
Abstract
1. Name of Disease (Synonyms)
Pontocerebellar hypoplasia type 9 (PCH9) and spastic paraplegia-63 (SPG63).
2. OMIM# of the Disease
615809 and 615686.
3. Name of the Analysed Genes or DNA/Chromosome Segments
AMPD2 at 1p13.3.
4. OMIM# of the Gene(s)
102771.
Similar content being viewed by others

1. Disease characteristics
1.1 Name of the disease (Synonyms)
Pontocerebellar hypoplasia type 9 (PCH9) and spastic paraplegia-63 (SPG63).
1.2 OMIM# of the disease
615809 and 615686.
1.3 Name of the analysed genes or DNA/chromosome segments
AMPD2 at 1p13.3.
1.4 OMIM# of the gene(s)
102771.
1.5 Mutational spectrum
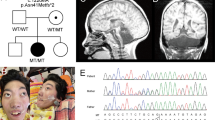
The autosomal recessive disorders PCH9 and SPG63 are caused by variants in AMPD2, a gene encoding adenosine monophosphate deaminase 2. AMPD2 has a critical role in energy metabolism as part of the purine nucleotide cycle by converting adenosine monophosphate to inosine monophosphate. Loss of AMPD2 function is associated with adenosine-mediated neurotoxicity and defective protein synthesis secondary to guanine nucleotide deficiency [1]. To date, 17 variants have been associated with PCH9: ten missense, three nonsense, three frameshift and one splice site variant. Of these, six were inherited as compound heterozygotes. There has been one variant associated with SPG63: a frameshift variant predicted to disrupt three of six putative AMPD2 isoforms. All but one missense variant localise to the catalytic domain of the protein.
1.6 Analytical methods
Sanger sequencing of the coding exons and flanking intronic regions of all AMPD2 transcripts (NCBI reference sequences: NM_004037.7, NM_001257360.1, NM_001308170.1, NM_203404.1, NM_001257361.1 and NM_139156.3). Massively parallel sequencing (MPS) approaches, including exome sequencing or a targeted gene panel, may be more cost effective than Sanger sequencing.
1.7 Analytical validation
Genomic DNA from a second, independent sample should be used for analytical validation. Ideally, variants identified using Sanger sequencing would be confirmed by a different chemistry or an alternative method. Variants identified using MPS approaches should be confirmed using bidirectional Sanger sequencing. Targeted techniques such as real-time quantitative PCR, enzymatic activity assays and enzyme-linked immunosorbent assay can also be utilised to confirm loss or reduction of AMPD2 using lysed fibroblasts or peripheral blood mononuclear cells. If possible, segregation would be confirmed by sequencing parental genomic DNA.
1.8 Estimated frequency of the disease
(Incidence at birth (“birth prevalence”) or population prevalence. If known to be variable between ethnic groups, please report):
Twenty-three and two genetically confirmed individuals with PCH9 or SPG63, respectively, have been reported to date [1,2,3,4,5,6,7,8,9]. The frequency and prevalence of these disorders are not known, although collectively pontocerebellar hypoplasia (PCH) syndromes are very rare, whereas hereditary spastic paraplegia (HSP) disorders are more common, with an estimated prevalence of 1.8 per 100,000 [10]. There is no known phenotypic disparity between ethnic groups; however, both disorders appear to be more prevalent in ethnicities that practice consanguineous marriage due to their recessive nature.
1.9 Diagnostic setting
Yes | No | |
A. (Differential) diagnostics | ⊠ | ☐ |
B. Predictive Testing | ☐ | ⊠ |
C. Risk assessment in Relatives | ⊠ | ☐ |
D. Prenatal | ⊠ | ☐ |
Comment: PCH syndromes are a group of rare, inherited neurodegenerative disorders with prenatal onset [11]. They share characteristic features of progressive degeneration of the ventral pons, inferior olive and cerebellum, with subsequent degeneration of supratentorial structures such as cerebral cortex [12], although perinatal or postnatal onset is sometimes observed and non-progressive forms do exist [13, 14]. PCH syndromes are associated with global developmental delay and a range of other neurological sequelae, depending on the subtype. To date, 16 genes have been linked to PCH (Table 1).
HSP are a group of inherited, neurodegenerative motor neuron diseases with a variable age of onset extending between early infancy and late adulthood. Affected individuals share characteristic features of slowly progressive, retrograde degeneration of the corticospinal tract beginning at the distal end of motor neurons [15]. HSP are clinically divided into pure (or uncomplicated) and complicated forms; pure HSP presents in isolation, whereas complicated HSP are observed alongside other neurological or non-neurological manifestations. Isolated and complicated HSP are both associated with pyramidal signs, namely lower limb weakness and spasticity, often with urinary dysfunction. Individuals with complicated HSP may additionally present with seizures, ataxia, intellectual disability, a thin corpus callosum, peripheral neuropathies or extrapyramidal signs, among others [16]. HSP are a genetically heterogeneous group of disorders and, to date, more than 75 loci and 55 genes have been linked to HSP via autosomal dominant, autosomal recessive, X-linked and mitochondrial modes of transmission [17, 18].
2. Test characteristics
Genotype or disease | A: true positives | C: false negative | ||
Present | Absent | B: false positives | D: true negative | |
Test | ||||
Pos. | A | B | Sensitivity: Specificity: | A/(A+C) D/(D+B) |
Neg. | C | D | Pos. predict. value: Neg. predict. value: | A/(A+B) D/(C+D) |
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
Approximately 100% employing the analytical methods and validation described. However, the limitations of genomic testing mean some variants may be missed due to allele dropout, large deletions and duplications or suboptimal coverage. A second analytical method should be used for variant-negative individuals with a highly suggestive PCH9 or SPG63 phenotype, particularly if no other candidate variants have been identified in other PCH or HSP genes.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
Approximately 100% employing the analytical methods and validation described. False positives may arise owing to misinterpretation of rare polymorphic variants.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
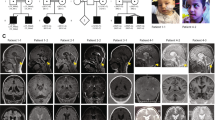
PCH9 is characterised by progressive atrophy of the cerebellum and brainstem, progressive postnatal microcephaly, small or absent corpus callosum and a distinctive midbrain “figure of 8” appearance on axial magnetic resonance (MR) imaging. Affected individuals manifest severe clinical features, including profound developmental delay, progressive spasticity, axial hypotonia, cortical visual impairment and intellectual disability, often with dysmorphic features and drug-resistant seizures. Less frequent findings include nystagmus, strabismus and axonal peripheral neuropathy in the second decade of life. Although PCH syndromes share several common characteristics, namely atrophy of the cerebellum and brainstem, the combination of a “figure of 8” appearance of the midbrain on axial MR imaging with severe hypoplasia or absence of the corpus callosum appears unique to PCH9. The proportion of AMPD2 variants is approximately 100% in individuals with classic PCH9, but assumedly less for individuals with a milder callosal and brainstem phenotype, whose imaging features would overlap with other PCH syndromes to a greater degree.
SPG63 is a complicated HSP subtype. As only two individuals from one family have been described to date [7], the phenotypic spectrum of SPG63 remains to be defined. The age of onset was reported to be 14 months, evidence by delayed walking and a scissoring gait. Affected individuals had increased deep tendon reflexes but no objective sensory loss and normal cognition. MR imaging of one individual revealed a thin, dysplastic corpus callosum and periventricular deep white matter changes with no brainstem or cerebellar abnormalities, distinct from the severe brain phenotype associated with PCH9. As only one family has been described, the clinical sensitivity for SPG63 remains unknown.
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
Approximately 100% for PCH9 and SPG63, but not other PCH and HSP subtypes.
2.5 Positive clinical predictive value (life time risk to develop the disease if the test is positive)
Approximately 100%, as PCH9 and SPG63 appear to be fully penetrant disorders.
2.6 Negative clinical predictive value
(Probability not to develop the disease if the test is negative). Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
Approximately 100%. PCH9 and SPG63 appear to be fully penetrant disorders with prenatal or infantile onset, respectively. However, it is unknown whether carriers are susceptible or have an increased risk of developing other metabolic disorders later in life.
Index case in that family had not been tested:
Variable, as PCH and HSP show allelic and locus heterogeneity associated with variable clinical presentations. As PCH syndromes generally have prenatal, perinatal or postnatal onset, there is minimal risk for a non-affected individual after infancy. However, as HSP syndromes exhibit a variable age of onset, ranging from early infancy to late adulthood, the risk to develop the disorder for a non-affected individual will depend on the observed mode of inheritance and age of onset for the index case.
3. Clinical Utility
3.1 (Differential) diagnostics: the tested person is clinically affected
(To be answered if in 1.9 “A” was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?
No. | ☐ (continue with 3.1.4) | |
Yes. | ⊠ | |
Clinically | ⊠ - SPG63 | |
Imaging | ⊠ - PCH9 | |
Endoscopy | ☐ | |
Biochemistry | ☐ | |
Electrophysiology | ☐ | |
Other (please describe): | ||
3.1.2 Describe the burden of alternative diagnostic methods to the patient
Clinically—low burden. SPG63, similar to other HSP subtypes, is clinically diagnosed by the presence of typical symptoms such as lower limb weakness, progressive spasticity, increased deep tendon reflexes, normal brain MR imaging, often a family history and diagnosis by exclusion. Differential diagnoses for HSP include a variety of metabolic disorders (e.g., adrenoleukodystrophy), autoimmune and infectious diseases (e.g., myelitis), and degenerative disorders (e.g., spastic ataxias), as well as structural damage (e.g., cerebral palsy) [17].
Imaging—low burden. The burden of imaging to the individual is low and predominantly relates to radiation exposure (for computerised tomography) and risks associated with general anaesthetic, which is usually required for children aged 3 months to 5 years, or older individuals with developmental or behavioural disorders.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Clinical testing may represent a more cost effective diagnostic method for making a clinical diagnosis of HSP; however, making a specific diagnosis of SPG63 requires genetic testing as the clinical features are not unique enough to distinguish SPG63 from other HSP. In addition, clinical testing does not provide accurate information on recurrence risk and reproductive options for family members.
As PCH9 cannot be suspected without prior neuroimaging, brain MR imaging is technically not an alternative diagnostic method. Computerised tomographic scanning does not usually provide adequate imaging of the brainstem and cerebellum, and thus MR imaging is required. The characteristic neuroradiological features of PCH9 do enable a diagnosis using brain MR imaging, although its specificity and sensitivity is relatively lower compared with genetic testing, which also provides important information on recurrence risk and reproductive options for family members. Although brain MR imaging with a general anaesthetic is less cost effective than a genetic test, it is critical for obtaining an accurate PCH9 diagnosis.
3.1.4 Will disease management be influenced by the result of a genetic test?
No. | ☐ | |
Yes. | ⊠ | |
Therapy (please describe) | Treatment for PCH9 and SPG63 is purely symptomatic and supportive. | |
Prognosis (please describe) | The prognosis for PCH9 is generally poor, as most affected individuals die during childhood or early adolescence. The prognosis of SPG63 is unknown but appears to be relatively favourable with survival into adulthood. | |
Management (please describe) | Management for both PCH9 and SPG63 is symptomatic. In PCH9, the primary symptoms are neurological and developmental. Management therefore involves treatment of epilepsy, ensuring adequate nutritional needs, which may require gastrostomy feeding, antibiotics for chest infections and speech and physical therapy. Due to the progressive and life limiting nature of this condition, family support including input from palliative care services may be required. In SPG63, the primary symptom is progressive spasticity. This is managed with monitoring for orthopaedic complications such as hip dysplasia, physical therapy, medications such as baclofen, diazepam and botulinum toxin, as well as occasionally intervention by surgical approaches including muscle and tendon release, intrathecal baclofen or selective dorsal rhizotomy. |
3.2 Predictive Setting: the tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 “B” was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe). Not applicable.If the test result is negative (please describe). Not applicable.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Not applicable.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 “C” was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes. If a disease-associated variant is identified in the index patient, genetic testing of the parents, unaffected offspring and extended family can be offered for genetic counselling, while options such as prenatal diagnosis and pre-implantation genetic diagnosis can be explored.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes, in theory. PCH9 and SPG63 are autosomal recessive disorders that appear to be invariably inherited through the germline. Although it can be assumed that the parents of an affected offspring are heterozygous carriers, cascade genetic testing should be completed in all family members to enable accurate genetic counselling, particularly for those individuals wanting children.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
No. PCH9 and SPG63 are fully penetrant disorders with prenatal or early infancy onset; hence, this situation is unlikely to arise.
3.4 Prenatal diagnosis (To be answered if in 1.9 “D” was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes. Prenatal diagnosis is possible by molecular analysis of amniotic fluid fetal cells or chronic villus sampling, and by fetal ultrasound or MR imaging in the case of PCH9. Two carriers have a 25% chance of having an affected offspring with PCH9 or SPG63.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
A genetic diagnosis will prevent unnecessary further investigations and provide important prognostic information for clinicians and families. Furthermore, a positive genetic test will facilitate accurate genetic counselling and carrier testing for families and enable early prenatal or pre-implantation genetic diagnosis for relatives wanting children. Although no curative treatment for PCH9 or SPG63 is available at this time, personalised or precision medicine treatment options may become available in the future for individuals with loss or reduction of AMPD2 function.
References
Akizu N, Cantagrel V, Schroth J, Cai N, Vaux K, McCloskey D, et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell. 2013;154:505–17.
Accogli A, Iacomino M, Pinto F, Orsini A, Vari MS, Selmi R, et al. Novel AMPD2 mutation in pontocerebellar hypoplasia, dysmorphisms, and teeth abnormalities. Neurol Genet. 2017;3:e179.
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model–based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2014;17:578.
Kortüm F, Jamra RA, Alawi M, Berry SA, Borck G, Helbig KL, et al. Clinical and genetic spectrum of AMPD2-related pontocerebellar hypoplasia type 9. Eur J Hum Genet. 2018;26:695–708.
Marsh AP, Lukic V, Pope K, Bromhead C, Tankard R, Ryan MM, et al. Complete callosal agenesis, pontocerebellar hypoplasia, and axonal neuropathy due to AMPD2 loss. Neurol Genet. 2015;1:e16.
Marsh AP, Yap P, Tan T, Pope K, White SM, Chong B, et al. A novel AMPD2 mutation outside the AMP deaminase domain causes pontocerebellar hypoplasia type 9. Am J Med Genet A. 2017;173:820–3.
Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–11.
Severino M, Zara F, Rossi A, Striano P. Teaching NeuroImages: figure of 8: the clue to the diagnosis of AMPD2 pontocerebellar hypoplasia (PCH9). Neurology. 2017;89:e172–3.
Vanderver A, Simons C, Helman G, Crawford J, Wolf NI, Bernard G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol. 2016;79:1031–7.
Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42:174–83.
Rudnik-Schoneborn S, Barth PG, Zerres K. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet. 2014;166c:173–83.
Namavar Y, Barth PG, Poll-The BT, Baas F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis. 2011;6:50.
Marin-Valencia I, Gerondopoulos A, Zaki MS, Ben-Omran T, Almureikhi M, Demir E, et al. Homozygous mutations in TBC1D23 lead to a non-degenerative form of pontocerebellar hypoplasia. Am J Hum Genet. 2017;101:441–50.
Ben-Zeev B, Hoffman C, Lev D, Watemberg N, Malinger G, Brand N, et al. Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, mental retardation, and spastic quadriplegia. J Med Genet. 2003;40:e96.
DeLuca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004;30:576–84.
Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–28.
Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev Neurol (Paris). 2015;171:505–30.
Noreau A, Dion PA, Rouleau GA. Molecular aspects of hereditary spastic paraplegia. Exp Cell Res. 2014;325:18–26.
Acknowledgements
This work was supported by EuroGentest2 (Unit 2: “Genetic testing as part of health care”), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. We acknowledge the participation of the patients and their families in these studies, as well as the generous financial support of the Lefroy and Handbury families. APLM was supported by an Australian Postgraduate Award. PJL is supported by an NHMRC Career Development Fellowship (GNT1032364). RJL is supported by a Melbourne Children’s Clinician Scientist Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Marsh, A.P.L., Novarino, G., Lockhart, P.J. et al. CUGC for pontocerebellar hypoplasia type 9 and spastic paraplegia-63. Eur J Hum Genet 27, 161–166 (2019). https://doi.org/10.1038/s41431-018-0231-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0231-2
This article is cited by
-
Pontocerebellar hypoplasia type-9 due to a novel p.Arg503Ter truncating variant in AMPD2: a report from India
Acta Neurologica Belgica (2023)
