synthesis and Quantitative realtime PCR (QPCR)
RNA was extracted using RNA extraction kits with on-column DNase digestion (Qiagen, Limburg, Netherlands; Bioline, London, UK) or TriSure reagent (Bioline). cDNA was synthesised from total RNA using a Tetro cDNA synthesis kit (Bioline) or AffinityScript cDNA synthesis kit (Agilent Technologies, Santa Clara, CA, USA). Gene expression was quantified on a Stratagene Mx3000P QPCR system (Agilent) using Brilliant II SYBR green QPCR master mix (Agilent) with primers specific to genes of interest (Primer sequences in Table S1). Relative expression was quantified using the comparative CT method (2-(Gene Ct – Normalizer Ct)). Samples were amplified in duplicate.
Western blotting
Protein lysates were prepared in RIPA buffer (50 mM Tris-HCl pH7.4, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, 50 mM NaF). Protein (10–25 µg) was electrophoresed on 10% Bis Tris or 4–12% Bis-Tris gradient NuPAGE Novex protein gels (Life Technologies) and transferred to PVDF membrane (Merck Millipore, Billerica, MA, USA). Membranes were blocked in 5% skim milk in TBST (20 mM Tris, 150 mM NaCl, 0.1% Tween-20) for 1 h before incubation with primary antibodies diluted in 5% skim milk in TBST overnight a 4 °C, or for 1 h at room temperature in the case of pan-ACTIN. All antibodies, p53 (1C12; Cell Signaling 2524), Phospho-CREB ((Ser133) Cell Signaling Technologies, #9198), Anti-CREB1 ChIP grade (ab31387), and CBP (C-1, Santa Cruz, sc-7300) were used at 1:2000, except pan-actin (Ab-5, Thermo Scientific, Waltham, MA, USA) that was used at 1:3000. Cleaved caspase 3 Asp175 [5A1E] was from Cell Signaling Technologies 9664 S. Following four 10 min washes with TBST, membranes were exposed to ECL Prime (GE Healthcare Life Sciences, Piscataway, NJ, USA) and exposed to X-ray film to detect the expression levels of proteins.
Immunoprecipitation
1 × 106 cells for each subtype (Three independent isogenic p53WT/WT and p53KO/KO) were seeded and allowed to proliferate for 24 h. p53WT/WT and p53KO/KO cells were treated with less than 0.1% DMSO, 10 µM forskolin or 100 nM doxorubicin and in combination. Cells were also treated with 0.1% DMSO, 10 µM forskolin or 10 µM Nutlin and in combination). Post treatment the cells were scraped on ice cold PBS and snap frozen until further use. For immunoprecipitation, non-ionic lysis buffer containing 1% NP-40, Tris-HCl at pH8.0, 137 mM NaCl and 2 mM EDTA with protease inhibitors (Roche, Burlington, NC, USA) was used. 2–3 × 106 million cells were lysed in non-ionic lysis buffer and left on ice for 30 min, and the cells were further sonicated using a UCD-200 Bioruptor (Diagnenode, Denville, NJ, USA) on high at 4 °C for a total shearing time of 2 min (60 min of 10 s on and 50 s off). Cell debris was cleared by centrifugation at 13,000 rpm for 10 min at 4 °C and supernatants were diluted 10-fold in 1:10 diluted non-ionic lysis buffer. After removing 1% input for the total number of cells of each sample as an input control, samples were incubated with either 2 µg CREB1 antibody (Abcam: ab31387), 2 µg of phospho-CREB1 antibody (Cell Signaling: 9198), 2 µg of p53 antibody (1C12, Cell Signaling 2524), 2 µg of CBP (C-1, Santa Cruz: sc-7300) and 2 µg of control rabbit IgG (Merck Millipore), or no antibody overnight at 4 °C with rotation. Complexes were collected for 1 h at 4 °C with rotation with 60 µl of protein A sepharose beads (Invitrogen) that had been pre-equilibrated in 1:10 Non-Ionic lysis buffer for 1 h. Beads were washed one time each with Low Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.1, 150 mM NaCl), High Salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.1, 500 mM NaCl) and LiCl buffer (0.25 M LiCl, 1% NP40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH8.1), followed by two washes with TE buffer. Protein-Protein complexes were eluted from the beads adding 30 μL of 4X Laemilli buffer. The samples were ready for western blot analysis.
CREB-CBP inhibitor treatment
p53WT/WT and p53KO/KO cells and the corresponding OS cells derived from cell lines made from primary tumors were plated at 2500 cells per well in 96-well plates. After 24 h, the cells were treated with 666–15 (Tocris Bioscience) in an 11-point dose response ranging from 0.01 to 10 mmol/L. All assays were performed in duplicate. At 48 and 72 h after treatment, the cell viability for each well was quantified on the basis of the direct measurement of intracellular ATP using the ATP-Lite luminescent assay (PerkinElmer). All luminescent measurements were recorded on the EnSpire plate reader (PerkinElmer). Data were plotted and the IC50 value calculated using Prism 6 software.
Bioinformatics and data mining and selection of target genes
Creb1 target genes were selected from38,39,40. We only validated genes which were considered related to cAMP signaling. For validation of p53 direct targets we referred to41 that describes a genome-wide approach to detect lnc-RNAs and p53 transcriptional targets in human cells. We downloaded the ChIP-seq data for p53 in MEFs from42. We therefore compared the human p53 ChIP-seq data with previously published p53 ChIP-seq data from mouse embryonic fibroblasts (MEFs) treated with the DNA-damage drug doxorubicin. A subset of genes which were doxorubicin induced and were common in human and mouse datasets were selected.
Statistical analysis
Data were presented as mean ± SEM. Statistical comparisons were performed in Prism 6.0 unless otherwise indicated. Parametric Student’s t-test, area under the curve or 2-way ANOVA with multiple comparison test were used for comparisons with P < 0.05 considered as significant.
Results
Acute loss of p53 in primary osteoblasts activates PTHrP-cAMP-Creb1 signalling
We modelled the acute loss of p53, in isolation of other genetic changes, using primary osteoblastic cells isolated from long bones of R26-CreERT2ki/+Tp53fl/fl (referred to as KO herein) and R26-CreERT2ki/+Tp53+/+ (referred to as WT) animals. Cells were isolated and expanded for 5–7 days. The cells were then cultured in proliferative media and treated + /− tamoxifen to generate isogenic cultures. Tamoxifen treatment was continued until day 21 when it was stopped. The cells were genotyped to confirm they were wild-type or Tp53-deficient then used for the assays described. For all subsequent experiments, excluding those describing results from differentiation cultures, the cells were plated and allowed to reach ~70–80% confluence then used for experiments (for example, treated with IBMX or forskolin). For differentiation experiments, the cells were plated at the appropriate cell density (no tamoxifen added) and placed in differentiation inducing media for a further 21 days. Isogenic cell pairs (Tp53 WT and Tp53 KO) were made in order to allow comparison of cells from the same animal (Fig. 1a).

a Schematic representation of generation of independent p53WT/WT and p53KO/KO cells. b p53WT/WT and p53KO/KO cells assessed for expression of Sca-1, CD51, representative results shown, n = 3 independent experiments. c Cell surface markers from each cell line (n = 3 independent cultures). d mRNA for Osterix, Runx2, Pthr1, Pthlh, Osteocalcin, Sost, Pparg, Adiponectin by qPCR in p53 WT and p53 KO cells (normalized to β2m; mean ± SEM, n = 3). e PTHrP in lysates of p53WT/WT and p53KO/KO; pmol/µg of protein using radioimmunoassay. Data pooled from 3 independent cultures; mean ± SEM. f cAMP accumulation (pmol/µg of protein) after treatment with 100 μM IBMX for 30 mins in p53WT/WT and p53KO/KO. Data pooled from 3 independent cultures; mean ± SEM. g Western blot of p53, pCREB1, and Creb1, β-Actin used as a loading control. Data representative of 2 independent cell lines from each. See Figure S1. For all panels: *P < 0.05, **P < 0.01, ***P < 0.001
Analysis of proliferative cultures immediately after cessation of tamoxifen treatment demonstrated that the expression of p53 target genes was diminished in the KO cultures, compared to non-tamoxifen treated isogenic cultures8. By cell surface phenotype, expression of CD51 was increased and Sca1 was decreased on KO cells compared to WT counterparts (Fig. 1b, c). At the completion of tamoxifen treatment, when deletion of p53 was confirmed, there was increased expression of Osterix, Pthrp, and Sost in KO cells, with no significant increase in other osteoblast lineage markers Pthr1 and Runx2 (Fig. 1d). A clearer indication of the effect of Tp53 deficiency was obtained by examining differentiation potential of the WT and Tp53 deleted cells by culturing for a further 21 days in osteogenic differentiation media. Expression of osteoblastic lineage markers remained stable in the KO cells and generally at a higher level than in the p53 WT cells during differentiation (Supplemental Fig. 1A, B). Adipogenic markers Pparg and Adiponectin changed comparably between genotypes (Supplemental Fig. 1A, B). These findings are consistent with previous reports that p53 loss is associated with altered osteoblast differentiation32,43.
We assessed the effect of Tp53 deficiency on PTHrP-cAMP-Creb1 pathway status. KO cells had increased PTHrP protein levels and intracellular cAMP accumulation compared to p53 WT counterparts (Fig. 1e, f). Consistent with these findings, there were higher levels of basal phospho-Creb1 in p53-deficient cells (Fig. 1g). Examination of the kinetics of p53 protein expression during differentiation revealed that in WT cells, p53 protein level gradually reduced, concurrent with a reduction of Creb1 (Supplemental Fig. 1C, D). However, p53-deficient osteoblasts retained Creb1 expression over the differentiation time course, including phosphorylated active Creb1 (Supplemental Fig. 1C, D). This result is consistent with the persistence of Creb1 and phospho-Creb1 in OS cells induced to differentiate in vitro8.
Elevated cAMP induces apoptosis that is regulated by p53
As intracellular cAMP accumulation increased after p53 deletion from primary osteoblasts (Fig. 1f), we sought to determine the effect of acutely elevating cAMP levels on wild type osteoblastic cells. The cells were treated with DMSO (control) or IBMX (1 mM), which inhibits the phosphodiesterases (PDE) that degrade cAMP, to enable accumulation of the cells’ intrinsic cAMP. Within 1 h of IBMX treatment there was a major difference in cell morphology, with WT cells poorly attached to the culture dish compared to the KO (Fig. 2a). There was a significant increase in AnnexinV positive cells in the WT culture (Fig. 2b, c). Cell death/apoptosis was assessed by measuring the levels of cleaved caspase-344. Upon preventing breakdown of intracellular cAMP, cleavage of caspase-3 was rapidly induced in WT cells unlike the KO counterpart (Fig. 2d).

a p53WT/WT and p53KO/KO cells treated with 1 mM IBMX; representative image of 3 independent isogenic cells. b AnnexinV/7-AAD profiles of IBMX-treated p53WT/WT and p53KO/KO . c Percent apoptotic cells in each culture p53WT/WT and p53KO/KO post-IBMX treatment (mean ± SEM, n = 3). d Apoptosis shown by cleaved caspase-3 activation in 3 independent p53WT/WT and p53KO/KO isogenic cells. e Annexin V and 7AAD profiles of forskolin-treated p53WT/WT primary osteoblasts cells. f Percent apoptotic cells in each culture p53WT/WT pre- and post-forskolin treatment (mean ± SEM, n = 3). g Western blot of phospho-p53, phospho-Creb1 with β-Actin as a loading control. Data representative of 2 independent cell lines from each. h AnnexinV and 7AAD profiles of forskolin-treated p53WT/WT and p53KO/KO primary osteoblasts cells. i Presence of cleaved caspase-3 in 3 independent p53WT/WT and p53KO/KO isogenic cells. j Western blot of p53, phospho-Creb1 with β-Actin as a loading control. Data representative of 2 independent cell lines from each. k mRNA for p53, Cdkn1a1, Atp9a1, Mdm2, Creb1, Nr4a1, Nr4a2, Nr4a3 by qPCR in p53WT/W cells at 72 h after overexpression of p53. Expression levels normalized to β2m; mean ± SEM (n = 3). l Intracellular cAMP accumulation (pmol/µg protein; cells treated with 100 μM IBMX for 30 mins) in p53WT/WT cells overexpressed with p53; data pooled from 3 independent cultures; mean ± SEM. For all panels: *P < 0.05, **P < 0.01, ***P < 0.001
As an alternative approach to persistently elevate cAMP, cells were treated with the direct cAMP stimulus forskolin (Supplemental Figure 2A). After 24 h of forskolin treatment, p53 WT cells had an altered morphology similar to the IBMX-treated cells (data not shown). WT cells underwent cell death within 24 -hours of forskolin treatment as quantitated by AnnexinV/7AAD staining (Fig. 2e, f). Elevation of cAMP via forskolin resulted in increased phosphorylation of Creb1, as expected, and also of p53 (Fig. 2g). p53 KO cells were protected from cell death induced by forskolin treatment (Fig. 2h, i). This occurred despite the p53 KO cells generating higher intracellular cAMP levels from the same dose of forskolin than the WT cells (Supplemental Figure 2A).
The deletion of p53 alone resulted in an increase in intracellular cAMP and phosphorylation of Creb1 (Fig. 1f, g). We tested if overexpression of p53 could suppress the cAMP-Creb1 axis. Primary osteoblasts were infected with lentivirus overexpressing murine p53 (Fig. 2j, k). Downstream transcriptional targets of p53 were significantly increased in expression (Fig. 2k) and, strikingly, Creb1 targets were repressed (Fig. 2k). This result was confirmed independently in HEK293T cells overexpressing human P53 (Supplemental Fig. 2C, D). The overexpression of p53 also led to a profound reduction in intracellular cAMP formation in the cells (Fig. 2l), and a reduction of Creb1 phosphorylation (Fig. 2j). Thus, overexpression of p53 repressed cAMP accumulation and reduced Creb1 activity.
Accumulation of cAMP in p53-deficient cells activates proliferative networks
WT cells and isogenic KO counterparts were treated with DMSO (control) or 100 μM IBMX for 1 h and the expression/activation of the downstream cAMP-PKA pathway monitored. Analysis of independently derived cell cultures revealed increased levels of the PKA-catalytic subunit and phospho-Creb1 in KO cells. There was a basal activation of phospho-Creb1 in p53 KO cells as compared to p53 WT cells (Fig. 3a).

a Western blot of p53, phospho-Creb1 and Creb1, PKA (catalytic subunit); β-Actin as loading control. Data representative of three independent isolates of isogenic p53WT/WT and p53KO/KO. Cells were treated with 1 mM IBMX as indicated in methods. b Heat map of qPCR data. Expression of the p53 target genes between indicated cell types; three independent isogenic cell lines for each, expressed as mRNA normalized to β2m. c Heat map of qPCR data. Expression of Creb1 target genes between indicated cell types; > 3 independent isogenic cell lines for each, expressed as mRNA normalized to β2m. d Heat map of qPCR data for genes important in cellular proliferation. Expression of the Creb1 target genes between indicated cell types.; > 3 independent isogenic cell lines for each, expressed as mRNA normalized to β2m. e Heat map of qPCR data for genes that are negative regulators of cAMP. Expression of the genes between indicated cell types; three independent isogenic cell lines for each, expressed as mRNA normalized to β2m. f Heat map of qPCR data for genes important in development. Expression of the genes between indicated cell types; > 3 independent isogenic cell lines for each, expressed as mRNA normalized to β2m. Data pooled from 3 independent cultures; mean ± SEM. See Figure S3. For all panels: *P < 0.05, **P < 0.01, ***P < 0.001
To confirm that the cAMP accumulated following IBMX treatment, as seen in Fig. 1f, resulted in activation of signaling qRT-PCRs were performed. The deletion of p53 resulted in an expected decreased expression of Tp53 and p53 target genes (Fig. 3b, Supplemental Figure 3A) and was associated with activation of Creb1 target genes as we previously described7,8 (Fig. 3c, Supplemental Fig. 3B). Loss of p53 was associated with an increased expression of Ras and Myc45,46. To address this finding, we selected several targets known to be regulated by Myc and Ras47,48. Transcriptional target genes of both Myc and Ras were significantly upregulated in p53 KO cells (Fig. 3d, Supplemental Figure 3C). In human OS, mutations in components of the cAMP pathway have been defined, including in the PDE family, A kinase anchoring proteins (AKAP) and protein phosphatases (PP)1. We tested whether loss of p53 directly affected the expression of these genes in WT and KO osteoblasts. The majority of genes were not different between the genotypes, apart from a complete loss of Pde4d mRNA in p53 KO cells (Fig. 3e, Supplemental Fig. 3D). Pde4d hydrolyzes cAMP and results in its breakdown49. Expression of markers of osteoblast differentiation were upregulated in p53 KO cells compared to the isogenic counterpart (Fig. 3f, Supplemental Fig. 3E).
Activation of p53 directly suppresses Creb1
We next asked if genome-wide gene signatures associated with cAMP-PKA were impacted by loss of p53 in silico. Creb1 and P53 target genes were identified from ChIP-seq studies39,41. The P53 target genes were further refined against a second independent dataset of p53 ChIP-seq from murine embryonic fibroblasts42. When overlapped, ~37% of Creb1 targets could also be directly regulated by p53 (Fig. 4a). Of the common p53-Creb1 targets, the genes were then separated into groups that were either up (n = 314) or down (n = 301) regulated by p53, respectively, and pathway analysis performed using DAVID50 (Fig. 4b). The functions of the genes repressed by p53 were over-represented in pathways related to protein kinase signaling (Fig. 4c), whereas the genes that could be upregulated by p53 were associated with classical p53 signatures including cell cycle arrest and apoptosis (Fig. 4d).

a Venn diagram depicting the overlap and outliers between common human and mouse p53 targets and Creb1 ChIP-seq dataset. b Table showing the bifurcation of up- and downregulated Creb1 targets regulated by p53. c Pathways governed by p53-mediated downregulated Creb1 targets. d Pathways governed by p53-mediated upregulation of Creb1 targets
To directly test the link between p53 and protein kinase signaling we used a pharmacogenetic approach to examine how these pathways interacted when activated individually or concurrently. WT osteoblasts were treated with the MDM2 inhibitor Nutlin-3a to activate WT p53 in a time course of 24 h. qRT-PCR demonstrated a significant activation of p53 target genes in a temporal fashion (Fig. 5a). There was a reciprocal relationship between p53 activation and the expression of Creb1 target genes (Fig. 5b). The downregulation of Creb1 activity was an early consequence of p53 activation, as Creb1 targets were significantly repressed with 2 h of Nutlin-3a treatment (Fig. 5c, d). Similar inhibition of Creb1 activity occurred when p53 was activated using doxorubicin, a genotoxic drug used to treat a number of cancer including OS (Supplemental Fig. 4A–C). Western blot analysis confirmed the increase in p53 by Nutlin-3a treatment in p53 WT cells (Fig. 5e). In assessing this further, Nutlin-3a mediated activation of p53 significantly decreased intracellular cAMP levels in WT osteoblasts and this effect was completely prevented in KO cells (Fig. 5f). The activation of p53 therefore leads to reduced formation of intracellular cAMP and a repression of Creb1 transcriptional activity.

a qPCR validation of p53 target gene expression following 10 μM Nutlin treatment over time course of 24 h; 3 independent isogenic p53WT/WT cells, mean ± SEM. b qPCR validation of Creb1 target gene expression following 10 μM Nutlin treatment over time course of 24 h; 3 independent isogenic p53WT/WT cells, mean ± SEM. c qPCR validation of p53 target gene expression following 10 μM Nutlin treatment over time course of 2 h; 3 independent isogenic p53WT/WT cells, mean ± SEM. d qPCR validation of Creb1 target gene expression following 10 μM Nutlin treatment over time course of 2 h; 3 independent isogenic p53WT/WT cells, mean ± SEM. e Western blot of p53, Phospho-Creb1 and Creb1 post-Nutlin-3a treatment for time course of 2 h; β-Actin used as a loading control; Data representative of 2 independent isogenic cell lines. f Intracellular cAMP formation (pmol/μg protein) in p53WT/WT and p53KO/KO (IBMX 100 μM) 30 mins post-Nutlin treatment; data pooled from 3 independent cultures; mean ± SEM. See figure S4. For all panels: *P < 0.05, **P < 0.01, ***P < 0.001
Simultaneous p53 activation and elevated cAMP levels induce apoptosis
In order to determine whether elevated intracellular cAMP can directly affect the cells’ response to p53 activation, we treated WT and KO osteoblasts with Nutlin-3a alone (p53 activator), forskolin alone (cAMP stimulus) or the combination of both for 24 h and evaluated the cellular effects and gene expression. Elevating cAMP together with activation of p53 led to apoptosis in p53 WT osteoblasts. qRT-PCR was performed revealing that simultaneous activation of p53 and cAMP resulted in the suppression of Creb1 targets, suggesting that the response by p53 was dominant over the cAMP-induced response (Fig. 6a, b). There was no significant effect on the expression of genes important for osteoblastic lineage development such as Osterix and Runx2 (Supplemental Fig. 5A). This effect was rapid, seen within 1 h. The transcriptional effect of p53 activation and Creb1 signature suppression was absent in p53 KO cells, which confirmed the specificity of this response to p53 (Fig. 6c). A comparable effect was observed when doxorubicin was combined with forskolin, with the cells undergoing apoptosis and Creb1 target genes being suppressed (Fig. 6d–f, Supplemental Fig. 5B). PI staining was performed to assess the cell cycle status of the cells and there was no significant change in cell cycle parameters (Supplemental Fig. 5C), suggesting that at least in the window of treatment there was no obvious changes in cell cycle parameters. This indicates that the ability to tolerate elevated and aberrant cAMP pathway activation in osteoblasts, as is seen in OS, is dictated by p53 status.

a AnnexinV/7-AAD profiles of p53WT/WT cells treated with less than 0.1% DMSO, 10 μM Nutlin, 10 μM Forskolin, and combined treatment with Forskolin and Nutlin. b Percent apoptotic cells in each treatment group; mean ± SEM. c Heat map of qPCR data. Expression of the p53 and Creb1/pro-proliferative targets, developmental targets between cell types; Data from 3 independent isogenic cell lines for each, expressed as relative expression normalized to β2m. Data expressed as mean ± SEM (n = 3). d AnnexinV/7-AAD profiles of p53WT/WT cells treated with less than 0.1% DMSO, 500 nM Doxorubicin, 10 μM Forskolin and combinatorial treatment of Forskolin and Doxorubicin. e Percent apoptotic cells in each treatment group; 3 independent cell lines. f Heat map of qPCR data. Expression of the p53 and Creb1/pro-proliferative targets, developmental targets between cell types; Data from 3 independent isogenic cell lines for each, expressed as relative expression normalized to β2m. Data expressed as mean ± SEM (n = 3). For all panels: *P < 0.05, **P < 0.01, ***P < 0.001. See figure S5. For all panels: *P < 0.05, **P < 0.01, ***P < 0.001
Creb1, Cbp, and p53 can occur in complex
Phosphorylation of CREB mediates recruitment of co-activator associated complex of Creb1 (CBP) to p53-responsive promoters, through potentially a direct interaction with p5336. In whole cell lysates from p53 WT and KO cells, steady state Creb1 and phospho-Creb1 were significantly higher (Figs. 1, 2). To assess if there was a direct interaction between p53 and Creb1, Creb1 was immunoprecipitated and the Creb1-containing complexes from p53 WT cells assessed with p53 KO cells used as a control. In the WT cells, p53 was co-immunoprecipitated with both Creb1 and phospho-Creb1-containing protein complexes (Fig. 7a). We assessed whether Cbp was also associated with Creb1 complexes in these cells. When Creb1 was immunoprecipitated in both cell types, it could be found associated with Cbp (Fig. 7b). Interestingly, when Cbp containing complexes were immunoprecipitated from p53 WT and KO cells, it revealed a different interaction. In the presence of p53, Cbp could interact with p53, but there was greatly reduced interaction with Creb1 (Fig. 7a). However, with the loss of p53 there was enhanced interaction of Creb1 with Cbp (Fig. 7b). Whilst the consequences of this are not immediately clear, they suggest that p53 status may regulate the extent of interaction of Creb1 and Cbp.

a Western blot following immunoprecipitation of Creb1 and immunoblot with Cbp, p53 and Creb1 from 3 independent isogenic p53WT/WT and p53KO/KO. b Western blot following Cbp immunoprecipitation and immunoblot with Cbp, p53, and Creb1 from 3 independent isogenic p53WT/WT and p53KO/KO. c Western blot analysis of p53 immunoprecipitation and immunoblot with Cbp, p53, Creb1, and phospho-Creb1 from isogenic p53WT/WT and p53KO/KO cells treated with DMSO, Nutlin, Forskolin, and Forskolin + Nutlin
We further sought to understand how the dynamics of the p53-Creb1-Cbp interactions changed in response to activation of p53 or cAMP. WT and KO cells were treated with DMSO, Nutlin-3a, forskolin, or forskolin/Nutlin-3a followed by immunoprecipitation with p53. Following p53 activation there was a decreased amount of Creb1 associated with p53 (Fig. 7c). When cAMP levels were increased in isolation of p53 activation, there was evidence of the p53-Creb1-Cbp interaction, however there was less p53 associated with this complex. When both p53 and cAMP pathways were activated concurrently, the strongest association between these proteins could be seen.
Inhibition of the Creb1-Cbp interaction as a potential therapeutic strategy for osteosarcoma
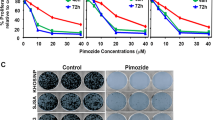
Several new chemical inhibitors of the CREB pathway have been described, including the CREB inhibitor 666–1551. We tested 666–15 against normal osteoblasts (p53+/+), primary p53 KO osteoblasts and primary OS cell cultures isolated from both fibroblastic and osteoblastic OS mouse models16,22. A dose response assay demonstrated genotype dependent toxicity after 48 h, which was specific for p53 KO osteoblastic cells and the p53 null OS (Fig. 8a). Normal osteoblasts tolerated high doses of 666–15, unlike OS cells and p53-deficient osteoblasts. Incubating the cells for 72 h with 666–15 led to a more pronounced effect on the OS cells (Fig. 8b). We therefore determined how the cells were dying by measuring the levels of cleaved caspase-3. Inhibition of Creb1-Cbp interaction led to apoptosis in the OS cells but not in normal osteoblasts (Fig. 8c). This result suggested that the Creb1-Cbp interaction is required for the maintenance of proliferation of p53-deficient osteoblasts and, more importantly, the viability of OS cells.

a Dose response curves of cells treated with 666–15 Creb1-Cbp inhibitor for 48 h. b Dose response curves of cells treated with 666–15 Creb1-Cbp inhibitor for 72 h. c Western blot analysis for cleaved caspase-3 from the indicated cells treated for 48 h with the Creb1-Cbp interaction inhibitor 666–15. d Graphical summary of the differences between lineages committed pre-osteoblasts and the sequence of expression of paracrine action of PTHrP on differentiating cells expressing PTHR1. Pre-osteoblasts cannot tolerate increased cAMP levels as compared to p53-deficient osteosarcoma cells
Discussion
P53 is a negative regulator of key cell fate-mediated programs which, when perturbed, can lead to tumor formation7,16,20,21,22,52. OS, unlike most tumor types, frequently harbors null alleles of TRP531,3. The reason for this mutational preference in osteoblastic cells, the lineage of origin of OS22, is not clear. Evidence for elevated and persistent signalling through the cAMP/PKA axis in OS has come from mouse genetic models, including those we have developed6,7,8,9, as well as from mutational analysis of human OS1, all leading to the view that deregulation of this pathway plays a critical role in OS6,7,8. In the present work, we have studied the nexus between p53 and cAMP pathways in normal osteoblastic cells. Persistently elevated cAMP levels induce cellular stress in osteoblastic cells, and p53 counteracts this by inducing apoptosis (Fig. 8d). In the absence of p53, a state sufficient to initiate OS, osteoblastic cells become tolerant to elevated and otherwise toxic levels of cAMP/Creb1 pathway activity (Fig. 8d).
We have utilized a reductionist cellular model to understand the consequences of the key mutation in OS, p53 loss, in primary osteoblasts. In this model there was a coordinated increase in cAMP and activated Creb1 levels and transcriptional activity as osteoblastic cells became p53-deficient, prior to transformation into OS. In addition, there was increased expression of Pthlh and PTHrP, which we have shown previously to be a critical autocrine stimulus of the cAMP-Creb1 pathway in OS6,7,8. When WT primary osteoblasts were prompted to undergo osteogenic differentiation in culture, p53 and Creb1 expression declined during differentiation. However, in p53 KO primary osteoblasts, the decrease in Creb1 expression during differentiation was delayed. The loss of p53 thus induces an aberrant differentiation program and depresses the cAMP pathway in osteoblasts.
Undifferentiated osteoblastic cells expressed PTHrP but have low to undetectable levels of PTHR1. In normal osteoblast lineage cells, immature osteoblasts make PTHrP but do not sense it, instead it acts in a paracrine manner on more mature osteoblastic lineage cells10,14. However, in OS we previously reported that OS cells make PTHrP and have functional PTHR1, with an autocrine axis maintaining elevated and persistent intracellular cAMP levels in OS (Fig. 8d)6,8. To investigate the consequence of maximal accumulation of cAMP in osteoblastic cells, we treated cells with either IBMX (prevents breakdown of cAMP) or used forskolin (directly elevates intracellular cAMP). This demonstrated that excessive intracellular cAMP was poorly tolerated by normal osteoblastic cells, resulting in apoptosis. The loss of p53 was sufficient to enable the cells to survive and tolerate the increased cAMP levels. Strikingly, overexpression of p53 in osteoblastic cells was able to reduce intracellular cAMP formation and subsequent activation of the cAMP-Creb1 pathway. We assessed the interaction of these pathways in WT osteoblasts by simultaneously treating cells with forskolin and Nutlin-3a, alone or in combination. p53 activation (Nutlin-3a) is sufficient to repress downstream target gene activation within the cAMP pathway. Our data indicate that osteoblastic cells do not normally tolerate sustained elevations of intracellular cAMP. Loss of p53 imparts an advantage to osteoblasts to tolerate accumulated cAMP levels. Increased expression/activity of the cAMP-Creb pathway is an early event following p53 loss in osteoblasts. Thus, abrogation of p53 activity is necessary before cells can tolerate elevated cAMP pathway activity and utilize it for tumor initiation. Our data also indicate that OS cells tolerate elevated cAMP levels due to the abrogation of the p53-dependent apoptotic response that normal osteoblastic cells incur following activation of this pathway.
Restoration of p53 tumor suppressor pathway function is a promising anti-cancer strategy53. However, such approaches are not feasible in p53 null settings such as OS. New treatment options for OS are sorely needed, with the advances in patient outcome over the last 30 years coming from optimizing application of existing therapies rather than the introduction of new agents. The rapid activation of the cAMP pathway following p53 deletion, and its persistence and amplification in OS, may highlight a therapeutic vulnerability in OS cells. We identified that p53 can interact with Creb1 and its co-activator Cbp. Loss of p53 increased the association of Creb1 with Cbp, further enhancing the cellular reprogramming by enabling the elevated cAMP levels to be reflected transcriptionally. Based on these findings, we explored the option of inhibiting the Creb1-Cbp complex interaction. We used an inhibitor, 666–15, which acts by blocking the interaction of CREB1 with CBP51. 666–15 or its preceding compounds have demonstrated preclinical efficacy in vivo in leukemia models54. Strikingly, inhibition of the Creb1-Cbp interaction had a selective effect on p53 null cells, with a graded response of increasing sensitivity as cells transition from p53-deficient osteoblasts to OS, sparing normal osteoblasts. This result demonstrates that sustained Creb1-Cbp activity is required for survival of OS cells. Collectively our results demonstrate a close relationship between loss of p53 and tolerance to elevated and persistent cAMP/Creb1 signaling in osteoblasts. These pathways are central to the biology of OS and here we find that they are coupled from the earliest events in the initiation of this tumor. Importantly, our results also demonstrate that this pathway may be therapeutic vulnerable in p53-deficient tumors such as OS.
References
Chen, X. et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7, 104–112 (2014).
Perry, J. A. et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl Acad. Sci. USA 111, E5564–E5573, https://doi.org/10.1073/pnas.1419260111 (2014).
Ribi, S. et al. TP53 intron 1 hotspot rearrangements are specific to sporadic osteosarcoma and can cause Li-Fraumeni syndrome. Oncotarget 6, 7727–7740, https://doi.org/10.18632/oncotarget.3115 (2015).
Karlsson, E. K. et al. Genome-wide analyses implicate 33 loci in heritable dog osteosarcoma, including regulatory variants near CDKN2A/B.Genome. Biol. 14, R132 (2013).
Savage, S. A. et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat. Genet. 45, 799–803, https://doi.org/10.1038/ng.2645 (2013).
Ho, P. W. et al. Knockdown of PTHR1 in osteosarcoma cells decreases invasion and growth and increases tumor differentiation in vivo. Oncogene 34, 2922–2933, https://doi.org/10.1038/onc.2014.217 (2015).
Walia, M. K., Castillo-Tandazo, W., Mutsaers, A. J., Martin, T. J. & Walkley, C. R. Murine models of osteosarcoma: a piece of the translational puzzle. J. Cell. Biochem. 119, 4241–4250, https://doi.org/10.1002/jcb.26601 (2017).
Walia, M. K. et al. Activation of PTHrP-cAMP-CREB1 signaling following p53 loss is essential for osteosarcoma initiation and maintenance. eLife 5, e13446, https://doi.org/10.7554/eLife.13446 (2016).
Molyneux, S. D. et al. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J. Clin. Invest. 120, 3310–3325, https://doi.org/10.1172/JCI42391 (2010).
Martin, T. J. Parathyroid hormone-related protein, its regulation of cartilage and bone development, and role in treating bone diseases. Physiol. Rev. 96, 831–871, https://doi.org/10.1152/physrev.00031.2015 (2016).
Massfelder, T. et al. Opposing mitogenic and anti-mitogenic actions of parathyroid hormone-related protein in vascular smooth muscle cells: a critical role for nuclear targeting. Proc. Natl Acad. Sci. USA 94, 13630–13635 (1997).
Miao, D. et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J. Clin. Invest. 115, 2402–2411, https://doi.org/10.1172/JCI24918 (2005).
Miao, D. et al. Parathyroid hormone-related peptide is required for increased trabecular bone volume in parathyroid hormone-null mice. Endocrinology 145, 3554–3562, https://doi.org/10.1210/en.2003-1695 (2004).
Ansari, N. et al. Autocrine and paracrine regulation of the murine skeleton by osteocyte-derived parathyroid hormone-related protein. J. Bone Miner. Res. 33, 137–153, https://doi.org/10.1002/jbmr.3291 (2018).
Juppner, H. Receptors for parathyroid hormone and parathyroid hormone-related peptide: exploration of their biological importance. Bone 25, 87–90 (1999).
Mutsaers, A. J. et al. Modeling distinct osteosarcoma subtypes in vivo using Cre:lox and lineage-restricted transgenic shRNA.Bone 55, 166–178 (2013).
Hainaut, P. & Plymoth, A. Targeting the hallmarks of cancer: towards a rational approach to next-generation cancer therapy. Curr. Opin. Oncol. 25, 50–51, https://doi.org/10.1097/CCO.0b013e32835b651e (2013).
Bieging, K. T., Mello, S. S. & Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370, https://doi.org/10.1038/nrc3711 (2014).
Ng, A. J., Mutsaers, A. J., Baker, E. K. & Walkley, C. R. Genetically engineered mouse models and human osteosarcoma. Clin. Sarcoma Res. 2, 19, https://doi.org/10.1186/2045-3329-2-19 (2012).
Mutsaers, A. J., & Walkley, C. R. Cells of origin in osteosarcoma: mesenchymal stem cells or osteoblast committed cells?. Bone 62, 56–63 (2014).
Janeway, K. A., & Walkley, C. R. Modeling human osteosarcoma in the mouse: From bedside to bench. Bone 47, 859–865 (2010).
Walkley, C. R. et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 22, 1662–1676, https://doi.org/10.1101/gad.1656808 (2008).
Vogelstein, B., Lane, D. & Levine, A. J. Surfing the p53 network. Nature 408, 307–310, https://doi.org/10.1038/35042675 (2000).
Miyashita, T., Harigai, M., Hanada, M. & Reed, J. C. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res. 54, 3131–3135 (1994).
Murphy, M., Hinman, A. & Levine, A. J. Wild-type p53 negatively regulates the expression of a microtubule-associated protein. Genes Dev. 10, 2971–2980 (1996).
Hoffman, W. H., Biade, S., Zilfou, J. T., Chen, J. & Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 277, 3247–3257, https://doi.org/10.1074/jbc.M106643200 (2002).
Pietrzak, M., & Puzianowska-Kuznicka, M. p53-dependent repression of the human MCL-1 gene encoding an anti-apoptotic member of the BCL-2 family: the role of Sp1 and of basic transcription factor binding sites in the MCL-1 promoter. Biol. Chem. 389, 383–393 (2008).
Werner, H., Karnieli, E., Rauscher, F. J. & LeRoith, D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Natl Acad. Sci. USA 93, 8318–8323 (1996).
Astanehe, A. et al. Mechanisms underlying p53 regulation of PIK3CA transcription in ovarian surface epithelium and in ovarian cancer. J. Cell Sci. 121, 664–674, https://doi.org/10.1242/jcs.013029 (2008).
Aylon, Y. & Oren, M. The Paradox of p53: what, how, and why? Cold Spring Harb. Perspect. Med. 6, a026328, https://doi.org/10.1101/cshperspect.a026328 (2016).
Miled, C., Pontoglio, M., Garbay, S., Yaniv, M. & Weitzman, J. B. A genomic map of p53 binding sites identifies novel p53 targets involved in an apoptotic network. Cancer Res. 65, 5096–5104, https://doi.org/10.1158/0008-5472.CAN-04-4232 (2005).
Artigas, N. et al. p53 inhibits SP7/Osterix activity in the transcriptional program of osteoblast differentiation. Cell Death Differ. 24, 2022–2031 (2017).
Dobroff, A. S. et al. Silencing cAMP-response element-binding protein (CREB) identifies CYR61 as a tumor suppressor gene in melanoma. J. Biol. Chem. 284, 26194–26206, https://doi.org/10.1074/jbc.M109.019836 (2009).
Son, J., Lee, J. H., Kim, H. N., Ha, H., & Lee, Z. H. cAMP-response-element-binding protein positively regulates breast cancer metastasis and subsequent bone destruction. Biochem. Biophys. Res. 398, 309–314 (2010).
Wu, D. et al. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene 26, 5070–5077, https://doi.org/10.1038/sj.onc.1210316 (2007).
Giebler, H. A., Lemasson, I. & Nyborg, J. K. p53 recruitment of CREB binding protein mediated through phosphorylated CREB: a novel pathway of tumor suppressor regulation. Mol. Cell. Biol. 20, 4849–4858 (2000).
Junk, D. J. et al. Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia 10, 450–461 (2008).
Ravnskjaer, K. et al. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 26, 2880–2889, https://doi.org/10.1038/sj.emboj.7601715 (2007).
Everett, L. J. et al. Integrative genomic analysis of CREB defines a critical role for transcription factor networks in mediating the fed/fasted switch in liver. BMC Genom. 14, 337, https://doi.org/10.1186/1471-2164-14-337 (2013).
Zhang, X. et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl Acad. Sci. USA 102, 4459–4464, https://doi.org/10.1073/pnas.0501076102 (2005).
Sanchez, Y. et al. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 5, 5812, https://doi.org/10.1038/ncomms6812 (2014).
Kenzelmann Broz, D. et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031, https://doi.org/10.1101/gad.212282.112 (2013).
Wang, X. et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J. Cell Biol. 172, 115–125, https://doi.org/10.1083/jcb.200507106 (2006).
Nicholson, D. W. et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376, 37–43, https://doi.org/10.1038/376037a0 (1995).
Fridman, J. S. & Lowe, S. W. Control of apoptosis by p53. Oncogene 22, 9030–9040, https://doi.org/10.1038/sj.onc.1207116 (2003).
Wang, C., Lisanti, M. P. & Liao, D. J. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle 10, 57–67, https://doi.org/10.4161/cc.10.1.14449 (2011).
Ma, Z., Webb, D. J., Jo, M. & Gonias, S. L. Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. J. Cell Sci. 114, 3387–3396 (2001).
Rajalingam, K., Schreck, R., Rapp, U. R., & Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 1773, 1177–1195 (2007).
Zhang, K. Y. et al. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol. Cell 15, 279–286 (2004).
Jiao, X. et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 28, 1805–1806, https://doi.org/10.1093/bioinformatics/bts251 (2012).
Xie, F. et al. Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J. Med. Chem. 58, 5075–5087, https://doi.org/10.1021/acs.jmedchem.5b00468 (2015).
Ng, A. J. et al. The DNA helicase recql4 is required for normal osteoblast expansion and osteosarcoma formation. PLoS Genet. 11, e1005160, https://doi.org/10.1371/journal.pgen.1005160 (2015).
Lain, S. et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13, 454–463 (2008).
Mitton, B. et al. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia 30, 2302–2311 (2016).
Acknowledgements
We thank the SVH BioResources Centre for animal care. This work was supported by grants: NHMRC Project Grant APP1084230 (to C.W., M.W., and T.J.M.); Victorian Cancer Agency Mid-Career Research Fellowship (CRW; MCRF15015); in part by the Victorian State Government Operational Infrastructure Support Program (to St. Vincent’s Institute).
Author information
Authors and Affiliations
Contributions
M.W., T.J.M. and C.W. conceived study; M.W., S.T. and P.H. performed experiments, analysed and interpreted data; T.J.M and C.W. provided intellectual input and conceptual advice; M.W., T.J.M. and C.W. wrote the manuscript; all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by R. Pearson
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Walia, M.K., Taylor, S., Ho, P.W.M. et al. Tolerance to sustained activation of the cAMP/Creb pathway activity in osteoblastic cells is enabled by loss of p53. Cell Death Dis 9, 844 (2018). https://doi.org/10.1038/s41419-018-0944-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0944-8
This article is cited by
-
cAMP-PKA/EPAC signaling and cancer: the interplay in tumor microenvironment
Journal of Hematology & Oncology (2024)
-
Loss of p53 in mesenchymal stem cells promotes alteration of bone remodeling through negative regulation of osteoprotegerin
Cell Death & Differentiation (2021)
-
The HBx–CTTN interaction promotes cell proliferation and migration of hepatocellular carcinoma via CREB1
Cell Death & Disease (2019)
-
Osteosarcoma in the Post Genome Era: Preclinical Models and Approaches to Identify Tractable Therapeutic Targets
Current Osteoporosis Reports (2019)

{kind=link}
{kind=link}