
Abstract
Iodine is a reactive trace element in atmospheric chemistry that destroys ozone and nucleates particles. Iodine emissions have tripled since 1950 and are projected to keep increasing with rising O3 surface concentrations. Although iodic acid (HIO3) is widespread and forms particles more efficiently than sulfuric acid, its gas-phase formation mechanism remains unresolved. Here, in CLOUD atmospheric simulation chamber experiments that generate iodine radicals at atmospherically relevant rates, we show that iodooxy hypoiodite, IOIO, is efficiently converted into HIO3 via reactions (R1) IOIO + O3 → IOIO4 and (R2) IOIO4 + H2O → HIO3 + HOI + (1)O2. The laboratory-derived reaction rate coefficients are corroborated by theory and shown to explain field observations of daytime HIO3 in the remote lower free troposphere. The mechanism provides a missing link between iodine sources and particle formation. Because particulate iodate is readily reduced, recycling iodine back into the gas phase, our results suggest a catalytic role of iodine in aerosol formation.

Similar content being viewed by others

Main
Iodine is a trace constituent of the atmosphere that is particularly efficient at forming new particles. While sulfuric acid (H2SO4)1,2,3, methanesulfonic acid1,4 and nitric acid5 all require an additional vapour (ammonia, NH3 or dimethylamine (DMA)) to form particles, highly oxygenated organic molecules (HOMs)6 and iodine7,8,9 can do so alone. Iodine nucleation rates exceed those of H2SO4 (in excess NH3) at comparable concentrations of iodic acid (HIO3)10. Furthermore, HIO3 growth rates of nanoparticles are both charge- and dipole-enhanced, exceeding the neutral collision rate10,11.
Currently, iodine particle formation is rarely represented in atmospheric models—such models form most particles from the nucleation of H2SO4 and include iodine primarily because of its ozone-destroying potential12. While sulfur emissions are projected to decrease due to pollution control measures (probably to a few tens of teragrams of SO2 per year by 2100 (ref. 13), iodine emissions have been increasing due to human activity. Iodine is primarily emitted from oceans by the reaction of O3 with iodide (I−) dissolved in surface waters, which liberates volatile iodine species (hypoiodous acid (HOI) and iodine (I2)) to the atmosphere14,15. This marine source is enhanced as a result of O3 pollution on local and hemispheric scales16,17 as well as the thinning of sea ice18, and now accounts for iodine emissions of ~3 Tg yr−1 (refs. 19,20). Over the past 70 years, iodine concentrations have tripled in ice-core records in Greenland18, Alpine glaciers17 and tree-ring records in Tibet21.
Iodine is highly reactive and participates in catalytic reaction cycles that enhance its atmospheric impact. A catalytic role is well known for O3 loss, but has, as of yet, not been suggested for particle formation. Iodine in the lower stratosphere has a 6–15 and 400–1,000 times higher O3 destruction potential per atom than bromine and chlorine22. Extremely low mixing ratios of iodine oxide (IO) radicals (for example, ~0.1 parts per trillion by volume (pptv); IO = 10−13 volume mixing ratio) can therefore affect the lifetime of climate-active gases (for example, O3 and CH4)19,23,24. This chemical reactivity extends to heterogeneous reactions involving aerosol iodide (I−)14,15 and iodate (IO3−) (refs. 25,26 and references therein), which is the thermodynamically most stable form of iodine. The efficient multiphase chemistry of IO3− is markedly different from that of inert aerosol sulfate (SO42−), which accumulates without further chemical conversion until it is scavenged from the atmosphere by wet or dry deposition.
Iodine is ubiquitous in the atmosphere22,23,27,28, and HIO3 has been detected in coastal marine air9,10,29, the Arctic and Antarctic boundary layer9,10,30,31,32, various continental sites10 and in the lower free troposphere10,33. Several precursors for HIO3 have been suggested: hydrated iodine atoms10,34, hydrated IO radicals34, iodine dioxide (OIO) radicals35 and larger iodine oxides (I2O3, I2O4 and I2O5; refs. 34,36,37,38). However, these mechanisms remain speculative and have not been demonstrated experimentally, leaving atmospheric HIO3 observations unexplained. Recent field observations of iodine-induced nucleation over remote oceans31 and of IO3− in stratospheric aerosols22 suggest a widespread role of iodine particle formation, but the conundrum of the missing HIO3 source mechanism blocks our ability to connect iodine sources to particle formation in atmospheric models.
Results and discussion
CLOUD measurements
In this Article we report iodine chemistry and particle formation experiments under marine boundary layer conditions at the CERN CLOUD chamber (Methods). Because of the large chamber volume (26.1 m3) and associated long wall-loss lifetime (~8 min; comparable to typical condensation rates in the atmosphere), precursor gas-phase concentrations do not need to be increased above atmospheric levels (Supplementary Table 1). Experiments were conducted at 283 K and 263 K, with I2 at a typical volume mixing ratio of 8 pptv (range of <0.5–330 pptv), 40% relative humidity (RH, <3–90%) and 40 ppbv O3 (<1–80 parts per billion by volume (ppbv)). The chemistry is driven by photolysis of I2, which is measured by cavity-enhanced differential optical absorption spectroscopy (CE-DOAS; Methods) and bromide chemical ionization mass spectrometry (Br−-MION-CIMS). HIO3 is measured quantitatively by NO3−-CIMS, and HOI by Br−-MION-CIMS. Both instruments also allow insights into the evolution of other iodine species (IO, OIO, I2O2, I2O4 and so on; Methods).
The measurements are accompanied by chemical box modelling, building on state-of-the-art iodine chemistry (Methods). The model is constrained by measurements of I2 concentrations, actinic fluxes, temperature, humidity and losses of molecules to the chamber walls (stainless steel, characterized via H2SO4) and chamber dilution (~2 h). Established iodine chemistry only contains a single reaction predicted from theory35 that could form HIO3 from OIO + OH. This reaction does not form HIO3 in the HOx-free conditions when I2 is photolysed by green light10. Even if OH radicals were present, they would be predominately scavenged by other species. The model base case does not form any HIO3 or HOI under the experimental conditions probed (Fig. 1). Based on the comprehensive experimental evidence of this work, and supported by theoretical calculations, the base case model is extended to include the following two reactions:
and considers an update to the thermal lifetime of IOIO (extended model, Supplementary Section 3).

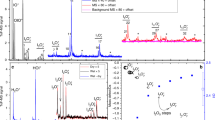
a–g, Time-resolved measurements of key iodine species (a,b,d show precursors to HIO3 (f) and HOI (g), and c and e show higher-oxide routes) are compared with model predictions after the start of I2 photolysis at green wavelengths within the CERN CLOUD chamber. Measured concentrations (grey lines) of HIO3 and HOI exceed 107 molecules per cm3 (molec cm−3) within minutes. Established gas-phase iodine chemistry (model base case, dashed blue lines) forms neither HIO3 nor HOI, contrary to the observations, and overestimates the concentrations of IOIO and I2O4. The extended model (solid red line), including reactions (R1) and (R2) and considering a longer thermal lifetime of IOIO, achieves good mass and temporal closure for HIO3, HOI, IOIO and I2O4.
Figure 1 shows that HIO3 and HOI concentrations rapidly increase to exceed 1 × 107 molecules per cm3 (molec cm−3) within a few minutes of the onset of I2 photolysis by green light (grey lines). While zero HIO3 and zero HOI are predicted by the base case model (current state of the art), the extended model achieves excellent agreement with regard to the measured concentrations and the timing of HIO3 and HOI formation. The extended model also improves the closure of timing and concentrations measured for OIO, IOIO and I2O4. Measured HIO3 concentrations reach a steady state after ~8 min, consistent with the wall-loss lifetime of other sticky molecules3 measured at CLOUD (Extended Data Fig. 1). HOI continues to accumulate due to a lower effective wall uptake. Notably, the IO radical concentrations closely resemble those in the remote marine boundary layer (compare Supplementary Table 1) and do not exceed 1 pptv (1 pptv = 2.68 × 107 molec cm−3 at 273 K and 1 atm pressure). The timing of IO radicals is predicted very well from both the base case and the extended model, reflecting the high level of trust in the gas-phase chemical kinetics during the early stages of the iodine photolysis experiments. Interestingly, iodine oxide clusters IxOy (x ≥ 2, y ≥ 3) larger than IOIO are formed too late to explain the rapid formation of HIO3 as an early generation product (Extended Data Fig. 2 and Supplementary Table 2).
Figure 2 shows that the extended model accurately predicts the measured HIO3 production rates, pHIO3, over a wide range of I radical production rates, pI (104–106 molec cm−3 s−1). Here, pHIO3 is calculated from HIO3 concentration measurements and the well-known loss rates to the chamber walls, and pI is calculated from the photolysis of I2. The HIO3 yield, defined as the ratio of pHIO3 and pI, is a function of the experimental conditions and varies between 10 and 20%. This variability is most pronounced for low pI (<105 molec cm−3 s−1) and is quantitatively explained by the wall loss of HIO3 precursors becoming progressively more relevant at lower gas concentrations. We corroborated that HIO3 formation from I atoms is a multistep process by carrying out an experiment with enhanced stirring (by two fans at the top and bottom of the chamber), thereby decreasing the wall accommodation lifetime of HIO3 from the standard ~8 min to ~2 min, while holding all other parameters constant. The HIO3 concentration decreased by more than one order of magnitude, indicating that the HIO3 suppression exceeds that expected from a change in lifetime alone (Extended Data Fig. 1). The extended model reproduces this superlinear response under the reasonable assumption of efficient reactive uptake of IO radicals on the chamber walls (red dashed line, Fig. 2). Indeed, if the extended model is run while disregarding IO wall loss (blue dashed line, Fig. 2), a constant and high yield of ~20% applies over the full pI range probed.

The HIO3 production rate pHIO3 scales in first order with the I atom production rate pI (median (solid line) and 25–75% and 5–95% inter-percentile ranges (dark and light grey shading)). The yield η is substantial (~20%) and near constant for pI larger than 105 molec cm−3 s−1. At smaller pI, losses of intermediates to chamber walls reduce η. This effect is captured by the model (red line (median)) and is explained by IO radical wall losses (compare blue dashed and red dotted lines (medians)). If larger IxOy clusters were the HIO3 precursor, a higher-order yield would be expected—this is not consistent with the observations.
That HIO3 formation is first order in pI (Fig. 2) explains the presence of HIO3 over remote oceans, where pI is low (Supplementary Table 1)10,31. This finding also carries key mechanistic information, in that it is incompatible with the hypothesis that larger IxOy (x ≥ 3) species are HIO3 precursors34 at CLOUD. If such IxOy were the precursor, the HIO3 yield would not be constant, but would increase progressively with pI, and pHIO3 would follow a higher-order rate law (Supplementary Fig. 2). This is not observed. We regularly detect I2O2 and I2O4, in agreement with predictions by the extended model (Fig. 1 and Extended Data Fig. 3), indicating that there is no fundamental limitation to our analytical capabilities to detect IxOy species. Interestingly, I2O3 is generally not detected, except in experiments that employ extremely high I2 concentrations (ppbv levels), which can bias reaction pathways to favour the formation of larger IxOy species (Supplementary Table 1 and Supplementary Section 5). Quantum chemical calculations support that the \({{{{\rm{I}}}}}_{2}{{{{\rm{O}}}}}_{3}\cdot {{{{{\rm{NO}}}}}_{3}}^{-}\) cluster is thermally stable (Supplementary Fig. 3) and should be observable. Including the formation of HIO3 from IOIO in the extended model reduces the predicted I2O3 by approximately a factor of two (Extended Data Fig. 3), and improves predictions about IOIO, in close agreement with observations (Fig. 1). The remaining discrepancy for I2O3 reflects the uncertainty in larger IxOy chemistry39. We conclude that IxOy species larger than IOIO are not needed as precursors for HIO3 under typical conditions at CLOUD.
HIO3 formation from IOIO is robust against variations in O3, H2O and temperature (Extended Data Fig. 4 and Supplementary Fig. 1). This suggests that neither O3 nor H2O are rate-limiting to HIO3 formation under the conditions probed. The rate-limiting step is the formation of IOIO, which is fully converted into HIO3 (Extended Data Fig. 4). We observe excellent closure between pHIO3 and pIOIO during the O3 ramps, where pIOIO is based on the well-known IO + IO rate coefficients40.
At O3 concentrations below a few ppbv, the chemistry slows down sufficiently that other sinks become relevant for IOIO (for example, wall loss and thermal decomposition), resulting in a slight dependence of the pHIO3-to-pIOIO ratio on O3. That slight dependence is captured by the extended model (assuming an IOIO wall uptake coefficient γwall(IOIO) = 1). In contrast, a pronounced O3 sensitivity would be expected if IO⋅H2O or OIO were HIO3 precursors (Supplementary Fig. 1). The absence of an O3 and H2O sensitivity is difficult to reconcile with any mechanism that does not quantitatively convert a single precursor. The comprehensive evidence (Supplementary Table 2 and Supplementary Section 2) strongly supports a rapid and quantitative conversion of IOIO into HIO3 and HOI.
Quantum chemical calculations
We employed quantum chemical calculations (density functional theory (DFT) methods M062X/aug-cc-pVTZ-PP, followed by coupled-cluster single-point energy corrections; Methods) to explore the reactivity of IOIO with O3, H2O and other available reactants to form HIO3 and HOI. IOIO reacts reasonably quickly with O3 to form HIO3, HOI and singlet oxygen via reaction sequences (R1) and (R2).
Figure 3 shows the reaction coordinate. The reactions (R1) and (R2) are exothermic and free of prohibitively large barriers. Accurately predicting energies and rate coefficients for iodine is challenging because of the inherent complexity of iodine atoms (atom size, number of electrons and relativistic effects). The strong sensitivity towards varying levels of theory is illustrated by comparing bond dissociation energies (BDEs) and proton affinities for simple iodine oxides where measurements are available (Table 1). The method used in this study has improved skill in the coupled-cluster part of the calculations, primarily due to a more balanced description of the basis set on iodine and the other atoms (Methods), and is found to reproduce experimental values within ~3 kcal mol−1 (with the exception of the OIO BDE), which translates into approximately one order of magnitude uncertainty in rate constants.

Reaction coordinate for the gas-phase reactions (R1) and (R2) as free energy ΔG(T = 298 K). The energies are calculated using theory at the CCSD(T)/CBS(T,Q)//M062X/aug-cc-pVTZ-PP level of theory. ΔG(TS3) (not rate-limiting) is calculated at the CCSD(T)/aug-cc-pVTZ-PP//M062X/aug-cc-pVTZ-PP level, due to memory limitations. The reaction coordinate supports that atmospheric concentrations of O3 and H2O lead to a quantitative conversion of IOIO into HIO3, HOI and singlet O2.
The transition states in Fig. 3 translate into the rate coefficients for reactions (R1) and (R2) at 298 K as shown in Table 1 (for temperature dependencies see Supplementary Fig. 6). Notably, the experimentally derived k1 ≥ 1.5 × 10−13 molec−1 cm3 s−1 is supported within the error margins of theory and maintains the quantitative conversion of IOIO into HIO3 even at low O3 concentrations (Supplementary Fig. 1). Our results led to a reassessment of the thermal lifetime of IOIO, which is predicted to be substantially longer than previously thought (Table 1), consistent with observations of IOIO (Extended Data Fig. 3), and its persistently quantitative conversion into HIO3 even at extremely low O3 concentrations at 263 K (Supplementary Fig. 5 and Supplementary Section 3). Reaction (R2) is predicted to proceed with k2 = 5.7 × 10−16 molec−1 cm3 s−1 at 298 K (Table 1 and Supplementary Fig. 6), corresponding to a typical conversion of IOIO4 into HIO3 within fractions of a second. Competing pathways of IOIO4 into other products than HIO3 were investigated (Supplementary Fig. 4, Supplementary Table 4 and Supplementary Section 3), but found to be unlikely. The marginal detection of IOIO4 (Extended Data Fig. 3 and Supplementary Section 2.4) is consistent with a value of k2 ≈ 2.0 × 10−16 molec−1 cm3 s−1 at 263 K. The detection of IOIO4 at the observed levels suggests that reaction (R2) is enhanced by water reacting with hot IOIO4 (Supplementary Section 3.3); assuming a lower k2 from thermalized IOIO4 leads to IOIO4 accumulation in the extended model that is not observed. We recommend temperature-dependent rate coefficients for k1 and k2 for the development of atmospheric models (Supplementary Section 3). Overall, the theory-predicted rates support the experimentally derived rates within the uncertainty of the calculations.
Atmospheric observations
The laboratory-derived mechanism can explain field measurements of HIO3 concentrations in the remote free troposphere. We use concurrent measurements of HIO3 (in situ, NO3−-CIMS), IO radicals (near-observatory, MAX-DOAS) and particle surface area measurements at the Maïdo observatory41 to assess the relevance of CLOUD findings in the real world. The observatory is located in the southern Indian Ocean on Réunion Island at an elevation of 2,200 m, and is frequently exposed to lower free tropospheric air (mornings) and anabatic orographic flows from the ocean (afternoons). The laboratory conditions at CLOUD closely match the conditions at the Maïdo observatory (Supplementary Table 1 and Methods) regarding IO concentrations (single pptv), condensational sink (~10−3 s−1) and temperature (~283 K).
Figure 4 shows pHIO3 in the field and laboratory on a common IO radical concentration axis. pHIO3 is calculated from HIO3 concentrations and the condensation sink surface area, assuming a steady state. IO radical concentrations are measured directly at the Maïdo observatory, and taken from the extended model at CLOUD. The solid line shown in Fig. 4 is not a fit to the data; it corresponds to pIOIO at 283 K and serves as a transfer standard to propagate the mechanistic finding of quantitative IOIO conversion into HIO3 from CLOUD (Extended Data Fig. 4) to the field observations. The excellent consistency between the laboratory experiments and field observations demonstrates the atmospheric relevance of the proposed HIO3 mechanism.

Good consistency is observed between HIO3 production rates measured in the CLOUD laboratory (red) and at the Maïdo field site (blue). IO radical concentrations at CLOUD overlap with those found in the remote lower free troposphere. The solid black line is the IOIO formation rate from IO radicals (at 283 K), and corresponds to the rate-limiting step of HIO3 formation under both field and laboratory conditions.
The ability of our HIO3-formation mechanism to predict simultaneous field measurements of HIO3 and IO radicals in the remote free troposphere is anything but trivial (Supplementary Fig. 7), and demonstrates the ability to approximate atmospherically relevant experimental conditions at CLOUD. Interestingly, HIO3 concentrations at Maïdo increase rapidly already under twilight conditions during sunrise (Supplementary Fig. 7 and Supplementary Section 4). He and colleagues10 had predicted the efficient formation of iodine oxoacids under cloudy daylight conditions, and Supplementary Fig. 7 provides field evidence in support of the rapid activation of iodine reservoir species into iodine oxoacids in the absence of ultraviolet irradiation.
Atmospheric implications
The mechanism provides a source of HIO3 that is effective even at low iodine concentrations, and will allow atmospheric models to test HIO3 field observations. Such model development will also help guide future laboratory experiments and field observations. The near-linear rate law of pHIO3 in pI also enables HIO3 formation and subsequent particle formation beyond hotspots at lower iodine concentrations in the background atmosphere31,42,43.
The gas-phase formation mechanism of HIO3 we present here facilitates a missing connection between iodine sources and particle formation in atmospheric models, as illustrated in Fig. 5. The activation of iodine reservoir species (Fig. 5, step 1) liberates iodine radicals, which rapidly form IO radicals and HIO3 (step 2) via reactions (R1) and (R2). Iodine oxoacid particle formation and growth (step 3) is driven by HIO3 in most atmospheric environments. Indeed, recent field observations of particle formation events over the remote Arctic Ocean indicate that all of the observed events were driven by HIO3 (ref. 31). IxOy species may also contribute locally in coastal hotspots with extremely high iodine concentrations. Freshly nucleated iodine particles are composed almost entirely of HIO3 (ref. 10); HIO3 is a strong acid (pKa = 0.8; ref. 44) that dissociates to form IO3−. IO3− is known to undergo reduction reactions that ultimately form more volatile iodine species (for example, HOI, I2 and IO), which are re-emitted to the gas phase (step 4). Field observations and laboratory experiments show that IO3− is reduced via iron redox chemistry, H2O2, nitrite, photosensitized reactions, photolysis and numerous other species (refs. 25,26 and references therein), with the overall effect of recycling iodine to the gas phase. The HIO3 formation mechanism thus completes a catalytic iodine reaction cycle, by which a single iodine atom can repeatedly form HIO3, driving particle formation. For each HIO3 molecule produced from I, three O3 molecules are consumed. The re-emission of reduced iodine species thus constitutes a multiphase reaction cycle that destroys O3.

After activation of iodine reservoirs (step 1), HIO3 is efficiently formed (step 2) and subsequently nucleates and grows particles extremely efficiently (step 3). Iodate (IO3−) can be reduced and re-emitted to the gas phase (step 4), closing an iodine-catalysed reaction cycle forming particles and destroying O3. HIO3 formation from IOIO links iodine sources and new particle formation even at lower IO concentrations. This mechanism is currently missing from atmospheric models.
That iodine recycling controls the iodine partitioning between the gas and particle phases is corroborated by field measurements of the size-resolved iodine activity in radioactive fallout45. Among the primary radioactive elements, 132Te, 137Cs and 103Ru abundances were found to correlate with the aerosol volume distribution, whereas 131I correlated with the aerosol surface area distribution instead. These empirical observations hint at efficient recycling occurring on timescales of hours to days, consistent with rapid HIO3 formation. Notably, although the reactive uptake of HOI on aerosols is known to be fast46, this reaction de facto removes halides from aerosols to the gas phase. A gas-phase source of HIO3 adds iodine to particles and, in conjunction with iodine recycling, provides a plausible explanation for the correlation of particulate 131I with the aerosol surface area distribution at the molecular level. Particulate IO3− is the primary reservoir of total (gas and particle) iodine in the stratosphere22. Whether HIO3 forms in the stratosphere—and controls iodine partitioning between the gas and particle phases—deserves further study. The HIO3-formation mechanism fills a gap in the representation of the geochemical iodine cycle in current atmospheric models.
Iodine particle formation has heretofore been considered to have only limited global importance19. This deserves re-evaluation in light of efficient HIO3 formation even at low concentrations, the catalytic role of iodine in particle formation, and the increased global iodine source in recent decades. Iodine particle formation is probably already relevant on global scales today, and will become even more important in view of decreasing global sulfur emissions and increasing iodine emissions in a future climate.
Methods
CLOUD experiments
Laboratory experiments were carried out at the CERN CLOUD chamber3,50 in Geneva, Switzerland as part of the CLOUD12 and CLOUD13 campaigns during 2017 and 2018. The CLOUD chamber is a temperature-controlled, electropolished stainless-steel reaction vessel with a volume of 26.1 m3. Experiments were carried out at temperatures of 283 and 263 K. The chamber was operated as a continuous-flow reactor, and ultra-pure N2 and O2 at 250–300 l min−1 were continuously replenished at a pressure of 1 atm, resulting in an air exchange time of ~80 min. Two fans at the top and bottom of the chamber established near-homogeneous mixing (mixing time ~2 min). Trace gases were added at the bottom of the chamber. I2 was produced from sublimating iodine crystals (Sigma-Aldrich, 99.999% purity), and concentrations inside the chamber were varied in the range 0.5 pptv < [I2] < 330 pptv (typically ~8 pptv). O3 was generated from UV irradiation of dry synthetic air, and the chamber was humidified using ultrapurified water, resulting typically in [O3] = 40 ppbv (range < 1–80 ppbv) and RH = 40% (<3–90%).
A typical experiment explored the formation of HIO3 following the selective photolysis of I2 using green light (light-emitting diodes (LEDs) centred at 523 nm, I2 photolysis frequencies \({j}_{{{{{\rm{I}}}}}_{2}}\le {6.5}\times {10}^{-3}\,{{{{\rm{s}}}}}^{-1}\)) in the presence of O3 and humidity (Fig. 1). Actinic frequencies were spectrally determined using a spectrometer and dedicated iodine actinometry experiments (Supplementary Section 6.3). Actinic fluxes of light sources at variable intensity were monitored during actual experiments by photodiodes. Sensitivity studies during individual experiments followed the response in the HIO3 concentration to variations in O3 (for example, Supplementary Fig. 1), chamber wall loss during variations of the fan mixing speed (for example, Extended Data Fig. 1) and by varying selected environmental parameters. The typical duration of individual experiments varied from a few tens of minutes to a few hours, depending on the experimental conditions.
I2 was measured by closed-path CE-DOAS51 using the unique ro-vibronic absorption bands between 508 and 554 nm. CE-DOAS is inherently calibrated from knowledge of the absorption cross-section. The I2 limit of detection is 8 pptv for an integration time of 10 min. Median I2 concentrations were below 8 pptv during most experiments, but elevated to up to 1.7 ppbv to calibrate the Br−-MION-CIMS, which also provides precise I2 measurements at low concentrations. The Br−-MION-CIMS is composed of an atmospheric-pressure interface–time of flight mass spectrometer (APi-TOF) coupled to a chemical ionization unit, using dibromomethane (CH2Br2) as the reagent gas. The CH2Br2 is fed into the sheath flow of the inlet and illuminated by a soft X-ray source. The produced bromide anions are directed into the sample flow by a negative electric field, and cluster with neutral molecules (I2) in the sample air. The overall uncertainty of the resulting I2 time series is estimated to be better than 30%52. The I2 constraint imposed to the model assimilates the lower bound of the measured I2 time series (within the 30% uncertainty), which results in the best closure between measured and predicted HIO3. Iodine radical production rates, pI, are calculated from the photolysis rate of I2 concentrations.
HIO3 was measured by a NO3−-CIMS system comprising an APi-TOF coupled to a chemical ionization unit that uses nitric acid as the reagent gas. It is used extensively for detecting H2SO4, highly oxygenated organic molecules and HIO3. Details of the instrument used in the present study are provided in ref. 53. The NO3−-CIMS has an ion filter integrated into its sampling line to avoid confusion with ions and charged clusters from the CLOUD chamber. It thus measures only neutral molecules and clusters in CLOUD. The uncertainty of the HIO3 measurement is estimated to be 50%.
The characteristic time for the deposition of sticky molecules to the chamber walls is 440 s with standard mixing by the fans (Extended Data Fig. 1), as characterized via H2SO4 loss rates. The loss to walls is the well-defined dominant sink of HIO3. Experiments that formed a large particle surface area (measured by nSEMS, nano-SMPS or long-SMPS) competitive to chamber wall loss were discarded in this study to avoid introducing uncertainty due to the other less-well-defined sinks for HIO3 and other iodine species. The HIO3 production rates were calculated from measured concentrations under the assumption of a steady state. Periods with rapid changes of HIO3 concentration are not considered in, for example, Fig. 2.
Box modelling
The photochemical box model builds on the framework described in refs. 22,23,24 and represents state-of-the-art iodine chemistry and HOx chemistry25,48. Here, the model is extended by the chamber auxiliary mechanism, which includes losses of gases to the chamber walls and to particles, losses by dilution and the actinic fluxes of the chamber lights. IO, OIO, IOIO, I2O3, I2O4, HI and HIO3 are assumed to be lost to the walls with the same rate constant as H2SO4, the prototypical sticky molecule. Accommodation of molecules to the CLOUD chamber walls is limited by transport, not by diffusion. Thus, the effective wall accommodation coefficient of molecules (most iodine species are reasonably sticky54,55,56, with accommodation coefficients of multiple tens of percent or even unity) used in the model is enhanced over the accommodation coefficient for individual collisions57. Extended Data Fig. 1 provides evidence for the efficient loss of iodine species to the chamber walls. The model is constrained by measurements of I2, O3 and H2O, photolysis frequencies (I2, IO, OIO, HOI, I2O2, I2O3 and I2O4), temperature and the aforementioned loss mechanisms. HOI is both lost to the walls and produced on the chamber walls through heterogeneous chemistry14, which also proceeds in dark conditions. This study did not make an attempt to describe the uptake and release of HOI at the molecular level. An empirical uptake efficiency of 25%, relative to H2SO4, establishes closure in regard to the temporal evolution and concentrations of HOI (Extended Data Fig. 3). See Supplementary Section 6 for more details.
Quantum chemical calculations
For the reactants, intermediates, transition states and products in Fig. 3 with multiple possible conformers, a systematic conformer sampling was carried out using the MMFF method in the Spartan ’18 program. The conformer sampling algorithm in Spartan allows for pre-optimization and the elimination of duplicate structures, which is computationally more efficient than other conformer sampling approaches like MS-TOR. Geometry optimization and frequencies were calculated using DFT methods (M062X/aug-cc-pVTZ-PP) with the ultrafine grid, followed by coupled-cluster single-point energy corrections at the CCSD(T)//CBS/aug-cc-pV(T,Q)Z-PP level of theory. Iodine pseudopotentials were taken from the Environmental Molecular Sciences Laboratory (EMSL) basis set library58,59. The accuracy of the final energetics is critical to reliably estimate the rate of conversion of IOIO4 to HIO3, which was simulated using the master equation solver for multi-energy well reactions (MESMER) program.
Final product fractions were calculated using the MESMER program60. In the simulation, IOIO + O3 was modelled to directly lead to IOIO4 using the MesmerILT method with a pre-exponential value of 2.7 × 10−14 molec−1 cm3 s−1, which corresponds to the transition-state-theory-derived bimolecular rate. The unimolecular isomerization reactions of intermediate complexes were treated using the SimpleRRKM method with Eckart tunnelling. The MesmerILT method with a pre-exponential value of 2.0 × 10−10 molec−1 cm3 s−1 was used for the bimolecular reaction of IOIO4 with H2O, with the latter set as the excess reactant with a defined initial concentration. All intermediate complexes were assigned as ‘modelled’ with Lennard–Jones potentials of σ = 6.5 Å and ϵ = 300 K. These are identical to those used by Galvez and colleagues for their iodine systems61. MESMER uses the exponential down (ΔEdown) model for simulating the collisional energy transfer; a value of 225 cm−1 was used in the simulations, which is within the 175–275 cm−1 range recommended by MESMER for nitrogen bath gas.
The energetics of ozonolysis reactions are difficult to calculate accurately using single-reference methods. The inherent uncertainties are probably even more pronounced for complex iodine-containing systems. Although no experimental values are available for the gas-phase ozonolysis reaction of iodine systems, proton affinities (PAs) and BDEs of simple molecules such as HI, HOI, IO and OIO are available. Table 1 shows that the differences between the literature values and the theoretical values calculated in this work are less than 3 kcal mol−1 (with the exception of the BDE of OIO). Previous quantum chemical calculations on iodine oxide reactions34,47 are included in Table 1 for comparison, highlighting the improved skill of the method used in this study in the coupled-cluster part of the calculation, as benchmarked through comparisons with experimental PAs and BDEs. Previous studies used a double-zeta basis set (LanL2DZ) for I atoms, but a larger triple-zeta basis set (aug-cc-pVTZ) for O and H atoms, leading to substantial overestimation of the exothermicity of bond-forming reactions involving iodine. Our approach uses a large basis set for all atoms, substantially reducing this overestimation.
Field measurements
The field data were collected during an intensive operating period in April 2018 at the Maïdo observatory41, Réunion island, southern Indian Ocean (21° S, 55° E). The observatory is located at 2,200 m above sea level and is frequently exposed to lower free tropospheric air (mornings) and flows from the ocean (afternoons). Near-instrument altitude volume mixing ratios of IO radicals were retrieved from CU MAX-DOAS scattered sunlight observations. The retrieval62,63 leverages the high sensitivity of the limb viewing geometry to the atmospheric layers nearest to the instrument altitude, allowing for the parameterization of aerosol effects on the observed light path. Gas-phase HIO3 was measured directly by a NO3−-CIMS system using a methodology similar to that used in the laboratory experiments. The instrument was calibrated in the field in its actual field campaign sampling configuration by in situ-produced H2SO4, which resulted in a calibration factor of c = 1.7 × 1010 molec cm−3. This same calibration factor was used for all quantifications, so the determined concentrations here represent lower limits. The uncertainty of the determined [HIO3] was estimated similarly as [H2SO4], at −50% and +100% following the work in ref. 64. Particles were size-selected by a differential mobility particle sizer and counted with a condensation particle counter to determine the available particle surface area. The box modelling constraints are described in Supplementary Section 4.1. TUV calculated spectral fluxes65 were used to determine the photolysis frequencies of individual iodine species.
Data availability
The output files of quantum chemical calculations and a MESMER input file are provided in the public data repository at https://doi.org/10.5281/zenodo.6637910. The box model supporting the findings of this study is described in detail in the Supplementary Information (Supplementary Tables A5–A9 and text). Source data are provided with this paper.
References
Kreidenweis, S. M. & Seinfeld, J. H. Nucleation of sulfuric acid–water and methanesulfonic acid–water solution particles: implications for the atmospheric chemistry of organosulfur species. Atmos. Environ. 22, 283–296 (1988).
Kuang, C., McMurry, P. H., McCormick, A. V. & Eisele, F. L. Dependence of nucleation rates on sulfuric acid vapor concentration in diverse atmospheric locations. J. Geophys. Res. Atmos. https://doi.org/10.1029/2007JD009253 (2008).
Kirkby, J. et al. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature 476, 429–433 (2011).
Dawson, M. L. et al. Simplified mechanism for new particle formation from methanesulfonic acid, amines, and water via experiments and ab initio calculations. Proc. Natl Acad. Sci. USA 109, 18719–18724 (2012).
Wang, M. et al. Rapid growth of new atmospheric particles by nitric acid and ammonia condensation. Nature 581, 184–189 (2020).
Kirkby, J. et al. Ion-induced nucleation of pure biogenic particles. Nature 533, 521–526 (2016).
Hoffmann, T., O’Dowd, C. D. & Seinfeld, J. H. Iodine oxide homogeneous nucleation: an explanation for coastal new particle production. Geophys. Res. Lett. 28, 1949–1952 (2001).
O'Dowd, C. D. et al. Marine aerosol formation from biogenic iodine emissions. Nature 417, 632–636 (2002).
Sipilä, M. et al. Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3. Nature 537, 532–534 (2016).
He, X.-C. et al. Role of iodine oxoacids in atmospheric aerosol nucleation. Science 371, 589–595 (2021).
He, X.-C. et al. Determination of the collision rate coefficient between charged iodic acid clusters and iodic acid using the appearance time method. Aerosol Sci. Technol. 55, 231–242 (2021).
Solomon, S., Garcia, R. R. & Ravishankara, A. R. On the role of iodine in ozone depletion. J. Geophys. Res. Atmos. 99, 20491–20499 (1994).
IPCC Climate Change 2014: Synthesis Report (eds Core Writing Team, Pachauri, R. K. & Meyer L. A.) (IPCC, 2014).
Garland, J. A. & Curtis, H. Emission of iodine from the sea surface in the presence of ozone. J. Geophys. Res. Ocean. 86, 3183–3186 (1981).
Carpenter, L. J. et al. Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 6, 108–111 (2013).
Gaudel, A. et al. Aircraft observations since the 1990s reveal increases of tropospheric ozone at multiple locations across the Northern Hemisphere. Sci. Adv. 6, eaba8272 (2020).
Legrand, M. et al. Alpine ice evidence of a three-fold increase in atmospheric iodine deposition since 1950 in Europe due to increasing oceanic emissions. Proc. Natl Acad. Sci. USA 115, 12136–12141 (2018).
Cuevas, C. A. et al. Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century. Nat. Commun. 9, 1452 (2018).
Sherwen, T. et al. Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem. Atmos. Chem. Phys. 16, 12239–12271 (2016).
Carpenter, L. J. et al. Marine iodine emissions in a changing world. Proc. R. Soc. A Math. Phys. Eng. Sci. 477, 20200824 (2021).
Zhao, X., Hou, X. & Zhou, W. Atmospheric iodine (127I and 129I) record in spruce tree rings in the Northeast Qinghai-Tibet Plateau. Environ. Sci. Technol. 53, 8706–8714 (2019).
Koenig, T. K. et al. Quantitative detection of iodine in the stratosphere. Proc. Natl Acad. Sci. USA 117, 1860–1866 (2020).
Dix, B. et al. Detection of iodine monoxide in the tropical free troposphere. Proc. Natl Acad. Sci. USA 110, 2035–2040 (2013).
Wang, S. et al. Active and widespread halogen chemistry in the tropical and subtropical free troposphere. Proc. Natl Acad. Sci. USA 112, 9281–9286 (2015).
Koenig, T. K. et al. Ozone depletion due to dust release of iodine in the free troposphere. Sci. Adv. 7, eabj6544 (2021).
Cuevas, C. A. et al. The influence of iodine on the Antarctic stratospheric ozone hole. Proc. Natl Acad. Sci. USA 119, e2110864119 (2022).
Saiz-Lopez, A. et al. Atmospheric chemistry of iodine. Chem. Rev. 112, 1773–1804 (2012).
Volkamer, R. et al. Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2-O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements. Atmos. Meas. Tech. 8, 2121–2148 (2015).
Yu, H. et al. Iodine speciation and size distribution in ambient aerosols at a coastal new particle formation hotspot in China. Atmos. Chem. Phys. 19, 4025–4039 (2019).
Jokinen, T. et al. Ion-induced sulfuric acid–ammonia nucleation drives particle formation in coastal Antarctica. Sci. Adv. 4, eaat9744 (2018).
Baccarini, A. et al. Frequent new particle formation over the high Arctic pack ice by enhanced iodine emissions. Nat. Commun. 11, 4924 (2020).
Baccarini, A. et al. Low-volatility vapors and new particle formation over the Southern Ocean during the Antarctic Circumnavigation Expedition. Earth Sp. Sci. Open Arch. https://doi.org/10.1002/essoar.10506899.1 (2021).
Frege, C. et al. Chemical characterization of atmospheric ions at the high altitude research station Jungfraujoch (Switzerland). Atmos. Chem. Phys. 17, 2613–2629 (2017).
Gómez Martín, J. C. et al. A gas-to-particle conversion mechanism helps to explain atmospheric particle formation through clustering of iodine oxides. Nat. Commun. 11, 4521 (2020).
Plane, J. M. C., Joseph, D. M., Allan, B. J., Ashworth, S. H. & Francisco, J. S. An experimental and theoretical study of the reactions OIO + NO and OIO + OH. J. Phys. Chem. A 110, 93–100 (2006).
Khanniche, S., Louis, F., Cantrel, L. & Černušák, I. Computational study of the I2O5 + H2O = 2 HOIO2 gas-phase reaction. Chem. Phys. Lett. 662, 114–119 (2016).
Xia, D. et al. Formation mechanisms of iodine-ammonia clusters in polluted coastal areas unveiled by thermodynamics and kinetic simulations. Environ. Sci. Technol. 54, 9235–9242 (2020).
Gómez Martín, J. C., Lewis, T. R., James, A. D., Saiz-Lopez, A. & Plane, J. M. C. Insights into the chemistry of iodine new particle formation: the role of iodine oxides and the source of iodic acid. J. Am. Chem. Soc. 144, 9240–9253 (2022).
Gómez Martín, J. C. & Plane, J. M. C. Determination of the O-IO bond dissociation energy by photofragment excitation spectroscopy. Chem. Phys. Lett. 474, 79–83 (2009).
Saiz-Lopez, A. et al. Iodine chemistry in the troposphere and its effect on ozone. Atmos. Chem. Phys. 14, 13119–13143 (2014).
Baray, J.-L. et al. Maïdo observatory: a new high-altitude station facility at Reunion Island (21° S, 55° E) for long-term atmospheric remote sensing and in situ measurements. Atmos. Meas. Tech. 6, 2865–2877 (2013).
Atkinson, H. M. et al. Iodine emissions from the sea ice of the Weddell Sea. Atmos. Chem. Phys. 12, 11229–11244 (2012).
Allan, J. D. et al. Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA. Atmos. Chem. Phys. 15, 5599–5609 (2015).
Holleman, A. F. & Wiberg, N. in Anorganische Chemie Band 1: Grundlagen und Hauptgruppenelemente 102 edn. (Walter de Gruyter, 2016).
Jost, D. T., Gäggeler, H. W., Baltensperger, U., Zinder, B. & Haller, P. Chernobyl fallout in size-fractionated aerosol. Nature 324, 22–23 (1986).
Tham, Y. J. et al. Direct field evidence of autocatalytic iodine release from atmospheric aerosol. Proc. Natl Acad. Sci. USA 118, 2009951118 (2021).
Kumar, M., Saiz-Lopez, A. & Francisco, J. S. Single-molecule catalysis revealed: elucidating the mechanistic framework for the formation and growth of atmospheric iodine oxide aerosols in gas-phase and aqueous surface environments. J. Am. Chem. Soc. 140, 14704–14716 (2018).
Burkholder, J. B. et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19. Technical Report (Jet Propulsion Laboratory, 2019); http://jpldataeval.jpl.nasa.gov
Ghanty, T. K. & Ghosh, S. K. Proton affinity and acidity of hypohalous acids: a density functional study. J. Phys. Chem. A 101, 5022–5025 (1997).
Duplissy, J. et al. Effect of ions on sulfuric acid-water binary particle formation: 2. Experimental data and comparison with QC-normalized classical nucleation theory. J. Geophys. Res. Atmos. 121, 1752–1775 (2016).
Thalman, R., Zarzana, K. J., Tolbert, M. A. & Volkamer, R. Rayleigh scattering cross-section measurements of nitrogen, argon, oxygen and air. J. Quant. Spectrosc. Radiat. Transf. 147, 171–177 (2014).
Wang, M. et al. Measurement of iodine species and sulfuric acid using bromide chemical ionization mass spectrometers. Atmos. Meas. Tech. 14, 4187–4202 (2021).
Kürten, A. et al. Neutral molecular cluster formation of sulfuric acid-dimethylamine observed in real time under atmospheric conditions. Proc. Natl. Acad. Sci. USA 111, 15019–15024 (2014).
Sommariva, R., Bloss, W. J. & von Glasow, R. Uncertainties in gas-phase atmospheric iodine chemistry. Atmos. Environ. 57, 219–232 (2012).
Vogt, R., Sander, R., von Glasow, R. & Crutzen, P. J. Iodine chemistry and its role in halogen activation and ozone loss in the marine boundary layer: a model study. J. Atmos. Chem. 32, 375–395 (1999).
von Glasow, R., Sander, R., Bott, A. & Crutzen, P. J. Modeling halogen chemistry in the marine boundary layer 2. Interactions with sulfur and the cloud-covered MBL. J. Geophys. Res. Atmos. 107, ACH2-1–ACH2-12 (2002).
McMurry, P. H. & Grosjean, D. Gas and aerosol wall losses in teflon film smog chambers. Environ. Sci. Technol. 19, 1176–1182 (1985).
Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 17, 1571–1586 (1996).
Peterson, K. A., Figgen, D., Goll, E., Stoll, H. & Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 119, 11113–11123 (2003).
Glowacki, D. R., Liang, C.-H., Morley, C., Pilling, M. J. & Robertson, S. H. MESMER: an open-source master equation solver for multi-energy well reactions. J. Phys. Chem. A 116, 9545–9560 (2012).
Galvez, O., Gomez Martín, J. C., Gomez, P. C., Saiz-Lopez, A. & Pacios, L. F. A theoretical study on the formation of iodine oxide aggregates and monohydrates. Phys. Chem. Chem. Phys. 15, 15572–15583 (2013).
Sinreich, R., Merten, A., Molina, L. & Volkamer, R. Parameterizing radiative transfer to convert MAX-DOAS dSCDs into near-surface box-averaged mixing ratios. Atmos. Meas. Tech. 6, 1521–1532 (2013).
Verreyken, B. et al. Characterisation of African biomass burning plumes and impacts on the atmospheric composition over the south-west Indian Ocean. Atmos. Chem. Phys. 20, 14821–14845 (2020).
Kürten, A., Rondo, L., Ehrhart, S. & Curtius, J. Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid. J. Phys. Chem. A 116, 6375–6386 (2012).
Madronich, S. TUV software package version 4.1 a (1993); https://www.acom.ucar.edu/Models/TUV/Interactive_TUV/
Acknowledgements
We thank the European Organization for Nuclear Research (CERN) for supporting CLOUD with important technical and financial resources. H.F. is recipient of a NASA Earth and Space Science Fellowship (NASA-80NSSC17K0369) (H.F. and R. Volkamer). This research has received support from the US National Science Foundation (AGS-1801280, AGS-1620530, AGS-2027252 (R. Volkamer); AGS-1447056, AGS-1439551, AGS-1531284, AGS-1801574, AGS-1801897 and AGS-2132089 (R.C.F. and N.M.D.)); the Academy of Finland (projects 346369 (T.K.); 331207 (M.R.); 1325656, 316114, 325647, 337549 and 302958 (M.K.); 296628 (M. Sipilä)); Russian Mega Grant project 075-15-2021-574 (M.K.); Jane and Aatos Erkko Foundation (2020-220-08-5835, X.-C. H.); Samsung PM2.5 SRP (M.K.); European Research Council under the European Union’s Horizon 2020 research and innovation programme (projects 714621 (M. Sipilä); 742206 and 895875 (M.K.); 616075 (M.V.-P.); 101002728 (M.R.)); Innovative Training Networks–ITN (CLOUD-Motion H2020-MSCA-ITN-2017 no. 764991, J.C.); German Ministry of Science and Education (CLOUD-16, 01LK1601A, J.C.); Wallace Research Foundation, Carnegie Mellon University Scott Institute for Energy Innovation (N.M.D.); Jenny and Antti Wihuri Foundation (X.-C.H.); Swiss National Science Foundation (200020_172602, BSSGI0_155846 and 20FI20_172622, C.P.L.); Ministry of Science and Higher Education of the Russian Federation (V.M.); National Natural Science Foundation of China (42175118, Y.J.T.); and the Portuguese National Funding Agency for Science, Research and Technology-CERN/FIS-COM/0028/2019 (A.T.). The Maïdo IOP was performed in the framework of the OCTAVE project of the ‘Belgian Research Action through Interdisciplinary Networks’ 5 (BRAIN-be) research programme (2017-2021) through the Belgian Science Policy Office (BELSPO; contract no. BR/175/A2/OCTAVE, J.B.) with ACTRIS-2 TNA support from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 654109 (M.R.), and support by UAR3365 of OSU-Réunion (J.B.).
Author information
Authors and Affiliations
Contributions
H.F., X.-C.H., V.M., J.C., N.M.D., M.K., J. Kirkby, M. Sipilä and R. Volkamer conceived and planned the experiments. H.F., X.-C.H., M. Simon, T.K.K., R.B., A.B., D.M.B., L.C., D.C., B.C., L.D., J.D., M.H., C.K., A. Kürten, A. Kvashnin, H.L., C.P.L., K.L., Z.L., V.M., H.E.M., G.M., R.M., R.L.M., B.M., T.M., T.P., M.P., B.R., J.S., D.S., Y.J.T., A.T., M.V.-P., A.C.W., Y.W., D.S.W., M.W., S.K.W., Y.W., M.X., Q.Y., M.Z.-W., J. Krechmer, M.R. and R. Volkamer prepared facilities or instrumentation. H.F., S.I., X.-C.H., M. Simon, T.K.K., A.A., A.B., L.B., D.M.B., D.C., R.C., B.C., L.D., J.D., M.H., D.K., C.K., H.L., C.P.L., Z.L., V.M., G.M., R.L.M., B.M., A.R., J.S., D.S., C.T., Y.J.T., A.T., A.C.W., Y.W., S.K.W., W.N., Y.W., M.X., Q.Y., J.B., J. Krechmer and M.R. collected data. H.F., S.I., X.-C.H., M. Simon, T.K.K., C.F.L., R. Valiev, M.H., C.K., H.L., G.M., R.L.M., J.S., S.K.W., N.M.D., M.R., T.K. and R. Volkamer analysed data. H.F. performed box modelling with help from T.K.K. and R. Volkamer. S.I. performed quantum chemical simulations with help from T.K., R. Valiev and M.R. H.F. and R. Volkamer wrote the manuscript with contributions from S.I., M.R., X.-C.H., J. Kirkby and comments from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Response in the HIO3 concentration to varying the mixing fan speed.
A strong sensitivity of the HIO3 concentration to changes in the wall loss lifetime twall (dashed black line) is observed. While other parameters are held constant, stirring of the CLOUD atmospheric simulation chamber is reduced at 19:57 UTC, increasing the wall loss lifetime by a factor of four. HIO3 concentrations recover by a factor 12. The superlinear response is evidence for a reasonably long-lived precursor (that is, IO) that gets lost to the chamber walls. At 20:25 UTC, light is turned off, HIO3 production stops, and the HIO3 concentration is efficiently lost to the chamber walls. The model reproduces the observed behaviour if IO is considered to efficiently get lost to the chamber wall.
Extended Data Fig. 2 Time and mass closure of hypothetical HIO3 formation mechanisms.
Sensitivity studies assume hypothetical mechanisms that form HIO3 from different precursors in the model. After the start of I2 photolysis (Δt = 0), ΔHIO3 is defined as HIO3(Δt) - HIO3(Δt = 0). HIO3 measurements (thick black line, grey shading indicates 50 % uncertainty) and simulated time profiles assuming different hypothesised mechanisms in the model (coloured thin lines). The four panels a-d show the closure at different temperatures, and HIO3 concentrations. The formation of HIO3 via reactions R1 and R2 is the only mechanism compatible with observations regarding temporal and mass closure.
Extended Data Fig. 3 Detection of iodine oxide radicals and IxOy species, including the key species IOIO, IOIO4, HOI, and HIO3.
Concentrations of iodine species as measured by the \({{{{\rm{NO}}}}}_{3}^{-}\)-CIMS and the Br−-MION-CIMS, and as modelled by the base-case and extended model. The bottom panel shows the loss rate of sticky molecules to the chamber walls, to particle surfaces, and to dilution. The grey shaded period shows an experiment with extremely high IOx concentrations, where IOIO4 is clearly detected, but extreme particle concentrations and chamber inhomogeneities lead to higher model-measurement differences. The base-case model does not form any HOI or HIO3 in UV-dark conditions. The extended model reproduces both and improves the closure also for other molecules. Calibration factors are given in Supplementary Table 3. T = 263 K.
Extended Data Fig. 4 Sensitivity studies of the HIO3 production towards changes in O3, H2O, and temperature.
For the ranges probed there is no pronounced sensitivity of HIO3 production (normalised by IOIO production) observed. The linear rate order lines (long dashes) assume either O3 or H2O were controlling the rate limiting step towards HIO3 formation. No such dependence is observed. The robustness in HIO3 formation is evidence that neither O3 nor H2O (nor temperature) control the rate limiting step under the conditions probed. Measurements and predictions of the extended model agree within uncertainties. Measurements: 5-95 % whiskers, 25-75 % boxes, median. Model: median only. The grey shading indicates the combined measurement model uncertainty (65 %, 2 − σ standard deviation).
Supplementary information
Supplementary Information
Supplementary discussion, Figs. 1–11 and Tables 1–11.
Source data
Source Data Fig. 1
Plain text data.
Source Data Fig. 2
Plain text data.
Source Data Fig. 4
Plain text data.
Source Data Extended Data Fig. 1
Plain text data.
Source Data Extended Data Fig. 2
Plain text data.
Source Data Extended Data Fig. 3
Plain text data.
Source Data Extended Data Fig. 4
Plain text data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Finkenzeller, H., Iyer, S., He, XC. et al. The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source. Nat. Chem. 15, 129–135 (2023). https://doi.org/10.1038/s41557-022-01067-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-022-01067-z
