
Abstract
Two-dimensional (2D) materials are considered for numerous applications in microelectronics, although several challenges remain when integrating them into functional devices. Weak adhesion is one of them, caused by their chemical inertness. Quantifying the adhesion of 2D materials on three-dimensional surfaces is, therefore, an essential step toward reliable 2D device integration. To this end, button shear testing is proposed and demonstrated as a method for evaluating the adhesion of 2D materials with the examples of graphene, hexagonal boron nitride (hBN), molybdenum disulfide, and tungsten diselenide on silicon dioxide and silicon nitride substrates. We propose a fabrication process flow for polymer buttons on the 2D materials and establish suitable button dimensions and testing shear speeds. We show with our quantitative data that low substrate roughness and oxygen plasma treatments on the substrates before 2D material transfer result in higher shear strengths. Thermal annealing increases the adhesion of hBN on silicon dioxide and correlates with the thermal interface resistance between these materials. This establishes button shear testing as a reliable and repeatable method for quantifying the adhesion of 2D materials.
Similar content being viewed by others
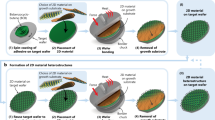


Introduction
The integration of two-dimensional (2D) materials into semiconductor devices is gaining momentum as the technology matures1,2,3. One of the remaining challenges is the adhesion, or lack thereof, of the 2D films to adjacent layers due to their van der Waals (vdW) nature4,5. Adhesion ultimately affects the device reliability, but also 2D layer transfer methods6,7. Although there are approaches to generally improve adhesion8,9, a quantitative and reliable method for measuring and evaluating the adhesion of 2D materials is lacking. Existing methodologies for measuring absolute adhesion energy values like blister tests10,11,12,13,14,15,16, nanoparticles17,18,19, and atomic force microscopy (AFM)20,21,22,23,24,25,26 are predominantly rather time-consuming. A shearing technique for interlayer cleavage energy of graphite was proposed by Wang et al.27. Other methods like substrate streching28,29,30,31,32 are restricted to specific substrates. The double cantilever beam method33,34,35 is applicable to gather information on the average adhesion of large area interfaces in the millimeter range but cannot be applied for local measurements in the micrometer range. A promising method for some applications is scratch testing36,37,38. However, the mechanical properties of all materials in a sample stack, such as hardness, influence the critical load in a scratch test39. This limits the comparability of multilayer systems or of measurements on different substrates. Four-point bending as an established adhesion measurement method40,41 was recently used to assess the adhesion of transition metal dichalcogenides (TMDC)42. All methods can be categorized by their ratio of forces perpendicular (mode I) and parallel (mode II) to the surface, or a mix.
Here, we introduce button shear testing as a quantitative method for determining the shear strengths of 2D materials. Button shear testing is an established method for adhesion measurements in typical semiconductor technologies and materials43,44,45,46,47,48, which yields rapid and conclusive results based on vast existing knowledge. We demonstrate the feasibility of the method for different 2D materials and substrate combinations and its application in evaluating the effect of sample treatments on adhesion.
Results
Button shear testing
The graphene samples were based on commercial chemical vapor deposited (CVD) monolayer graphene on copper. This was transferred onto three different substrates by a wet-etching process49 before button fabrication, (a) 250 nm silicon dioxide (SiO2) deposited with oxygen and tetraethyl orthosilicate precursors (TEOS SiO2), (b) 250 nm silicon nitride deposited from ammonia and dichlorosilane (Si3N4), and (c) 90 nm silicon dioxide grown by thermal oxidation of silicon (thermal SiO2). A subset of TEOS SiO2 and thermal SiO2 substrates was treated with oxygen (O2) plasma before graphene transfer. CVD hexagonal boron nitride (hBN) was also transferred from a copper growth substrate onto O2 plasma-treated thermal SiO2 substrates by a wet-etching process49, and subsets with hBN were annealed up to 1000 °C between the transfer process and button fabrication. Metal-organic chemical-vapor-deposited (MOCVD) molybdenum disulfide (MoS2)50,51 and MOCVD tungsten diselenide (WSe2)52 was transferred from the sapphire growth substrate onto O2 plasma-treated thermal SiO2 substrates. Schematic cross-sections of all samples are shown in Supplementary Fig. 1.
Buttons were fabricated by spin-coating polymethyl methacrylate (PMMA) onto the samples to create a 5 µm PMMA film. A 20 nm thick aluminum hard mask deposition followed by a lithography process step led to structures (buttons) with typical lateral dimensions of 60 ×100 µm. Anisotropic dry etching of the PMMA film in an oxygen (O2) plasma produced the desired buttons (Fig. 1a, b). Polystyrene as an alternative button material did not lead to a cuboid button cross-section after O2 plasma-etching and was excluded from the shear experiments (Supplementary Fig. 2).

a Colored scanning electron microscopy cross-section of a fabricated polymethyl methacrylate (PMMA) button (red). SiO2 (blue) and graphene (green) are indicated. b Colored microscope image of a PMMA button (red) on SiO2 (blue) before (left) and after (right) button shear testing. The shifted position due to the shear process is visible and the original button position before shear testing is indicated by white dashed lines on the right side. Button length and button width are assigned to the button dimensions. The white arrow on the upper right corner indicates the direction of the force F acting onto the button and the direction of the relative movement along x between shear head and button. c Schematic of the button shear test with graphene at the interface of the PMMA button and a SiO2 substrate. The shear head approaches the button along x and will measure F. d Influence of button length on the critical shear force FC. Measurements were performed with PMMA on SiO2 without 2D material and a fixed button width of 100 µm. Each data point represents a measurement on a separate button. A reliable result can be achieved at 60 µm button length.
Button shear test measurements were performed with a DAGE4000Plus pull-shear tester. The stage with the sample moved with a defined speed to create contact between the fixed shear head and the button (Fig. 1c). The lateral displacement of the stage was then recorded while the force that acts on the shear head was measured by the cartridge. The force increased as the shear head made contact with the buttons. The force reached a maximum at a certain point where the shearing of the button started, moving it slightly on the target (see Fig. 1b). After the initiation of the shear process, less force was required to maintain the shear process, and the measurement was stopped. The maximum recorded force corresponds to the force that is required to initiate the shear process. This is expected to occur at the weakest spot of the interface underneath the button and is labeled as critical shear force FC within this work.
Reference button shear tests were performed on samples with PMMA on thermal SiO2 and button lengths of 20, 40, and 60 µm. Typically button shear tests use button sizes in the millimeter range and measure FC in the Newton range43. However, 2D materials today must be expected to have cracks, holes, or other defects53,54,55 that influence their mechanical properties32,56,57 in such millimeter-range contact areas. We therefore implemented a measurement routine based on smaller buttons to overcome this challenge. A button length of 60 µm was established as a minimum for reproducible results by these initial experiments because smaller buttons with 20 µm and 40 µm button length show very low and scattered FC (Fig. 1d). PMMA is a soft material with low elastic modulus compared to other assistance layers in shear testing experiments with 2D materials58. That leads to button deformation near the contacted button edge. Hence, the ratio of forces perpendicular (mode I) and parallel (mode II) to the surface varies along the shear path43,59. The pronounced mixed mode near the contacted edge can be the reason for the very low FC at small button lengths60. The influence of plasma on PMMA edges61,62 is an additional possible reason for the FC variability with smaller button lengths.
Shear tests were further performed on a calibration sample that consists of a step in the (100) silicon surface. This step can be considered as a button that cannot be sheared off to assess the accuracy of the force and displacement measurement. Ideally, the measured force before contacting the stable obstacle is zero and jumps to a very high value upon contact. Here, we chose a shear speed of 1 µm s−1 to be able to stop the measurement in time and prevent damage to the tool. A large cartridge with high internal stiffness and a small cartridge for small forces were compared (Fig. 2a). The large cartridge delivered correct displacement values, but the noise in the force data before contact made reliable force measurements impossible. The small cartridge produced no noise in the force data and was chosen for the experiments. However, the pronounced force-dependent stiffness of the small cartridge leads to non-reliable displacement measurements. A multipoint calibration may reduce this effect, but there are several other system-dependent influences on the displacement measurement45. Therefore, no quantitative evaluation of the displacement was performed, but FC was extracted only from the force data. Dividing FC by the button area 100 µm x 60 µm leads to the area-independent shear strength τC.

a Force vs. displacement curves of measurements with two different cartridges contacting a stable obstacle. The small cartridge (blue) delivers a lower noise in the force signal at the cost of no reliable displacement data compared to the large cartridge (red). b Force vs. displacement curves and c extracted critical shear force FC and shear strength τC on samples with polymethyl methacrylate (PMMA) on SiO2 at different shear velocities from 1 µm s−1 (blue) to 50 µm s−1 (red). The black arrow in b indicates the increase in FC and reduction of the displacement at FC at higher shear velocities. The dotted gray line in c is a guide to the eye connecting the data points. The velocity 10 µm s−1 (orange) is highlighted and was used in further experiments with 2D materials.
The influence of the shear speed was evaluated on samples with PMMA on thermal SiO2 because shear speed effects strongly influence τC31,35,48. τC at low shear speeds below 5 µm s−1 was significantly lower than at high shear speeds (Fig. 2b, c). Similar trends were observed in previous studies48,63,64,65,66 and can be assigned to viscoelastic properties of PMMA67,68,69,70. We chose a shear speed of 10 µm s−1 for the experiments with 2D materials to prevent a significant influence of the viscoelastic properties of PMMA.
Graphene
The shear strength of graphene on the three substrates TEOS SiO2, Si3N4, and thermal SiO2 was measured with the optimized parameters of a small cartridge and 10 µm s−1 shear speed. Figure 3a compares τC of graphene on as deposited TEOS SiO2 (1.55 ± 0.31 MPa), Si3N4 (2.88 ± 0.31 MPa), and thermal SiO2 (2.68 ± 0.11 MPa). We correlated these results with the surface roughness sa of the substrates and the surface roughness of graphene on the substrates as the roughness is known to influence the adhesion of a 2D material21,71. AFM roughness data show that bare TEOS SiO2 has a roughness of sa = 0.76 nm and graphene on TEOS SiO2 has a lower roughness of sa = 0.64 nm. On the smoother substrate thermal SiO2 the roughness remains at sa = 0.22 nm without and with graphene. On Si3N4 the roughness increases from sa = 0.30 nm without graphene to sa = 0.42 nm with graphene (see Fig. 3b–d and Table 1). This increase in roughness may be attributed to PMMA residues from the wet etching transfer process72,73 and limits the validity of roughness measurements on transferred CVD-grown graphene. However, the lower roughness of graphene on TEOS SiO2 compared to bare TEOS SiO2 is an indication that the graphene does not fully follow the TEOS SiO2 morphology. This behavior on rough substrates is predicted in theoretical studies74,75 and leads to a smaller effective interface area and finally to a smaller adhesion76.

a Shear strength τC of graphene on TEOS SiO2 (dark green, number of buttons n = 11), Si3N4 (red, n = 9), and thermal SiO2 (dark blue, n = 5). The right blue box plot represents the τC of hBN on thermal SiO2 (n = 8). Box plots in light green (n = 6) and light blue (n = 9) show measurements of samples with O2 plasma treatment before the transfer of 2D material. Box plots indicate median (middle line), 25th, 75th percentile (box) and minimum and maximum (whiskers). b–d Atomic force microscopy (AFM) roughness measurements of TEOS SiO2 (sa = 0.76 nm), Si3N4 (sa = 0.30 nm), and thermal SiO2 (sa = 0.22 nm) without O2 plasma, respectively. e-g Contact angle CA measurements with water on TEOS SiO2 (CAwater = 7.3 ± 1.5 °), Si3N4 (CAwater = 19.3 ± 0.3 °), and thermal SiO2 (CAwater = 42.7 ± 0.3 °) without O2 plasma, respectively. h X-ray photoelectron spectroscopy (XPS) measurement of C 1 s peak on TEOS SiO2 before (dark green) and after (light green) O2 plasma treatment. i XPS measurement of C 1 s peak on thermal SiO2 before (dark blue) and after (light blue) O2 plasma treatment.
The TEOS SiO2 and thermal SiO2 samples were further exposed to O2 plasma. This was not the case for the Si3N4 sample, because an O2 plasma would have modified the surface to an undefined silicon-oxide-nitride composition8. The O2 plasma treatment increased τC of graphene on TEOS SiO2 to 2.21 ± 0.33 MPa, but we did not observe a significant change on thermal SiO2 (2.77 ± 0.32 MPa, Fig. 3a). AFM measurements show that the roughness of our oxides did not change significantly after the O2 plasma, and, hence, this was ruled out as the reason for the different behavior (Table 1). However, several other potential explanations for the influence of the plasma treatment on adhesion remain: O2 plasma reduces the water (H2O) contact angle (CAwater), increases the surface energy8,21,23, and reduces carbon contamination on the surface8. Contact angle measurements showed that the applied O2 plasma reduced the CAwater from 7.3° to 3.0° for TEOS SiO2 and from 42.7° to 2.7° for thermal SiO2, (Fig. 3e-g and Table 1). Since the reduction of CAwater is more pronounced on thermal SiO2, which showed no significant change in τC, we rule this effect out as the main contributor. X-ray photoelectron spectroscopy (XPS) measurements of C 1 s peak on TEOS SiO2 (Fig. 3h) and thermal SiO2 (Fig. 3i) before and after O2 plasma show that the reduction of carbon contamination on the surface is more pronounced on TEOS SiO2, which confirms previous investigations8. Therefore, we conclude that the adhesion increase is dominated by the effect of surface cleaning. We note that only buttons with visibly intact graphene underneath were included in the assessment. This is because the high CAwater caused cracking and partial delamination of graphene during the drying process in some cases. Buttons on areas with partially delaminated graphene were excluded to evaluate only pure graphene-substrate-interfaces (see also Supplementary Fig. 3). In all cases, delamination at the substrate-graphene interface occurred. This was confirmed by optical microscopy (Supplementary Fig. 4) and Raman measurements (Supplementary Fig. 5). Minor graphene residues are found predominantly near the contacted button edge. The pronounced mixed mode at the contacted button edge is a likely reason for this observation60.
Hexagonal boron nitride
hBN is of interest as a 2D dielectric for encapsulating electrically active 2D materials like graphene or semiconducting TMDCs1,77,78. In many integration schemes, hBN is therefore in contact with a three-dimensional substrate like SiO2. We have investigated the adhesion of hBN on thermal SiO2 with O2 plasma cleaning. The τC of 4.43 ± 0.95 MPa is significantly higher on this substrate compared to graphene, which was 2.77 ± 0.32 MPa, but also shows a higher standard deviation (Fig. 3a). This is in contrast with the work by Rokni and Lu25, who compared the adhesion of graphene and hBN on a silicon oxide substrate and measured a larger interfacial adhesion energy in the case of graphene. However, the high adhesion energy was only apparent when the graphene was pressed onto the substrate by a pressure of at least 3 MPa before measurement, and their experiments were conducted with (nearly) defect-free exfoliated 2D materials. They hypothesized that the large adhesion energy values for graphene on silicon oxide arose from non-vdW forces, i.e. hydrogen bonds (e.g. C-H…O-Si) or maybe even covalent bonds (e.g. C-O-Si)25. In our case, we expect a higher defect density of the CVD-grown hBN compared to the CVD-grown graphene because the growth processes are less mature53. As an indication of the defect density of graphene and hBN used in this work, we provide laser scanning microscope images, AFM measurements, and Raman measurements in Supplementary Fig. 6. A higher defect density can drastically increase the adhesion, e.g. by hydrogen bonds as shown for graphene on polymers30,32. This finding could be used to engineer 2D materials with defects at well-defined locations as a reasonable integration scheme to achieve sufficient adhesion of 2D-3D heterostructures if one can tolerate the altered electronic properties of the material at the defect sites.
Thermal annealing is an established post-process treatment to reduce polymer residues79,80, remove interfacial water residues81, and increase the adhesion9,37. Thermal annealing is also expected to lead to a more conformal contact of 2D materials with the respective substrates9,82 and to a larger effective interaction area of the van der Waals forces. We performed button shear tests with hBN on plasma-treated thermal SiO2 samples before and after thermal annealing in N2 atmosphere. The focus was on temperatures from 100 °C to 400 °C, which are typical annealing temperatures compatible with the back end of the line in silicon technology. Anneals up to 1000 °C were additionally executed to investigate possible effects above this regime. The button shear test showed that annealing up to 300 °C – 400 °C significantly increases the adhesion, in line with recent results for graphene9,38. Higher temperatures have only a minor additional influence on the adhesion, which may also lie within the standard deviation of the single measurements (Fig. 4a).

a Shear strength τC of monolayer hBN on thermal SiO2 after different thermal annealing temperatures in N2 atmosphere. A significant increase of the adhesion until 300 – 400 °C anneal temperature is visible. The average values with the corresponding error bars representing standard deviation are shifted slightly for clarity. The dashed line is a guide to the eye connecting the average values. The number of measured buttons n at the temperatures 100 °C, 200 °C, 300 °C, 400 °C, 800 °C, and 1000 °C is 5, 8, 8, 7, 6, and 8, respectively. b Thermal signal of scanning thermal microscopy (SThM) measurements on multilayer hBN on thermal SiO2 on Si stack before anneal (dark blue), after 400 °C (orange), and after 1000 °C (dark red). The signal is normalized to the signal of SiO2 on Si. The thermal resistance is reduced after a 400 °C anneal and remains unchanged after an additional anneal at 1000 °C. Two measurements per temperature are shown to indicate the reproducibility. c Exemplary topography map and d thermal signal map. These maps were collected on sample 1 after a 400 °C anneal. e Full-width at half-maximum (FWHM) \({{\varGamma }}_{{E}_{2{{{{{\rm{g}}}}}}}}\) of hBN Raman signal on monolayer hBN on thermal SiO2 after different thermal anneal temperatures. Higher anneal temperatures result in a shift towards higher FWHM. The number of Raman measurements on hBN without anneal, after 100 °C anneal, 200 °C, and 400 °C anneal are 1105, 145, 262, and 115, respectively.
We performed scanning thermal microscopy (SThM) to determine the thermal conductivity (or thermal resistance)83,84,85 of the multilayer hBN on thermal SiO2 samples before and after annealing as a measure of the conformal contact of the 2D material with its substrate9,82. In SThM, the measured thermal signal in Volt corresponds to the thermal interface resistances (TIRs) between the layers for a system of ultrathin layers86. This, in turn, correlates with high adhesion as shown for other material systems87,88,89. The thermal signal (V) in the hBN samples decreases significantly from 4.0 V to 3.1 V after a 400 °C anneal (Fig. 4b), but a subsequent anneal at 1000 °C does not further decrease the thermal signal, which remained at 3.2 V. We assume that the TIR between the Si and the thermal SiO2 remained unaffected by annealing at 400 °C and 1000 °C, and attribute the TIR reduction to a change in the hBN and thermal SiO2 interface. This is in good agreement with the adhesion measurement data. Measurements with different probe tips and comparative measurements on a similar sample demonstrate the reproducibility of the results. Additional SThM measurements for hBN on bulk Si samples show a similar behavior (Supplementary Fig. 7).
Neuman et al. showed for graphene that slight deformations occur when the 2D material follows a substrate’s roughness, leading to nanometer strain variations90. We therefore employed Raman measurements to corroborate our hypothesis that the hBN forms a more conformal contact with the SiO2 substrate upon annealing. Our Raman data of E2g peaks of the hBN on thermal SiO2 before and after thermal annealing show a slight increase of the full-width at half-maximum (FWHM) \({{\varGamma }}_{{E}_{2{{{{{\rm{g}}}}}}}}\) with higher anneal temperatures (histograms in Fig. 4e). This is a signature of increased strain variation, which influences the position of the E2g peak of hBN. The peak broadening is caused by overlapping subpeaks of different strain values in different directions91, and serves to explain the increased adhesion upon annealing.
Molybdenum disulfide and tungsten diselenide
We transferred MoS2 and WSe2 with lateral dimensions of approximately one by one centimeter onto thermal SiO2 with O2 plasma cleaning and created buttons on top of and next to the TMDCs. Supplementary Fig. 8 shows Raman spectra of MoS2 and Wse2 films on SiO2. Figure 5 correlates the measured shear strength with the button position on, partly on, or next to the TMDC film. On the WSe2 sample, button column six, row five is on WSe2 and has a τC = 3.39 MPa, button column six, row six is partly on WSe2 (τC = 15.76 MPa), and button column six, row seven is next to WSe2 and directly on SiO2 (τC = 34.43 MPa).

Optical microscope stitching images of a MoS2 sample and b WSe2 sample. A nine-by-nine array of buttons is measured on both samples. The images are taken after resist development and aluminum etching but before PMMA and 2D material etching. The MoS2 and WSe2 films can be identified underneath the PMMA. τC measurements on c MoS2 and d WSe2 sample. τC of each button is displayed in a logarithmic color scale shown on the right side.
The shear strength on thermal SiO2 with O2 plasma cleaning is τC = 2.66 ± 0.09 MPa and τC = 3.47 ± 0.27 MPa for MoS2 and WSe2, respectively. Adhesion energy values for 2D materials on SiO2 in the literature vary widely. Therefore, only comparisons of two materials with the same measurement method can be used as references. Few publications compare the adhesion of MoS2 on SiO2 with graphene on SiO2. They consistently conclude lower adhesion energy for MoS2 on SiO2 compared to graphene on SiO2 by blister test10,14, spontaneously formed blisters92 intercalation of nanoparticles18, and AFM techniques25,93. The button shear test also assigns a slightly lower shear force to MoS2 compared to graphene (τC = 2.77 ± 0.32 MPa) on thermal SiO2 with O2 plasma cleaning. τC of WSe2 is significantly higher and exhibits a gradient over the film. τC of WSe2 is highest at columns three to five and gradually decreases to columns eight and nine. The reason for this observation remains unclear, although one possible explanation may be non-uniform drying after wet transfer. This data shows that button shear testing can reveal nonuniformities in 2D material samples and may be used to optimize deposition, transfer, and subsequent device fabrication process steps regarding uniformity and adhesion on a wafer scale.
Control of the mechanical properties of the button material is crucial for reproducible results. As discussed previously, the relatively soft PMMA leads to button deformation and pronounced mixed mode near the contacted button edge. Variations in button fabrication can lead to changes in the PMMA properties and, hence, to changes in the measured shear strength60. The shear strength of PMMA on thermal SiO2 is τC = 31.84 ± 2.61 MPa on the MoS2 sample and τC = 30,00 ± 7,18 MPa on the WSe2 sample. Previous measurements of PMMA on thermal SiO2 in Fig. 1d with 60 µm buttons (FC = 189.75 ± 1.99 mN, τC = 31.63 ± 0.33 MPa) and in Fig. 2c with 10 µm s−1 (τC = 34.60 MPa) performed months before these measurements led to similar τC, confirming the reproducibility of button shear testing with PMMA as button material.
Table 2 compares the button shear testing method with other adhesion measurement methods for 2D materials in the literature. It compares favorably in terms of accuracy and measurement effort and provides fraction mode data for 2D materials on standard semiconductor substrates like silicon.
Discussion
We established button shear testing as a viable and quantitative technique for adhesion measurements of 2D materials parallel to the surface. We established suitable fabrication and measurement parameters for button dimensions and shear speed. The examples of graphene, hBN, MoS2, and WSe2 demonstrate that this approach is suitable for different 2D materials and on various substrates like Si3N4 and different silicon dioxides. Our results indicate that low surface roughness is beneficial for the strong adhesion of graphene on its substrate. We further used the method to explore means of enhancing adhesion, in particular, O2 plasma treatments of SiO2 surfaces before graphene transfer or post-transfer annealing after a wet transfer of hBN. Our work thus introduces a reliable adhesion measurement technique for 2D materials and provides measurement parameters that can be utilized for quantifying and enhancing the adhesion of 2D materials on rigid substrates. Button shear testing may thus play a crucial role in the development of 2D materials for commercial semiconductor production.
Methods
Reference sample
A reference sample without 2D material was fabricated by spin-coating polymethyl methacrylate in chlorobenzene onto an oxidized silicon wafer to create a 5 µm PMMA film. A 20 nm thick aluminum (Al) hard mask was deposited by thermal evaporation. S1805 resist was used to define structures (buttons) with typical lateral dimensions of 60 × 100 µm. The alkaline developer in this process simultaneously etched the Al hard mask. A myplas-III© tool from Plasma Electronic was used to structure the PMMA in an O2 plasma. The buttons were tested both after the removal of the Al film as the last processing step and with the Al film remaining on top of the buttons. We observed no differences in the delamination behavior. The presence of Al provided a higher optical contrast and, hence, faster identification of buttons, so the process option with Al on top of the buttons was chosen for our experiments. A schematic cross-section of the reference sample can be found in Supplementary Fig. 1a. The reference sample was used for button shear test measurements with different button dimensions in Fig. 1d and with different shear speeds in Fig. 2b, c.
Calibration sample
A calibration sample was fabricated by a lithography process step on blank (100) silicon followed by anisotropic wet etching in potassium hydroxide (KOH) solution. This creates a step in the silicon that can be considered as button that cannot be sheared away. A schematic cross-section of the calibration sample can be found in Supplementary Fig. 1b. The calibration sample was used for button shear test measurements with different cartridges in Fig. 2a.
Graphene samples for button shear testing
Our graphene samples were based on commercial chemical vapor deposited (CVD) monolayer graphene on copper. This was transferred onto three different substrates by a wet-etching process before button fabrication, (a) 250 nm silicon dioxide deposited with oxygen and tetraethyl orthosilicate precursors (TEOS SiO2), (b) 250 nm silicon nitride deposited from ammonia and dichlorosilane (Si3N4), and (c) 90 nm silicon dioxide grown by thermal oxidation of silicon (thermal SiO2). A subset of TEOS SiO2 and thermal SiO2 substrates was treated with O2 plasma before graphene transfer. A schematic cross-section of the graphene samples for button shear testing can be found in Supplementary Fig. 1c-g.
hBN samples for button shear testing
Our hBN samples were based on commercial CVD monolayer hBN, transferred onto O2 plasma-treated thermal SiO2 substrates. Thermal annealing of hBN samples was conducted in nitrogen atmosphere with a hold time of two hours in a Jipelec Jetfirst 300 tool between the transfer process and button fabrication. A schematic cross-section of the hBN samples for button shear testing can be found in Supplementary Fig. 1h-i.
MoS2 and WSe2 samples for button shear testing
The MoS2 and WSe2 samples were based on MOCVD-grown few-layer MoS2 and WSe2 on sapphire substrates. After spin-coating with PMMA, the TMDC was delaminated from sapphire by KOH/H2O and transferred onto O2 plasma-treated thermal SiO2 substrates. A schematic cross-section of the MoS2 and WSe2 samples for button shear testing can be found in Supplementary Fig. 1k and Supplementary Fig. 1l, respectively.
hBN samples for Scanning thermal microscopy (SThM) measurements
SThM samples were prepared by growing 15 nm of thermal oxide on silicon. A lithography step and wet-etching of SiO2 with hydrofluoric acid (HF) created areas with blank Si surface for referencing and normalization of the thermal signal. Multilayer hBN (15 nm) was transferred onto this substrate from copper foil using wet-etching transfer49. SThM measurements were performed at the edge of the transferred hBN area to be able to compare the thermal signal with and without hBN. A schematic cross-section and top view of the hBN samples for SThM measurements can be found in Supplementary Fig. 1j.
Button shear testing
Button shear test measurements were performed with a DAGE4000Plus pull-shear tester from Nordson Corporation. The samples with the processed buttons were placed into the sample holder of the tool. The shear head with a width of 100 to 120 µm was mounted into a cartridge, positioned 2 µm above the substrate surface, and a few tens of µm in front of a button. The stage with the sample moved with a defined speed to create contact between the fixed shear head and the button (Fig. 1c). The lateral displacement of the stage was then recorded while the force that acts on the shear head was measured by the cartridge. The force increased as the shear head made contact with the buttons. The force reached a maximum at a certain point where the shearing of the button started, moving it slightly on the target (see Fig. 1b). After the initiation of the shear process, less force was required to maintain the shear process, and the measurement was stopped. The maximum recorded force corresponds to the force that is required to initiate the shear process. This is expected to occur at the weakest spot of the interface underneath the button and is labeled as critical shear force FC within this work. Dividing FC by the button area 100 µm x 60 µm leads to the area-shear strength τC.
AFM roughness measurements
AFM roughness measurements were performed on the substrates TEOS SiO2 without O2 plasma, TEOS SiO2 with O2 plasma, Si3N4, thermal SiO2 without O2 plasma, and thermal SiO2 with O2 plasma and on graphene on TEOS SiO2 without O2 plasma, graphene on Si3N4, and graphene on thermal SiO2 without O2 plasma with a Bruker Dimension ICON AFM with Nanoscope V Controller. We used ScanAsyst-Air tips with a nominal radius of rtip,nom = 2 nm and a spring constant of knom = 0.4 N m−1 in ScanAsyst-Mode. Every sample was scanned at two different positions. Before and after scanning the samples, a calibration scan on a PA01 tip characterization sample from MikroMasch Europe was conducted to exclude tip degradation during the scan. The measurements were performed under ambient atmosphere (T = 21 °C, RH = 55 %).
XPS measurements
XPS measurements were performed on a Thermo Fisher Scientific ESCALAB Xi+ with an Al K alpha source at 1486.6 eV. TEOS SiO2 samples and thermal SiO2 samples were analyzed by XPS initially, exposed to the O2 plasma afterwards, and finally characterized by XPS again.
SThM measurements
SThM measurements were performed with a Bruker Dimension ICON AFM with the Nanoscope V Controller. Here, we used a VITA-DM-NANOTA-200 tip and a VITA Module to evaluate the thermal interface resistance (TIR) of hBN to both SiO2 and Si after thermal annealing. Multiple measurements were recorded before and after the annealing process. A plane correction was applied to remove thermal drifts that are common in SThM measurements. The mean value of the thermal signal of the respective area was determined by the software Gwyddion 2.58. This mean thermal signal was normalized such that the thermal signal of Si = 0 V and the thermal signal of SiO2 on Si = 1 V. Constant TIRs between Si and SiO2 are assumed.
Raman measurements
Raman measurements were performed on hBN before and after anneal with a 532 nm laser, 240 s integration time, and a 100x objective with a Horiba LabRAM Evolution HR system. A linear baseline correction and a fitting with a Lorentz-Gauss profile of the hBN E2g peak and the neon reference lines were performed to evaluate the hBN signal.
Data availability
Relevant data supporting the key findings of this study are available within the article and the Supplementary Information file. All raw data generated during the current study are available from the corresponding authors upon request.
References
Neumaier, D., Pindl, S. & Lemme, M. C. Integrating graphene into semiconductor fabrication lines. Nat. Mater. 18, 525–529 (2019).
Akinwande, D. et al. Graphene and two-dimensional materials for silicon technology. Nature 573, 507–518 (2019).
Lemme, M. C., Akinwande, D., Huyghebaert, C. & Stampfer, C. 2D materials for future heterogeneous electronics. Nat. Commun. 13, 1392 (2022).
Asselberghs, I. et al. Scaled transistors with 2D materials from the 300mm fab. in 2020 IEEE Silicon Nanoelectronics Workshop (SNW) 67–68 (IEEE, Honolulu, HI, USA, 2020). https://doi.org/10.1109/SNW50361.2020.9131651.
Zhai, Y. et al. Fabrication of graphene nanomesh FET terahertz detector. Micromachines 12, 641 (2021).
Quellmalz, A. et al. Large-area integration of two-dimensional materials and their heterostructures by wafer bonding. Nat. Commun. 12, 917 (2021).
Wittmann, S. et al. Assessment of wafer‐level transfer techniques of graphene with respect to semiconductor industry requirements. Adv. Mater. Technol. 8, 2201587 (2023).
Lukose, R. et al. Influence of plasma treatment on SiO2/Si and Si3N4/Si substrates for large-scale transfer of graphene. Sci. Rep. 11, 13111 (2021).
Megra, Y. T., Lim, S., Lim, T., Na, S. R. & Suk, J. W. Enhancement of the adhesion energy between monolayer graphene and SiO2 by thermal annealing. Appl. Surf. Sci. 570, 151243 (2021).
Koenig, S. P., Boddeti, N. G., Dunn, M. L. & Bunch, J. S. Ultrastrong adhesion of graphene membranes. Nat. Nanotechnol. 6, 543–546 (2011).
Liu, X. et al. Observation of pull-in instability in graphene membranes under interfacial forces. Nano Lett 13, 2309–2313 (2013).
Kitt, A. L. et al. How graphene slides: measurement and theory of strain-dependent frictional forces between graphene and SiO2. Nano Lett 13, 2605–2610 (2013).
Boddeti, N. G. et al. Graphene blisters with switchable shapes controlled by pressure and adhesion. Nano Lett 13, 6216–6221 (2013).
Lloyd, D. et al. Adhesion, stiffness, and instability in atomically thin MoS2 bubbles. Nano Lett 17, 5329–5334 (2017).
Dai, Z. et al. Interface-governed deformation of nanobubbles and nanotents formed by two-dimensional materials. Phys. Rev. Lett. 121, 266101 (2018).
Calis, M., Lloyd, D., Boddeti, N. & Bunch, J. S. Adhesion of 2D MoS2 to graphite and metal substrates measured by a blister test. Nano Lett 23, 2607–2614 (2023).
Zong, Z., Chen, C.-L., Dokmeci, M. R. & Wan, K. Direct measurement of graphene adhesion on silicon surface by intercalation of nanoparticles. J. Appl. Phys. 107, 026104 (2010).
Torres, J., Zhu, Y., Liu, P., Lim, S. C. & Yun, M. Adhesion energies of 2D graphene and MoS2 to silicon and metal substrates. Phys. Status Solidi A 215, 1700512 (2018).
Dai, Z., Sanchez, D. A., Brennan, C. J. & Lu, N. Radial buckle delamination around 2D material tents. J. Mech. Phys. 137, 103843 (2020).
Jiang, T. & Zhu, Y. Measuring graphene adhesion using atomic force microscopy with a microsphere tip. Nanoscale 7, 10760–10766 (2015).
Spear, J. C., Custer, J. P. & Batteas, J. D. The influence of nanoscale roughness and substrate chemistry on the frictional properties of single and few layer graphene. Nanoscale 7, 10021–10029 (2015).
Suk, J. W. et al. Probing the adhesion interactions of graphene on silicon oxide by nanoindentation. Carbon 103, 63–72 (2016).
Zeng, X., Peng, Y. & Lang, H. A novel approach to decrease friction of graphene. Carbon 118, 233–240 (2017).
Li, Y., Huang, S., Wei, C., Wu, C. & Mochalin, V. N. Adhesion of two-dimensional titanium carbides (MXenes) and graphene to silicon. Nat. Commun. 10, 3014 (2019).
Rokni, H. & Lu, W. Direct measurements of interfacial adhesion in 2D materials and van der Waals heterostructures in ambient air. Nat. Commun. 11, 5607 (2020).
Ku, B. et al. AFM‐based Hamaker constant determination with blind tip reconstruction. Adv. Mater. Technol. 8, 2200411 (2023).
Wang, W. et al. Measurement of the cleavage energy of graphite. Nat. Commun. 6, 7853 (2015).
Gong, L. et al. Interfacial stress transfer in a graphene monolayer nanocomposite. Adv. Mater. 22, 2694–2697 (2010).
Jiang, T., Huang, R. & Zhu, Y. Interfacial sliding and buckling of monolayer graphene on a stretchable substrate. Adv. Funct. Mater. 24, 396–402 (2014).
Wang, G. et al. Tuning the interfacial mechanical behaviors of monolayer graphene/PMMA nanocomposites. ACS Appl. Mater. Interfaces 8, 22554–22562 (2016).
Xu, C., Xue, T., Qiu, W. & Kang, Y. Size effect of the interfacial mechanical behavior of graphene on a stretchable substrate. ACS Appl. Mater. Interfaces 8, 27099–27106 (2016).
Dai, Z. et al. Mechanical behavior and properties of hydrogen bonded graphene/polymer nano-interfaces. Compos. Sci. Technol. 136, 1–9 (2016).
Yoon, T. et al. Direct measurement of adhesion energy of monolayer graphene as-grown on copper and its application to renewable transfer process. Nano Lett 12, 1448–1452 (2012).
Na, S. R., Suk, J. W., Ruoff, R. S., Huang, R. & Liechti, K. M. Ultra long-range interactions between large area graphene and silicon. ACS Nano 8, 11234–11242 (2014).
Xu, C. et al. Rate‐dependent decohesion modes in graphene‐sandwiched interfaces. Adv. Mater. Interfaces 6, 1901217 (2019).
Das, S., Lahiri, D., Lee, D.-Y., Agarwal, A. & Choi, W. Measurements of the adhesion energy of graphene to metallic substrates. Carbon 59, 121–129 (2013).
Gu, M. et al. Effects of thermal treatment on the adhesion strength and osteoinductive activity of single-layer graphene sheets on titanium substrates. Sci Rep 8, 8141 (2018).
Ivanov, E., Batakaliev, T., Kotsilkova, R., Otto, M. & Neumaier, D. Study on the adhesion properties of graphene and hexagonal boron nitride monolayers in multilayered micro-devices by scratch adhesion test. J. Mater. Eng. Perform. 30, 5673–5681 (2021).
Wang, X. et al. A review on the mechanical properties for thin film and block structure characterised by using nanoscratch test. Nanotechnol. Rev. 8, 628–644 (2019).
Dauskardt, R. H., Lane, M., Ma, Q. & Krishna, N. Adhesion and debonding of multi-layer thin film structures. Eng. Fract. Mech. 61, 141–162 (1998).
Birringer, R. P., Chidester, P. J. & Dauskardt, R. H. High yield four-point bend thin film adhesion testing techniques. Eng. Fract. Mech. 78, 2390–2398 (2011).
Nguyen, V. L. et al. Wafer-scale integration of transition metal dichalcogenide field-effect transistors using adhesion lithography. Nat. Electron. 6, 146–153 (2023).
Pufall, R. et al. Degradation of moulding compounds during highly accelerated stress tests - a simple approach to study adhesion by performing button shear tests. Microelectron. Reliab. 52, 1266–1271 (2012).
Wang, H. et al. Low-temperature copper bonding strategy with graphene interlayer. ACS Nano 12, 2395–2402 (2018).
Pflügler, N. et al. Experimental determination of critical adhesion energies with the advanced button shear test. Microelectron. Reliab. 99, 177–185 (2019).
Fan, H. B., Wong, C. K. Y. & Yuen, M. M. F. A new method to predict delamination in electronic packages. in Proceedings Electronic Components and Technology, 2005. ECTC ’05. vol. 2 145–150 (IEEE, Lake Buena Vista, FL, USA, 2005).
Bernasko, P. K., Mallik, S. & Takyi, G. Effect of intermetallic compound layer thickness on the shear strength of 1206 chip resistor solder joint. Solder. Surf. Mt. Technol. 27, 52–58 (2015).
Shohji, I., Shimoyama, S., Ishikawa, H. & Kojima, M. Effect of shear speed on the ball shear strength of Sn-3Ag-0.5Cu solder ball joints. Jpn. Inst. Electron. Packag 1, 9–14 (2008).
Wagner, S. et al. Graphene transfer methods for the fabrication of membrane-based NEMS devices. Microelectron. Eng. 159, 108–113 (2016).
Marx, M. et al. Metalorganic vapor-phase epitaxy growth parameters for two-dimensional MoS2. J. Electron. Mater. 47, 910–916 (2018).
Cun, H. et al. Wafer-scale MOCVD growth of monolayer MoS2 on sapphire and SiO2. Nano Res 12, 2646–2652 (2019).
Grundmann, A. et al. Impact of synthesis temperature and precursor ratio on the crystal quality of MOCVD WSe2 monolayers. Nanotechnology 34, 205602 (2023).
Zhang, L., Dong, J. & Ding, F. Strategies, status, and challenges in wafer scale single crystalline two-dimensional materials synthesis. Chem. Rev. 121, 6321–6372 (2021).
Lin, L., Peng, H. & Liu, Z. Synthesis challenges for graphene industry. Nat. Mater. 18, 520–524 (2019).
Zhang, K., Feng, Y., Wang, F., Yang, Z. & Wang, J. Two dimensional hexagonal boron nitride (2D-hBN): synthesis, properties and applications. J. Mater. Chem. C 5, 11992–12022 (2017).
Rasool, H. I., Ophus, C., Klug, W. S., Zettl, A. & Gimzewski, J. K. Measurement of the intrinsic strength of crystalline and polycrystalline graphene. Nat. Commun. 4, 2811 (2013).
Chu, K., Wang, J., Liu, Y. & Geng, Z. Graphene defect engineering for optimizing the interface and mechanical properties of graphene/copper composites. Carbon 140, 112–123 (2018).
Huang, X. et al. Robust microscale structural superlubricity between graphite and nanostructured surface. Nat. Commun. 14, 2931 (2023).
Dudek, R. et al. Determination of interface fracture parameters by shear testing using different theoretical approaches. in 2012 13th International Thermal, Mechanical and Multi-Physics Simulation and Experiments in Microelectronics and Microsystems 1/10-10/10 (IEEE, Cascais, Portugal, 2012). https://doi.org/10.1109/ESimE.2012.6191793.
Durix, L., Dreßler, M., Coutellier, D. & Wunderle, B. On the development of a modified button shear specimen to characterize the mixed mode delamination toughness. Eng. Fract. Mech. 84, 25–40 (2012).
Vesel, A. & Mozetic, M. Surface modification and ageing of PMMA polymer by oxygen plasma treatment. Vacuum 86, 634–637 (2012).
Sikora, A. et al. Surface modification of PMMA polymer and its composites with PC61BM fullerene derivative using an atmospheric pressure microwave argon plasma sheet. Sci Rep 11, 9270 (2021).
Na, S. R. et al. Selective mechanical transfer of graphene from seed copper foil using rate effects. ACS Nano 9, 1325–1335 (2015).
Shohji, I., Yoshida, T., Takahashi, T. & Hioki, S. Tensile properties of Sn–Ag based lead-free solders and strain rate sensitivity. Mater. Sci. Eng. A 366, 50–55 (2004).
Feng, X. et al. Competing fracture in kinetically controlled transfer printing. Langmuir 23, 12555–12560 (2007).
Meitl, M. A. et al. Transfer printing by kinetic control of adhesion to an elastomeric stamp. Nat. Mater. 5, 33–38 (2006).
Gent, A. N. Adhesion and strength of viscoelastic solids. is there a relationship between adhesion and bulk properties? Langmuir 12, 4492–4496 (1996).
Christöfl, P. et al. Comprehensive investigation of the viscoelastic properties of PMMA by nanoindentation. Polym. Test. 93, 106978 (2021).
Ionita, D., Cristea, M. & Banabic, D. Viscoelastic behavior of PMMA in relation to deformation mode. J. Therm. Anal. Calorim. 120, 1775–1783 (2015).
Persson, B. N. J. & Brener, E. A. Crack propagation in viscoelastic solids. Phys. Rev. E 71, 036123 (2005).
Deng, S., Gao, E., Xu, Z. & Berry, V. Adhesion energy of MoS2 thin films on silicon-based substrates determined via the attributes of a single MoS2 wrinkle. ACS Appl. Mater. Interfaces 9, 7812–7818 (2017).
Li, R. et al. Determination of PMMA residues on a chemical-vapor-deposited monolayer of graphene by neutron reflection and atomic force microscopy. Langmuir 34, 1827–1833 (2018).
Choi, W., Shehzad, M. A., Park, S. & Seo, Y. Influence of removing PMMA residues on surface of CVD graphene using a contact-mode atomic force microscope. RSC Adv 7, 6943–6949 (2017).
Aitken, Z. H. & Huang, R. Effects of mismatch strain and substrate surface corrugation on morphology of supported monolayer graphene. J. Appl. Phys. 107, 123531 (2010).
Li, T. & Zhang, Z. Substrate-regulated morphology of graphene. J. Phys. D: Appl. Phys. 43, 075303 (2010).
Gao, W. & Huang, R. Effect of surface roughness on adhesion of graphene membranes. J. Phys. D: Appl. Phys. 44, 452001 (2011).
Agarwal, H. et al. 2D-3D integration of hexagonal boron nitride and a high-κ dielectric for ultrafast graphene-based electro-absorption modulators. Nat. Commun. 12, 1070 (2021).
Piacentini, A. et al. Stable Al2O3 encapsulation of MoS2‐FETs enabled by CVD grown h‐BN. Adv. Electron. Mater. 8, 2200123 (2022).
Ahn, Y., Kim, J., Ganorkar, S., Kim, Y.-H. & Kim, S.-I. Thermal annealing of graphene to remove polymer residues. Mat Express 6, 69–76 (2016).
Zhuang, B., Li, S., Li, S. & Yin, J. Ways to eliminate PMMA residues on graphene - superclean graphene. Carbon 173, 609–636 (2021).
Kumar, K., Kim, Y.-S. & Yang, E.-H. The influence of thermal annealing to remove polymeric residue on the electronic doping and morphological characteristics of graphene. Carbon 65, 35–45 (2013).
Zheng, F. et al. Critical stable length in wrinkles of two-dimensional materials. ACS Nano 14, 2137–2144 (2020).
Metzke, C. et al. On the limits of scanning thermal microscopy of ultrathin films. Materials 13, 518 (2020).
Zhang, Y. et al. A review on principles and applications of scanning thermal microscopy (SThM). Adv. Funct. Mater. 30, 1900892 (2020).
Metzke, C., Kühnel, F., Weber, J. & Benstetter, G. Scanning thermal microscopy of ultrathin films: numerical studies regarding cantilever displacement, thermal contact areas, heat fluxes, and heat distribution. Nanomaterials 11, 491 (2021).
Guen, E. et al. Scanning thermal microscopy on samples of varying effective thermal conductivities and identical flat surfaces. J. Appl. Phys. 128, 235301 (2020).
Sakami, D., Lahmar, A., Scudeller, Y., Danes, F. & Bardon, J. P. Thermal contact resistance and adhesion studies on thin copper films on alumina substrates. J. Adhes. Sci. Technol. 15, 1403–1416 (2001).
Pelzl, J., Kijamnajsuk, P., Chirtoc, M., Horny, N. & Eisenmenger-Sittner, C. Correlation between thermal interface conductance and mechanical adhesion strength in Cu-coated glassy carbon. Int. J. Thermophys. 36, 2475–2485 (2015).
Boddison-Chouinard, J., Scarfe, S., Watanabe, K., Taniguchi, T. & Luican-Mayer, A. Flattening van der Waals heterostructure interfaces by local thermal treatment. Appl. Phys. Lett. 115, 231603 (2019).
Neumann, C. et al. Raman spectroscopy as probe of nanometre-scale strain variations in graphene. Nat. Commun. 6, 8429 (2015).
Androulidakis, Ch. et al. Strained hexagonal boron nitride: Phonon shift and Grüneisen parameter. Phys. Rev. B 97, 241414 (2018).
Sanchez, D. A. et al. Mechanics of spontaneously formed nanoblisters trapped by transferred 2D crystals. Proc. Natl. Acad. Sci. U.S.A. 115, 7884–7889 (2018).
Li, Y. et al. Adhesion between MXenes and other 2D materials. ACS Appl. Mater. Interfaces 13, 4682–4691 (2021).
Acknowledgements
This work has received funding from the European Union’s Horizon 2020 research and innovation programme Graphene Flagship Core3 under grant agreement No 881603 (M.C.L., M.H.), from “Bayerisches Staatsministerium für Wirtschaft, Landesentwicklung und Energie StMWi“ under the project AlhoiS (DIE-2003-0002//DIE0111/01 (J.S.), DIE-2003-0004//DIE0111/02 (J.W., C.M.)), and from the German Ministry of Education and Research (BMBF) under the projects GIMMIK (03XP0210A (M.H.), 03XP0210F (M.C.L.)), NEUROTEC II (16ME0399 (M.C.L.), 16ME0400 (M.C.L.), 16ME0403 (M.H.)), NeuroSys (03ZU1106AA (M.C.L), 03ZU1106AD (M.H.)), and NobleNEMS (16ES1121K (M.C.L.)).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M.C.L., S.P., and J.S. designed the study. N.N., A.G., S.L., and J.S. fabricated the samples. N.N., J.Walter and J.S. were involved in the button shear test measurements. A.G., H.K., M.H., and A.V. provided MoS2 and WSe2. J.Weber, S.L., and C.M. conducted the AFM and SThM measurements and S.L. and T.S. the Raman and XPS measurements. M.C.L., J.Walter, S.P., F.S., and J.S. analyzed and discussed the data. The manuscript was written through contributions of all authors. All authors have given approval for the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Van Luan Nguyen, Hossein Rokni and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schätz, J., Nayi, N., Weber, J. et al. Button shear testing for adhesion measurements of 2D materials. Nat Commun 15, 2430 (2024). https://doi.org/10.1038/s41467-024-46136-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46136-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
