
Abstract
Despite two decades of intensified research to understand and cure tuberculosis disease, biological uncertainties remain and hamper progress. However, owing to collaborative initiatives including academia, the pharmaceutical industry and non-for-profit organizations, the drug candidate pipeline is promising. This exceptional success comes with the inherent challenge of prioritizing multidrug regimens for clinical trials and revamping trial designs to accelerate regimen development and capitalize on drug discovery breakthroughs. Most wanted are markers of progression from latent infection to active pulmonary disease, markers of drug response and predictors of relapse, in vitro tools to uncover synergies that translate clinically and animal models to reliably assess the treatment shortening potential of new regimens. In this Review, we highlight the benefits and challenges of ‘one-size-fits-all’ regimens and treatment duration versus individualized therapy based on disease severity and host and pathogen characteristics, considering scientific and operational perspectives.
Similar content being viewed by others
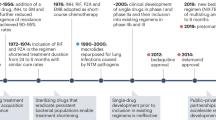
Introduction
The Nobel Prize-winning discovery of streptomycin has enabled the treatment of several infectious diseases, including tuberculosis (TB). Since then, many newer antibiotics have been combined into anti-TB drug treatment regimens that have saved millions of lives. However, despite advances in the past, until the emergence of SARS-CoV-2, TB was the leading cause of infectious disease mortality worldwide, with more than 1 million deaths annually.
Although the current standard treatment for TB is effective, it is also unwieldy. Patients with uncomplicated drug-susceptible TB are required to take multiple antibiotics for 6 months. Since compliance is inconsistent, WHO recommends that this be directly supervised, adding an enormous layer of infrastructure to an exceptionally long treatment programme. With the rise of drug resistance, treatment failure rates have increased along with more toxic therapies that are far more costly. Improved interventions could have a substantial effect on our ability to decrease the morbidity and mortality associated with the disease and to limit further spread, as treatment of active TB is the major modality for preventing transmission in most of the world.
We are at an exciting juncture in TB regimen development. For the first time in four decades, a 4-month regimen, containing rifapentine and moxifloxacin, was found non-inferior to the standard 6-month regimen in the treatment of drug-susceptible TB at the 12-month follow-up1. In 2019, the first 6-month regimen was approved for the treatment of multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB, comprising only three drugs with two novel mechanisms of action: bedaquiline, pretomanid and linezolid2. Yet, shorter, better tolerated and more successful treatments are needed for all patient populations. Accomplishing this will require both new antibiotics and new combinations of approved drugs and clinical candidates. Compared to a decade ago, the anti-TB drug pipeline is in healthy shape with both repurposed and repositioned antibiotic classes as well as drug candidates that act via novel mechanisms of action (Working Group on New TB Drugs). Because of this success, the most important research focus has become the prioritization of promising drug regimens. Although there is a considerable amount of active research and development in this area, one major limitation is the lack of validated in vitro and animal models that predict the success of drugs and drug combinations.
Here, we discuss the uneven distribution of TB burden and disease spectrum around the world and its implication on treatment strategies and challenges. We review new technological advances in mycobacteriology, their impact on TB drug discovery and development, and where knowledge gaps remain. Basic and preclinical research priorities are proposed to accelerate the development of curative drug regimens. Not included here are a survey of host-directed therapy approaches, an exhaustive summary of clinical drug candidates and their targets, nor a list of clinical trials of single drugs and drug regimens, all recently and comprehensively reviewed elsewhere3,4,5.
TB burden and spectrum of disease
Burden distribution and inequalities
TB disease is proceeding asynchronously around the world. In 2019, 30 countries with a high TB burden accounted for 87% of new TB cases and 8 countries accounted for two-thirds of the total cases (WHO tuberculosis)6. As disease burden is strongly associated with socio-economic conditions7, its incidence rates have different trajectories in different parts of the world. In the USA, TB has an incidence similar to amyotrophic lateral sclerosis; in other words, it is a very rare condition. China and the Russian Federation formerly suffered from high TB rates but, although still incurring substantial burden, they are on a better trajectory than most high-burden countries (Fig. 1). In other parts of the world, such as in South America, North Africa and parts of Asia, where incidence was moderate a few decades ago, the TB–diabetes syndemic has reverted the trend; diabetes being an important risk factor for developing TB and presenting more complicated disease, higher relapse rates and mortality8,9. Although several comorbidities can increase susceptibility to TB, HIV-1 is the leading determinant of reactivation risk and thus contributes to the TB burden in sub-Saharan Africa. The long-term repercussions of the COVID-19 pandemic10,11 combined with wealth inequalities will further widen the divide12. To a large extent, distinct regions and subcontinents face unique challenges that require tailored improvements in diagnostic and treatment strategies. Yet, treatment guidelines, diagnostics and research needs tend to be defined globally, based on the integration of worldwide statistics.

a | Incidence of tuberculosis (TB) per 100,000 population in 2020. Not applicable: WHO criteria for national prevalence survey not met. b | The top graphs represent the incidence of TB in South Africa, the Russian Federation and China from 1990 to 2020. Sub-Saharan Africa has been on an overall trajectory of increased incidence until 2010, mostly driven by the TB–HIV-1 epidemic and the 20-fold increased risk of reactivation in people positive for HIV-1. Initial increase in incidence and mortality in the Russian Federation coincides with the collapse of the Soviet Union and health-care system, which was brought under control after 2000. China has been on a consistent steady decline since 1990. The bottom graphs show the estimated impact of the COVID-19 pandemic on TB mortality in South Africa, the Russian Federation and China up to 2025. Plots were generated using publicly available TB burden data from WHO reports,5,6 and the World Bank database. Part b, top graphs, based on data from WHO global TB reports from 1990 to 2021 and adapted from the World Bank database, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Part a and part b, bottom, adapted with permission from ref.6, WHO.
Spectrum of disease
WHO defines latent TB infection (LTBI) as a state of persistent immune response to stimulation by Mycobacterium tuberculosis antigens without evidence of clinically manifested active TB and with bacillary replication absent or below some undefined threshold as a result of immunological control. LTBI affected approximately 1.7 billion individuals in 2014 (ref.13), just under one-quarter of the global population. The rate and risk of LTBI reactivation are greatest within 2 years from infection, then decline from 2 to 5 years and beyond14. Quantifying the rates of late reactivation versus early progression is complicated by reinfection in high-incidence areas15 and difficulties in validating starting assumptions16 such as the presumption that asymptomatic infection is lifelong17. The substantial gaps that remain in our understanding of reactivation hinder our efforts towards TB eradication as the burden of LTBI constitutes an enormous reservoir from which active TB cases can emerge. Identifying and triaging this population will drive our chances to meet the global targets of 90% reduction in TB incidence by 2035 and elimination of TB (less than 1 incident case per 1,000,000 per year) by 2050 (ref.18).
Active pulmonary TB disease either appears within 1–2 years after infection or following reactivation of latent infection in ~5–15% of infected individuals throughout their lifetime19. TB disease is defined by progressive bacterial replication and pulmonary necrosis and often but not always includes cavitary lesions that promote the transmission of bacteria20. Although the outcome of infection by M. tuberculosis has generally been represented as a bimodal distribution between active and latent TB based on the presence or absence of clinical symptoms, it is now increasingly viewed as a continuous spectrum generated by a varied immunopathology that supports bacterial replication, persistence or killing21,22,23,24, extending to within-host heterogeneity25.
The current drug regimen used to treat uncomplicated drug-susceptible TB is the outcome of several decades of clinical trials26. This short-course therapy with isoniazid, rifampicin, pyrazinamide and ethambutol for 2 months followed by 4 months of isoniazid and rifampicin was implemented four decades ago and has not changed since. By contrast, numerous clinical trials of drug-resistant TB were recently completed or are recruiting, providing a dynamic landscape and opportunities to improve cure rates and reduce therapy duration from 24 to 6 months2,27. The mandate of major drug discovery initiatives is to develop drugs that, in combination, create shorter, safer and simpler regimens to cure all patients with TB28.
The possibility that isoniazid could be used to prevent reactivation of LTBI was considered soon after it was introduced in 1952. Subsequent clinical trials in the 50s and 60s demonstrated a marked reduction in active TB in patients treated with isoniazid and a long-lasting protection29,30. Based on these results, preventive treatment of TB infection with isoniazid for 6 or 9 months became a widespread approach to TB control and is still in use today. More recently, a rifamycin was added to or substituted for isoniazid, which successfully reduced treatment duration31,32. Five regimens of various durations that include isoniazid and/or a rifamycin are recommended globally and considered equivalent by measure of effectiveness and hepatotoxicity, though they have not been directly compared in clinical trials32,33,34. Besides historic trials with pyrazinamide35, levofloxacin in children36 and adults37,38 and delamanid (PHOENIx MDR-TB) (ClinicalTrials.gov, NCT03568383) are currently being tested as prophylactic therapy in household contacts of patients with MDR-TB.
TB infection and disease present as a multifaceted spectrum that can be represented in three dimensions (Fig. 2). This multitude of manifestations and microbiological diversity creates formidable treatment challenges (see below). LTBI is now recognized as a dynamic continuum of response to infection21,23. At the ‘active’ end of the latency spectrum are individuals at high risk of developing active disease (‘progressors’), who would benefit from reactivation risk assessment and treatment. Identifying and curing this population is critical to meet the global targets for TB control and elimination by 2050 (ref.18). The spectrum of LTBI, from true latency to incipient disease, has been comprehensively reviewed elsewhere20,23. Due to the lack of validated immune and bacteriological markers, LTBI is only operationally defined as a state of persistent immune response to stimulation by M. tuberculosis antigens without evidence of clinically manifested active TB, hampering the triaging of those at greatest risk for progression to TB disease39. Overall, basic knowledge gaps are deeper for LTBI than for active disease.

Tuberculosis (TB) presents as a spectrum along three axes: disease pathology and severity, bacterial persistence and drug tolerance, and genetic resistance. The pathology of TB disease is a dynamic continuum from fully latent asymptomatic infection to active disease with high bacterial burden in open cavities, leading to transmission and more frequent treatment failure. Individuals with latent TB infection who are progressing towards incipient TB are at high risk of developing active disease and would benefit from reactivation risk assessment and treatment. The spectrum of immunopathology creates a diversity of microenvironments to which the pathogen responds with metabolic and physiological adaptations leading to drug tolerance or phenotypic drug resistance and persistent disease. Drug tolerance as well as other patient and pathogen factors lead to a spectrum of genetic resistance both in terms of the number of drugs a bacterium is resistant to and the level of resistance to each drug. Such variability along three axes creates a gradient of decreased drug efficacy and lesion sterilization within and across patients, constitutes a multidimensional challenge for health-care programmes and complicates clinical trials.
Active symptomatic TB presents with a range of severity, primarily classified as either cavitary disease or moderate non-cavitary disease. Decades of clinical trials consistently point towards poorer prognostic and treatment outcomes for cavitary disease40,41,42. Recent retrospective analyses have shown that patients with cavities and a high bacterial burden in sputum may require treatment durations of more than 6 months, whereas 4-month therapy was non-inferior to 6-month therapy in patients with non-cavitary minimal disease43. The identification of patients who might benefit from shorter treatment durations could therefore only require broadly available chest X-ray radiography and sputum smear methods.
Treatment of drug-susceptible and drug-resistant M. tuberculosis strains requires a minimum of three to four antibiotics in combination, leading to complex patterns of drug susceptibility and resistance. The infecting M. tuberculosis strain is classified as fully drug susceptible, mono-resistant, MDR or XDR. Within the latter two categories, individual drug susceptibility profiles guide the design of patient-tailored drug regimens, which are subject to national guidelines and institutional policies44,45.
In both LTBI and active disease, host immunity produces diverse microenvironmental niches that support suboptimal growth or complete growth arrest of M. tuberculosis. Many different lesions, with different immune effects, exist simultaneously in a single infected individual. The physiological state of non-replication in bacteria is associated with drug tolerance or phenotypic drug resistance. Indeed, the response to drug treatment is biphasic, with an initial phase of rapid though incomplete clearance of the bulk of infecting bacilli, followed by a longer phase required for complete sterilization of persisting populations46. In a defined bacterial population, drug tolerance is either induced by a variety of stresses or is conferred by pre-existing physiological heterogeneity47,48,49, both producing a persister subpopulation. Cell-to-cell variability creates an almost infinite spectrum of phenotypically heterogeneous subpopulations even under uniform conditions50,51,52,53,54,55, which is thought to ensure survival, as at least a small proportion of cells are predisposed to respond to potential threats such as antibiotics, a strategy coined as ‘bet-hedging’56. Cell-to-cell heterogeneity is viewed as the consequence of both stochastic noise and specific regulatory systems that couple cell cycle progression and adaptive bacterial metabolism with changing host-associated environmental stress. Inclusion of adaptive regulatory elements to the stochastic noise mechanism for the generation of phenotypically diverse bacteria within a population has led to a concept referred to as ‘phenomic potential’57. The phenomenon of drug tolerance is particularly acute at the sites of TB disease and is a hallmark of the disease as well as a major factor contributing to protracted therapy duration due to long-standing host–pathogen co-evolution58 and the resulting adaptations of M. tuberculosis in response to host immunity. There is broad consensus that phenotypic drug resistance contributes to individual variability in TB disease persistence59, relapse and reactivation. However, large knowledge gaps remain to determine how in vitro observations of these phenomena can be applied to predict clinical outcome. In the following section, we explore the treatment challenges that are associated with disease complexity.
Treatment challenges
Treatment of all forms of active TB disease requires multiple antibiotics administered for several months60. Approved first-line, second-line and third-line agents recommended to treat drug susceptible, MDR and XDR TB, respectively, are listed in Supplementary Table 1. The variability of disease progression, host response and drug resistance phenotypes complicates treatment and drug discovery but also creates opportunities to stratify patient populations and optimize preventive and therapeutic strategies. Because TB is largely a disease that occurs in resource-constrained countries, existing infrastructure only enables moderately complex interventions. This adds substantial operational and implementation challenges to the already daunting research mandate.
Identifying individuals with LTBI at high risk of reactivation
Currently, the WHO recommends preventive treatment for individuals with LTBI who are at high risk of TB reactivation; that is, people living with HIV-1, infants and children under 5 years of age who are household contacts of patients with pulmonary TB, and patients who receive immunosuppressive therapy32,33,34,39. Scientific breakthroughs have recently made it possible to identify patients at risk of reactivating LTBI based on immunobiology rather than on the mainly operational criteria described above61. These include non-invasive positron emission tomography–computed tomography (PET–CT) and PET–magnetic resonance imaging (PET–MRI), immune response markers identified by transcriptomics62 and other omics approaches, genome-wide association studies of host and pathogen, epigenetics, and the identification of differentially expressed genes in individuals who control LTBI and progressors in both patients and animal models (Supplementary Table 2). However, validation studies are needed to determine the utility of biomarkers in estimating the size of mycobacterial burden, and markers of protective immunity as surrogates of incipient TB63. Although some of these approaches are not compatible with the capacities in resource-poor countries, they critically contribute to our knowledge of reactivation and can form the basis for the development of surrogate markers adapted to the global health realities where TB burdens are highest. Once predictive markers are validated in prospective clinical studies, implementation hurdles will remain to translate ‘high risk’ determination into appropriate treatment of potential progressors64. This is further compounded by the caveats of current preventive therapy: the lack of a universal regimen active against drug-resistant M. tuberculosis32, the hepatotoxicity65 and limited sterilizing activity of isoniazid66, drug–drug interactions between rifamycins and anti-retroviral therapy67, and poor acceptance and completion rates globally68. Moreover, adding LTBI therapy to current TB treatment programmes will be a logistical challenge. However, the CDC does currently recommend treatment for every infected individual regardless of the risk of reactivation. The preferred regimens are identical to those recommended by WHO: three rifamycin-based regimens and two alternative monotherapy regimens with daily isoniazid, unless the infecting M. tuberculosis strain is presumptively resistant to both isoniazid and rifampin33. This treatment approach is manageable within the USA and, perhaps, other countries with either low or moderate prevalence of LTBI.
Assessing disease severity of patients with active TB
Cavitation status and bacterial burden in sputum are consistently emerging as correlates of disease outcome42,43. According to the current American Thoracic Society/CDC/Infectious Diseases Society of America Clinical Practice Guidelines69, therapy for 9 months is recommended to treat extensive cavitary TB as well as for patients with positive sputum cultures persisting at 2 months. Extended treatment duration is also considered for patients with low body mass index, smokers, individual with diabetes, and individuals with HIV-1. Recently, an in-depth analysis of clinical trial data has revealed that patients with non-cavitary TB, infected with drug-susceptible M. tuberculosis and with a low sputum burden could successfully complete therapy within 4 rather than 6 months. This opens up the possibility for a stratified approach that would tailor treatment duration based on initial disease severity (high bacterial burden in sputum and cavitary disease) as an alternative to the standard 6-month duration for treatment43. Shortening treatment when appropriate would improve completion rates and provide welcome relief to health-care systems. Markers such as presence of cavities and bacterial load in sputum can be assessed in resource-limited settings and could be rapidly validated if their correlation with treatment duration were systematically integrated in clinical trials70. Pending formal validation, the remaining challenge is the implementation of new flexible treatment guidelines on a global scale given burden inequalities and unique treatment challenges in distinct regions and subcontinents.
Understanding drug tolerance
Targeting drug-tolerant bacterial populations and persisters is the key to achieving faster durable cure71. These populations drive the drug recalcitrant nature of both active disease and LTBI. As a research community, we have a good understanding of the microenvironments and stress conditions that exist at the site of disease, but there remain uncertainties about the clinical relevance of each one and how they influence treatment outcome. We have developed assays that reproduce conditions of immune pressure and environment as well as drug-induced stress, in vitro and ex vivo, and we understand how M. tuberculosis adapts to these conditions and how this translates to reduced drug susceptibility in these models (Table 1). Ex vivo models, such as those relying on explanted cavity caseum from rabbits72 and differentially culturable M. tuberculosis retrieved from patient sputum53,73, most faithfully reproduce in vivo conditions but are inherently resource intensive and mostly suitable to profile approved drugs and clinical development compounds. Drug tolerance of intracellular M. tuberculosis remains a substantial knowledge gap for two reasons. First, a wide range of immortal cell lines and, occasionally, macrophages derived from primary blood or bone marrow monocytes are used for M. tuberculosis infection assays, leading to discrepant potency values. For example, M. tuberculosis is approximately 100-fold less susceptible to rifampicin within hypoxic lipid-loaded macrophages than in normoxic THP-1-derived macrophages74,75, yet macrophage-like immortalized cell lines are commonly used due to their low cost and ease of maintenance considerations. A comparative review of anti-TB drug potency within infected primary macrophages and common cell lines is lacking. Second, polymorphonuclear leukocytes are increasingly recognized as a crucial and permissive niche of bacterial survival and replication76,77,78,79. Rather than eliminating mycobacteria, neutrophils seem to provide a safe haven and transport mycobacteria to macrophages while they retain the potential to damage host tissue80. To date, no in vivo or ex vivo assay has been developed to assess the drug tolerance of M. tuberculosis residing within and around neutrophils and other granulocytes. Although the short lifespan of neutrophils may present a challenge, a recent study showed that granulocytes are retained in M. tuberculosis-infected mouse lungs at least as long as monocytes and macrophages, consistent with a model in which M. tuberculosis uses granulocytes as a replicative niche for intracellular growth81.
In vivo, we have optimized a wide range of animal models, none fully reproducing human TB, but each with features justifying its utility (Supplementary Table 3). It has been suggested that TB should be approached as a polymicrobial infection82. Human infection may consist of replicating, dormant (slow-growing or non-growing) and reactivated bacteria in all clinical stages82. The relative proportions of these cells may change in response to host immune responses and antibiotic therapy83. In other words, the range of phenotypic antibiotic susceptibility in a population is extreme, possibly exceeding that seen for genetic variants. Filling the knowledge gaps to achieve treatment shortening can thus be viewed as a two-step process: first, the inventory of available assays and models that collectively recapitulate micro-environmental conditions encountered in vivo and produce all potentially relevant pathophysiological states of the bacteria, validation of in vitro assay panels that predict clinical treatment shortening, and determination of which animal models are required to bridge in vitro assays and clinical efficacy for new drug candidates; second, the identification of missing assays, models, and bacterial profiles to refine the predictive power of the approach and determination of how they integrate within a patient to drive treatment outcome (that is, the response to treatment may be more complex than the sum of its parts, even if, collectively, assays and models faithfully capture all relevant in vivo conditions). Many have thought that the best way to identify successful therapies is to find agents active in all assay conditions; however, this might well not be true. Prioritizing in vitro assay conditions that have been validated as predictive of in vivo and clinical treatment is likely to yield better drug candidates and help focus resources. Though challenging, all this seems attainable with the ever-faster development of new technologies and a healthy drug pipeline.
Determining high risk of recurrence following therapy completion
Rather than complete bacterial eradication in all treated individuals, apparent bacterial sterilization during and following TB chemotherapy is now understood to include an antibiotic-induced persistent state that forms a relapsing reservoir in a subset of treated patients. Accordingly, ‘high resolution’ determination of the minimum inhibitory concentration (MIC) of isoniazid and rifampicin within the drug-susceptible range (that is, below standard resistance breakpoints) has shown that higher MIC values were associated with a greater risk of relapse than lower MIC values84. We lack validated markers to follow treatment response and distinguish, in real time, patients who need to remain on treatment from hyper-responders who can complete treatment earlier. Identifying patients who might require longer-than-standard treatment duration would minimize costly relapses. A five-gene signature that correlates with the pulmonary inflammatory state, as measured by PET–CT, identified patients at risk of treatment failure 1–4 weeks after start of therapy85. More recently, a 22-gene transcriptomic model that predicts cure-associated end-of-therapy was defined for patients infected with drug-susceptible and MDR M. tuberculosis strains86. Developing rapid and affordable point-of-care tests that quantify these biomarkers and deploying assays and adapted treatment guidelines are the major unmet needs.
Simplifying and improving treatment of drug-resistant disease
The clinical drug candidate pipeline is healthy, enabling a shift in favour of developing pan-TB or universal drug regimens to treat all forms of drug-susceptible and drug-resistant TB. The success of the NIX-TB trial (NCT02333799), a 6-month three-drug regimen that delivered high cure rates in select patient populations infected with MDR and XDR strains2, constitutes a promising step towards this aspirational goal. New diagnostic tools that either simplify or increase the accuracy of drug-resistance profiling are continuously improved to help physicians and health services interpret results and provide faster and more targeted treatment for patients87,88. Recently, the WHO published a catalogue of 17,000 M. tuberculosis mutations and their association with drug resistance89. The report provides a reference standard for the interpretation of mutations conferring resistance to all first-line and a variety of second-line drugs. It complements an elegant tool called SplitStrains, which leverages whole-genome sequencing data to identify and separate mixed M. tuberculosis infections with genetically different strains90, a phenomenon that can lead to hetero-resistance and the ensued treatment complications91,92. The new toolkit enables more precise detection, identification and quantification of multiple infecting strains within a sample. Besides the implementation of new improved diagnostics, the major challenge to eradicate drug-resistant TB is the discovery and development of universal regimens only comprising drugs with novel mechanisms of action and minimum side effects, which would not only be used to treat resistant TB but could also replace the first-line regimen. Towards this goal, the Tuberculosis Drug Accelerator was launched in 2011 as an experiment designed to facilitate collaboration in anti-TB drug discovery by breaking down barriers among competing laboratories and institutions. The Tuberculosis Drug Accelerator is a unique collaboration of academic, pharmaceutical, global non-governmental organizations and governmental organizations that pledge to make medicines affordable to those in need, build knowledge and seek to develop shortened, less toxic, universal drug regimens that can achieve rapid, durable cure irrespective of resistance to existing drugs28.
Drug development priorities
The shortcomings of TB treatment can be ascribed to four major challenges: curing TB takes considerably longer than any other bacterial infection of the lungs owing to a combination of drug, pathogen and host factors; drug tolerance fuels and synergizes with drug resistance; single drug and regimen development tested in sequence is inherently slow while tools are emerging to rationally prioritize regimens early in the cascade; and a surprisingly small number of drugs have been tested as preventive therapy of LTBI. Although the division into such broad areas may appear overly schematic, it provides a useful starting point to highlight where future research could focus and identify drug development priorities.
Addressing biological knowledge gaps to optimize treatment
Persistent TB disease is a consequence of immune evasion or the ability of M. tuberculosis to persist and multiply within the very host cells committed to eliminating bacterial pathogens93,94, suboptimal drug penetration at the sites of disease, and extreme drug tolerance of selected subpopulations, some located in necrotic granulomas and cavity caseum95,96. Such niche-specific pharmacokinetics and pharmacodynamics present a challenge to ensure optimal drug delivery to bacilli in dynamic physical loci and metabolic states49,97.
Host-directed approaches that complement antibacterial therapy to accelerate cure generally rely on four major concepts: modulation of pro-inflammatory mediators to dampen inflammation, curb immunopathology and improve lung function and integrity; enhancement of immune and memory response efficacy; enhancement of macrophage and neutrophil bactericidal mechanisms to counter the immune evasion mechanisms of M. tuberculosis; and disruption of granuloma structure to improve drug penetration and expose the bacilli to drug action. These approaches and corresponding therapies in clinical development are beyond the scope of this Review and have been comprehensively reviewed in recent years3,98.
A multipronged strategy centred on the pathogen, its metabolic and physiological adaptations, its precise location relative to immune cells and lesion structures, and its susceptibility to drugs and drug combinations in these niches will certainly increase our understanding and might contribute the knowledge required to develop shorter regimens. Recent efforts have focused on the identification of mycobacterial targets that are essential and vulnerable in persistence niches: sputum99,100, infected mouse lungs, ex vivo and in vitro macrophages, and other relevant environments (reviewed in ref.97). Although host transcriptome analyses are widespread and deliver increasingly robust data, the very low abundance of pathogen transcripts relative to host transcripts in biological samples constitutes a formidable challenge and a limitation of currently available technologies. However, this hurdle is likely to be overcome soon given the ever-improving performance of sequencing platforms and big data analysis. Multi-omics profiling of M. tuberculosis in caseum, foamy hypoxic macrophages, neutrophils and other persister populations in specific lesion compartments will identify pathways and functions that are critical to mycobacterial survival and might represent new antibiotic targets, thus possibly enabling shortening treatment duration. Likewise, a proven CRISPR interference-based functional genomics screen101 could be applied to persistence environments, such as ex vivo caseum, macrophages and mouse infections, to identify vulnerable targets as well as targets that synergize or antagonize drug treatment102.
Leveraging the potential of synergistic drug interactions in multidrug treatment regimens to accelerate durable cure is a growing field that successfully combines experimental, systems biology and computational tools in powerful platforms such as INDIGO and DiaMOND103,104,105,106. Collectively, they constitute a living repository of drug interactions on which researchers can build to refine their predictive value. However, although screening for positive and negative drug interactions in vitro is straightforward, understanding which model systems and in vitro assays predict synergies that might translate into the clinic is complex because TB is a polymicrobial disease spread across multiple sites of infection, whereas in vitro synergies are measured in a homogeneous environment using constant drug concentrations. Stochastic and adaptive cell-to-cell bet-hedging leading to a selective advantage for the whole bacterial population is likely to contribute to synergy and antagonism in ways that may be difficult to reproduce in vitro107. As a promising example, treatment with bedaquiline in vitro activates a regulatory network that coordinates multiple mechanisms to push M. tuberculosis into a tolerant state, which can be disrupted by knocking out its predicted transcription factors Rv0324 and Rv0880, increasing bedaquiline killing108. This finding enabled the prediction that pretomanid could synergize with bedaquiline through inhibition of the Rv0880-response regulon and potentiate killing by bedaquiline at the population level. Although the authors experimentally confirmed the predictions in vitro, identifying bacterial populations to which this applies in animal models and in patients with TB is the critical next step for bedaquiline–pretomanid as the backbone of the highly successful NIX-TB regimen2 and for all predicted drug–drug synergies in general. In vitro models, such as INDIGO, are pathway based, enabling the formulation of hypotheses to test the underlying biological mechanisms and adjust in vitro growth conditions to improve predictions. Recently, the predictive power of the DiaMOND platform was evaluated using drug combination dose responses measured under eight conditions that reproduce lesion microenvironments. Machine learning was applied to all two-drug and three-drug combinations of ten antibiotics to develop classifiers predictive of multidrug treatment outcome in a mouse model of disease relapse100. Identifying positive pharmacodynamics interactions in vitro that translate into the clinic is further complicated by the fluctuating drug concentrations seen by bacilli at the sites of infection as potency interactions inherently depend on the relative concentration of each drug in the combination. To better realize the potential of combination therapy, an interaction landscape is drawn over the full dose–response matrix to delineate synergistic and antagonistic dose regions and quantify the interactions. These can then be mapped back to the in vitro–in vivo correlation to refine in vivo predictions.
Elegant tools exist, and many more are developed, to probe the heterogeneity of bacterial behaviour inside the host niche, survey their metabolic state, and track them during therapy in animal models49,109,110 and inpatient bioaerosols111. One key pragmatic outcome of these activities is the detection and mapping of M. tuberculosis persisters that survive drug treatment, which would guide prioritization of future research. Validation of the findings from samples of the human lung is challenging although resected lung tissues from patients undergoing elective surgery for drug refractory disease may be an underexploited resource. As mentioned earlier, the contribution of M. tuberculosis bacilli that survive and multiply in and around neutrophils to persistent disease76,77,81 has been largely underappreciated, overshadowed by the role of macrophages, which are much more amenable to in vitro and ex vivo studies. However, in sputum and cavity caseum, M. tuberculosis bacilli are primarily extracellular or within neutrophils, with a smaller fraction replicating in macrophages79. Consistent with these observations, a signature of active TB is dominated by a neutrophil-driven interferon-inducible gene profile112 and cavity caseum is often infiltrated with infected neutrophils in both animal models and patients.
Cavitary disease has long been recognized as a predictor of poor treatment outcome and as a factor formally associated with the need for longer treatment duration, especially when associated with positive culture at 2 months43,69 (see above). In a series of new developments, pulmonary PET–CT and μCT imaging have shown that cavities spread M. tuberculosis to other parts of the lungs through the bronchi113,114,115, revealing wide networks of connected lesions with complex morphology, in stark contrast with the oversimplified view that TB granulomas and cavities are isolated spherical structures. The extent of bronchogenic spread, its contribution to disease dissemination, and its impact on treatment outcome in animal models remain to be defined to guide the manipulation of models that better recapitulate this aspect of TB116. Thus, targeting treatment not only to cavities but also to airway networks and the bronchial tree might be more important than previously realized to accelerate cure.
Prioritizing promising drug combinations
Owing to a growing pipeline of anti-TB drug candidates (Fig. 3a), a new paradigm of pan-TB regimen development has gained momentum over the past 5–10 years. New regimens to treat drug-susceptible, MDR and XDR M. tuberculosis infections have become conceivable due to the substantial proportion of drug candidates and recently approved drugs with novel mechanisms of action (Fig. 3b). Universal regimens would substantially accelerate global efforts to control TB. Given the overwhelming number of potential combinations of three, four and five drugs selected from ten major classes, whether approved or in clinical development, the number one priority is the development of a rationale to predict best performing regimens. So far, the ranking of regimens for clinical trials has been driven by limited preclinical data and could thus be optimized117.

a | Shown are promising drug candidates currently in preclinical and clinical development, including the development of regimens that combine repurposed, repositioned and new drug classes. Approved drugs are indicated by an asterisk (delamanid was approved by the EMA only, and pretomanid was approved by the FDA for use in the bedaquiline–pretomanid–linezolid regimen). Drugs are colour coded by chemical class and target pathway. For a complete list of published candidates currently in the pipeline, from early preclinical development to regulatory approval, and a review of their mechanism of action, see Working Group on New TB Drugs and ref.183. b | A simplified version of the cell envelope and the cytoplasmic membrane of Mycobacterium tuberculosis is shown with schematized versions of the targets of recently approved drugs and clinical candidates, with novel mechanisms of action, listed in part a. The majority of novel targets are membrane associated. The diarylquinolines bedaquiline, TBAJ-876 and TBAJ-587 target the ATP synthase. The nitroimidazoles pretomanid and delamanid exhibit a dual mode of action under low and normal oxygen tension, poison multiple essential pathways, and are bactericidal against replicating and non-replicating mycobacteria184. SQ109 and the MPL series are the most advanced among a broad panel of agents targeting MmpL3, involved in export of trehalose monomycolate, a mycolic acid component. Three chemically distinct series all target DprE1: OPC167832, TBA7371 and BTZ043 (ref.185). Both MmpL3 and DprE1 are unique to mycobacteria. GSK656 is the first oxaborole in clinical development targeting a mycobacterial tRNA synthetase186 and GSK286 is a new chemical entity with a novel mechanism of action related to cholesterol catabolism. Part b adapted from ref.187, Springer Nature Limited.
Several steps could contribute to a rationale for the prioritization of regimens combining drugs that optimally reach and kill all M. tuberculosis bacilli, thereby maximizing the potential to shorten treatment. We can measure how effectively each drug covers the diverse sites of infection, how much is required to inhibit or kill each mycobacterial subpopulation, and whether adequate concentrations are sustained during a typical dosing interval. Algorithms that integrate drug penetration into major lesion compartments, drug potency against intracellular and extracellular persisters at the sites of disease, host immunity, and efficacy in preclinical models can simulate lesion coverage118 and prioritize combinations predicted to accelerate sterilization and reduce relapse rates119,120,121,122. This approach, combined with predictions of synergistic drug combinations, could deliver an affordable number of promising drug combinations to be tested in resource-intensive animal models that reproduce features of human pathology not seen in mice123,124,125,126. Using an iterative strategy, researchers can identify specific features, read-outs and outcomes that predict treatment shortening and further refine the computational tool (Fig. 4). Concomitant testing of prioritized regimens in early clinical trials using biomarkers predictive of long-term outcome127 would therefore enable validation of animal models and refinement of the translational platform. This translational approach is in its infancy but holds promise to spare years of clinical development for new pan-TB drug regimens.

Schematic illustration of the large number of possible combinations if drug candidates selected from 10 drug classes shown in Fig. 3 are combined in 3, 4 or 5 drug regimens (a minimum of 482 combinations assumes only 1 drug per class). In addition, owing to the varying drug doses, varying treatment durations and varying dosing frequencies, the number of clinical trial arms is within the thousands. Resource considerations underline the need for prioritization, using validated in vitro assays, drug interaction platforms, such as INDIGO or DiaMOND, pharmacokinetic (PK) and pharmacodynamic (PD) studies in preclinical species, and translational modelling tools, to select drug combinations with the highest potential to reduce treatment duration and improve cure in patients with tuberculosis, thus reducing the number of clinical trial arms to practical dimensions. New strategies, such as adaptive trial designs, doses and treatment duration tailored to patient characteristics, and longitudinal biomarkers of efficacy are required to accelerate the learning cycle, validate the in vitro and in vivo prioritization tools, and refine the computational approaches. Middle panel, top right, adapted from ref.188, Springer Nature Limited. Middle panel, bottom left, adapted from ref.189, Springer Nature Limited. Middle panel, top left, reprinted from ref.190, Springer Nature Limited.
Conventional assessment of durable cure of active TB in clinical trials is inherently slow and involves very large patient cohorts followed by long follow-up periods. Adaptive trial designs that simplify and accelerate both phase II and phase III128 have been proposed to evaluate the growing pipeline of drug candidates and leverage the massive efforts dedicated to regimen prioritization using translational platforms that link in silico, in vitro and in vivo data (Supplementary Box 1). To maximize the impact of adaptive designs and rapidly identify drug combinations for progression from phase II to phase III, biomarkers predictive of long-term outcome are sorely needed, yet decades of research have not delivered a ‘magic bullet’. Improved bacteriological read-outs (quantitative, longitudinal and continuous measures that are more sensitive to differences than the traditional 8-week sputum conversion end point) have been proposed and are increasingly adopted in clinical trials129. Recent efforts have focused on PET–CT to distinguish lesions that resolve rapidly, slowly or not at all in response to conventional versus novel treatment regimens127. Key to the success of regimen prioritization, trials should include measurements of drug concentrations to validate the lesion coverage approach for predicting treatment shortening as well as risk stratification algorithms to tailor doses and treatment duration to the extent of disease, immune status, and other validated correlates of poor prognosis130. Overall, many innovative and exploratory development strategies remain to be harmonized and formally endorsed to accelerate and enhance our ability to learn.
Breaking the tolerance–resistance link
Bacteria can produce populations of dormant cells that are tolerant to killing by antibiotics. Drug tolerance conferred by slow growth can be genetically encoded or not and sets bacteria on the path to high-level resistance primarily mediated by ‘on-target’ mutations. The discovery of an evolutionary pathway from tolerance to resistance underscores the possibility that treatment strategies that can eliminate tolerant cells to prevent the development of genetic resistance from phenotypically resistant bacterial subpopulations could shorten therapy131,132. Bacteria can also acquire non-canonical mutations that confer survival advantages in the presence of a given antibiotic, that is, an increased level of tolerance to the drug, likely acting as stepping-stones for acquisition of canonical resistance133. In M. tuberculosis, several tolerance mechanisms have been suggested to facilitate the emergence of drug resistance: mistranslation134, alteration of propionate metabolism133, phase variation in glpK135,136 and hypoxia-induced carbon flux diversion137,138,139. RNase J is disproportionately mutated in drug-resistant clinical M. tuberculosis isolates133 and preliminary results indicate that deletion of the corresponding gene confers increased tolerance to lethal concentrations of several drugs by reducing their killing rate140. Disabling the DosRS dormancy regulon through chemical inhibition by artemisinin suppressed tolerance to isoniazid141; targeting asymmetric cell division leads to the loss of single-cell heterogeneity and faster killing by vancomycin and rifampicin54; and decreasing mistranslation with the aminoglycoside kasugamycin potentiates rifampicin activity in vitro and in mice142. Host immune responses may promote drug tolerance through several mechanisms: they apply pressure on bacterial growth, thereby altering drug susceptibility143; they shape the microenvironment to which drugs are exposed at the sites where M. tuberculosis resides and can lead to pH and oxygen concentrations144,145 that may reduce (or occasionally enhance) drug activity; and they alter granuloma histopathology in ways that impact drug access95,146. Host-directed therapies that target each of these mechanisms have the potential to reduce drug tolerance and resistance3. Additional mechanisms of drug tolerance are reviewed elsewhere147 such as the disproportionate number of toxin–antitoxin systems present in M. tuberculosis that mediate stress-induced adaptation, slowing of growth and drug tolerance. These are early-stage and exploratory observations, yet they lend support to the concept of tackling tolerance to prevent resistance.
Though not falling under the category of tolerance per se, other indirect mechanisms facilitate the emergence of resistance and could be targeted to prevent or limit it. For example, a preliminary study suggests that disruption of the ImuB–DnaN (β-clamp) interaction by griselimycin prevents DnaE2-mediated induced mutagenesis, suggesting that the mycobacterial mutasome might be a good target for novel ‘anti-evolution’ drugs aimed at protecting against emergent resistance148; inhibition of bacterial factors that promote mutagenesis as an anti-evolution strategy149; inhibition of intrabacterial metabolism by promiscuous cytochromes and other enzymes150,151 that may constitute a source of induced resistance; and inhibition of nonspecific drug efflux pumps shown to extrude anti-TB drugs152,153,154.
Preventing reactivation of LTBI
Once a validated biomarker signature of incipient and progressing TB has been established to triage patients with LTBI who would benefit from preventive therapy, TB control programmes will need single drugs or regimens that can be administered safely to individuals with LTBI who are not only at risk of reactivation but are also contacts of individuals who are infected with MDR M. tuberculosis. The five recommended regimens all contain isoniazid and/or a rifamycin and are thus inadequate for these important populations. In addition, individuals with HIV-1 and LTBI should not receive rifampicin if they are treated with antiretroviral agents that are substrates of CYP3A4. More so than for active TB, a universal drug or drug combination is critical as the MDR or XDR status cannot be assessed in LTBI and is only inferred from household contact susceptibility profiling. New pan-TB regimens that successfully shorten treatment of active TB may provide invaluable lessons to tackle persisters in latent TB granulomas and prevent reactivation of LTBI. A ‘sterilizing’ rationale would help design clinical trials with a better potential to cure subclinical, incipient and progressing TB (Box 1).
Conclusion
The repercussions of the COVID-19 outbreak have already made clear that we are unlikely to reach the targets of the WHO ‘end TB strategy’, which call for 80% reduction in TB incidence and 90% reduction of TB deaths by 2030 (ref.155). Even under more favourable circumstances, these goals are often considered an optimistic scenario given the logistical complexities of a 6-month multidrug regimen and the poorer treatment outcomes in the setting of antibiotic-resistant disease. Shorter and more effective therapies could get us a long way towards these millennium goals, but this will take decades if we are to rely on the traditional approach of iterative large clinical trials with small incremental variations in drug regimens. Breakthroughs will come from transformative trial designs and rationally designed regimens with novel mechanisms of action. The success of bedaquiline–pretomanid–linezolid for MDR and XDR M. tuberculosis is one promising step in that direction and has provided a backbone for limitless variations that can be explored. We are hopeful that the preclinical and clinical approaches we described in this Review can lead to the selection and evaluation of drug combinations earlier in the development cascade and greatly accelerate the approval of shorter treatment regimens.
Sociological changes, such as breaking down barriers among collections of academia, non-governmental organizations, funders and drug sponsors, favour collaborative actions that intensify the discovery of breakthrough drug candidates28. However, truly accelerating drug development will require changes at all levels, from sustainable funding of basic and applied research through modernized clinical trials. Additionally, ultimately, we might end up with more personalized therapy, which would entail broad changes in how we deliver care in most of the world. For now, big challenges remain but change has already come and there is cause for optimism with a healthy dose of realism.
References
Dorman, S. E. et al. Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N. Engl. J. Med. 384, 1705–1718 (2021).
Conradie, F. et al. Treatment of highly drug-resistant pulmonary tuberculosis. N. Engl. J. Med. 382, 893–902 (2020).
Young, C., Walzl, G. & Du Plessis, N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol. 13, 190–204 (2020).
Perveen, S., Kumari, D., Singh, K. & Sharma, R. Tuberculosis drug discovery: progression and future interventions in the wake of emerging resistance. Eur. J. Med. Chem. 229, 114066 (2022).
World Health Organization. Global tuberculosis report 2020 (WHO, 2020).
World Health Organization. Global tuberculosis report 2021 (WHO, 2021).
Marais, B. J., Hesseling, A. C. & Cotton, M. F. Poverty and tuberculosis: is it truly a simple inverse linear correlation? Eur. Respir. J. 33, 943–944 (2009).
Antonio-Arques, V., Franch-Nadal, J. & Cayla, J. A. Diabetes and tuberculosis: a syndemic complicated by COVID-19. Med. Clin. 157, 288–293 (2021).
Wilkinson, R. J. Tuberculosis and type 2 diabetes mellitus: an inflammatory danger signal in the time of coronavirus disease 2019. Clin. Infect. Dis. 72, 79–81 (2021).
Pai, M., Kasaeva, T. & Swaminathan, S. Covid-19’s devastating effect on tuberculosis care—a path to recovery. N. Engl. J. Med. https://doi.org/10.1056/NEJMp2118145 (2022).
Glaziou, P. Predicted impact of the COVID-19 pandemic on global tuberculosis deaths in 2020. Preprint at medRxiv https://doi.org/10.1101/2020.04.28.20079582 (2021).
McQuaid, C. F. et al. The potential impact of COVID-19-related disruption on tuberculosis burden. Eur. Respir. J. 56, 2001718 (2020).
Houben, R. M. & Dodd, P. J. The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med. 13, e1002152 (2016).
Zenner, D., Loutet, M. G., Harris, R., Wilson, S. & Ormerod, L. P. Evaluating 17 years of latent tuberculosis infection screening in north-west England: a retrospective cohort study of reactivation. Eur. Respir. J. 50, 1602505 (2017).
Dale, K. D. et al. Quantifying the rates of late reactivation tuberculosis: a systematic review. Lancet Infect. Dis. 21, e303–e317 (2021).
Menzies, N. A. et al. Progression from latent infection to active disease in dynamic tuberculosis transmission models: a systematic review of the validity of modelling assumptions. Lancet Infect. Dis. 18, e228–e238 (2018).
Behr, M. A., Edelstein, P. H. & Ramakrishnan, L. Is Mycobacterium tuberculosis infection life long? BMJ 367, l5770 (2019).
Uplekar, M. et al. WHO’s new end TB strategy. Lancet 385, 1799–1801 (2015).
Kiazyk, S. & Ball, T. B. Latent tuberculosis infection: an overview. Can. Commun. Dis. Rep. 43, 62–66 (2017).
Pai, M. et al. Tuberculosis. Nat. Rev. Dis. Primers 2, 16076 (2016).
Barry, C. E. 3rd et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845–855 (2009).
Boom, W. H., Schaible, U. E. & Achkar, J. M. The knowns and unknowns of latent Mycobacterium tuberculosis infection. J. Clin. Invest. 131, e136222 (2021).
Drain, P. K. et al. Incipient and subclinical tuberculosis: a clinical review of early stages and progression of infection. Clin. Microbiol. Rev. 31, e00021-18 (2018).
Lin, P. L. & Flynn, J. L. The end of the binary era: revisiting the spectrum of tuberculosis. J. Immunol. 201, 2541–2548 (2018).
Cadena, A. M., Fortune, S. M. & Flynn, J. L. Heterogeneity in tuberculosis. Nat. Rev. Immunol. 17, 691–702 (2017).
Fox, W., Ellard, G. A. & Mitchison, D. A. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946-1986, with relevant subsequent publications. Int. J. Tuberc. Lung Dis. 3(Suppl. 2), S231–S279 (1999).
Lee, M. et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med. 367, 1508–1518 (2012).
Aldridge, B. B. et al. The Tuberculosis Drug Accelerator at year 10: what have we learned? Nat. Med. 27, 1333–1337 (2021).
Glassroth, J., Robins, A. G. & Snider, D. E. Jr Tuberculosis in the 1980s. N. Engl. J. Med. 302, 1441–1450 (1980).
Debre, R., Perdrizet, S., Lotte, A., Naveau, M. & Lert, F. Isoniazid chemoprophylaxis of latent primary tuberculosis: in five trial centres in France from 1959 to 1969. Int. J. Epidemiol. 2, 153–160 (1973).
Menzies, D. et al. Four months of rifampin or nine months of isoniazid for latent tuberculosis in adults. N. Engl. J. Med. 379, 440–453 (2018).
World Health Organization. Latent tuberculosis infection: updated and consolidated guidelines for programmatic management (WHO, 2018).
Sterling, T. R. et al. Guidelines for the treatment of latent tuberculosis infection: recommendations from the National Tuberculosis Controllers Association and CDC, 2020. MMWR Recomm. Rep. 69, 1–11 (2020).
Ai, J. W., Ruan, Q. L., Liu, Q. H. & Zhang, W. H. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg. Microbes Infect. 5, e10 (2016).
Lobue, P. & Menzies, D. Treatment of latent tuberculosis infection: an update. Respirology 15, 603–622 (2010).
Seddon, J. A. et al. Levofloxacin versus placebo for the prevention of tuberculosis disease in child contacts of multidrug-resistant tuberculosis: study protocol for a phase III cluster randomised controlled trial (TB-CHAMP). Trials 19, 693 (2018).
Torre-Cisneros, J. et al. Tuberculosis prophylaxis with levofloxacin in liver transplant patients is associated with a high incidence of tenosynovitis: safety analysis of a multicenter randomized trial. Clin. Infect. Dis. 60, 1642–1649 (2015).
Fox, G. J. et al. Levofloxacin versus placebo for the treatment of latent tuberculosis among contacts of patients with multidrug-resistant tuberculosis (the VQUIN MDR trial): a protocol for a randomised controlled trial. BMJ Open. 10, e033945 (2020).
Shah, M. & Dorman, S. E. Latent tuberculosis infection. N. Engl. J. Med. 385, 2271–2280 (2021).
Aber, V. R. & Nunn, A. J. [Short term chemotherapy of tuberculosis. Factors affecting relapse following short term chemotherapy]. Bull. Int. Union Tuberc. 53, 276–280 (1978).
Savic, R. M. et al. Defining the optimal dose of rifapentine for pulmonary tuberculosis: exposure-response relations from two phase II clinical trials. Clin. Pharmacol. Ther. 102, 321–331 (2017).
Franke, M. F. et al. Culture conversion in patients treated with bedaquiline and/or delamanid. A prospective multicountry study. Am. J. Respir. Crit. Care Med. 203, 111–119 (2021).
Imperial, M. Z. et al. A patient-level pooled analysis of treatment-shortening regimens for drug-susceptible pulmonary tuberculosis. Nat. Med. 24, 1708–1715 (2018).
Lange, C. et al. Management of drug-resistant tuberculosis. Lancet 394, 953–966 (2019).
World Health Organization. WHO consolidated guidelines on drug-resistant tuberculosis treatment (WHO, 2019).
Horsburgh, C. R. Jr, Barry, C. E. 3rd & Lange, C. Treatment of tuberculosis. N. Engl. J. Med. 373, (2149–2160 (2015).
Balaban, N. Q. et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17, 441–448 (2019).
Gold, B. & Nathan, C. Targeting phenotypically tolerant Mycobacterium tuberculosis. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.TBTB2-0031-2016 (2017).
Dhar, N., McKinney, J. & Manina, G. Phenotypic heterogeneity in Mycobacterium tuberculosis. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.TBTB2-0021-2016 (2016).
Aldridge, B. B. et al. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335, 100–104 (2012).
Wakamoto, Y. et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95 (2013).
Manina, G., Griego, A., Singh, L. K., McKinney, J. D. & Dhar, N. Preexisting variation in DNA damage response predicts the fate of single mycobacteria under stress. EMBO J. 38, e101876 (2019).
Chengalroyen, M. D. et al. Detection and quantification of differentially culturable tubercle bacteria in sputum from patients with tuberculosis. Am. J. Respir. Crit. Care Med. 194, 1532–1540 (2016).
Rego, E. H., Audette, R. E. & Rubin, E. J. Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157 (2017).
Kieser, K. J. & Rubin, E. J. How sisters grow apart: mycobacterial growth and division. Nat. Rev. Microbiol. 12, 550–562 (2014).
Harms, A., Maisonneuve, E. & Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 354, aaf4268 (2016).
Slayden, R. A., Dawson, C. C. & Cummings, J. E. Toxin-antitoxin systems and regulatory mechanisms in Mycobacterium tuberculosis. Pathog. Dis. 76, fty039 (2018).
Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 16, 202–213 (2018).
Malherbe, S. T. et al. Persisting positron emission tomography lesion activity and Mycobacterium tuberculosis mRNA after tuberculosis cure. Nat. Med. 22, 1094–1100 (2016).
Sotgiu, G., Centis, R., D’Ambrosio, L. & Migliori, G. B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 5, a017822 (2015).
Barry, C. E. & Mayer-Barber, K. D. Signature required: the transcriptional response to tuberculosis. J. Exp. Med. 218, e20211665 (2021).
Singhania, A., Wilkinson, R. J., Rodrigue, M., Haldar, P. & O’Garra, A. The value of transcriptomics in advancing knowledge of the immune response and diagnosis in tuberculosis. Nat. Immunol. 19, 1159–1168 (2018).
Scriba, T. J. et al. Biomarker-guided tuberculosis preventive therapy (CORTIS): a randomised controlled trial. Lancet Infect. Dis. 21, 354–365 (2021).
Esmail, H., Cobelens, F. & Goletti, D. Transcriptional biomarkers for predicting development of tuberculosis: progress and clinical considerations. Eur. Respir. J. 55, 1901957 (2020).
Comstock, G. W. & Edwards, P. Q. The competing risks of tuberculosis and hepatitis for adult tuberculin reactors. Am. Rev. Respir. Dis. 111, 573–577 (1975).
Mitchison, D. A. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 66, 219–225 (1985).
Ignatius, E. H. & Swindells, S. Are we there yet? Short-course regimens in TB and HIV: from prevention to treatment of latent to XDR TB. Curr. HIV/AIDS Rep. 17, 589–600 (2020).
Alsdurf, H., Hill, P. C., Matteelli, A., Getahun, H. & Menzies, D. The cascade of care in diagnosis and treatment of latent tuberculosis infection: a systematic review and meta-analysis. Lancet Infect. Dis. 16, 1269–1278 (2016).
Nahid, P. et al. Executive summary: official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America clinical practice duidelines: treatment of drug-susceptible tuberculosis. Clin. Infect. Dis. 63, 853–867 (2016).
Romanowski, K. et al. Predicting tuberculosis relapse in patients treated with the standard 6-month regimen: an individual patient data meta-analysis. Thorax 74, 291–297 (2019).
Connolly, L. E., Edelstein, P. H. & Ramakrishnan, L. Why is long-term therapy required to cure tuberculosis? PLoS Med. 4, e120 (2007).
Sarathy, J. P. et al. Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob. Agents Chemother. 62, e02266-17 (2018).
Turapov, O. et al. Phenotypically adapted Mycobacterium tuberculosis populations from sputum are tolerant to first-line drugs. Antimicrob. Agents Chemother. 60, 2476–2483 (2016).
Daniel, J., Maamar, H., Deb, C., Sirakova, T. D. & Kolattukudy, P. E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 7, e1002093 (2011).
Lakshminarayana, S. B. et al. Comprehensive physicochemical, pharmacokinetic and activity profiling of anti-TB agents. J. Antimicrob. Chemother. 70, 857–867 (2015).
Kimmey, J. M. et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569 (2015).
Mishra, B. B. et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat. Microbiol. 2, 17072 (2017).
Dallenga, T. & Schaible, U. E. Neutrophils in tuberculosis — first line of defence or booster of disease and targets for host-directed therapy? Pathog. Dis. 74, ftw012 (2016).
Eum, S. Y. et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137, 122–128 (2010).
Parker, H. A., Forrester, L., Kaldor, C. D., Dickerhof, N. & Hampton, M. B. Antimicrobial activity of neutrophils against mycobacteria. Front. Immunol. 12, 782495 (2021).
Lovewell, R. R., Baer, C. E., Mishra, B. B., Smith, C. M. & Sassetti, C. M. Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol. 14, 229–241 (2021).
Evangelopoulos, D. & McHugh, T. D. Improving the tuberculosis drug development pipeline. Chem. Biol. Drug Des. 86, 951–960 (2015).
Chao, M. C. & Rubin, E. J. Letting sleeping dos lie: does dormancy play a role in tuberculosis? Annu. Rev. Microbiol. 64, 293–311 (2010).
Colangeli, R. et al. Bacterial factors that predict relapse after tuberculosis therapy. N. Engl. J. Med. 379, 823–833 (2018).
Thompson, E. G. et al. Host blood RNA signatures predict the outcome of tuberculosis treatment. Tuberculosis 107, 48–58 (2017).
Heyckendorf, J. et al. Prediction of anti-tuberculosis treatment duration based on a 22-gene transcriptomic model. Eur. Respir. J. 58, 2003492 (2021).
Zong, K., Luo, C., Zhou, H., Jiang, Y. & Li, S. Xpert MTB/RIF assay for the diagnosis of rifampicin resistance in different regions: a meta-analysis. BMC Microbiol. 19, 177 (2019).
Nathavitharana, R. R. et al. Accuracy of line probe assays for the diagnosis of pulmonary and multidrug-resistant tuberculosis: a systematic review and meta-analysis. Eur. Respir. J. 49, 1601075 (2017).
World Health Organization. Catalogue of mutations in Mycobacterium tuberculosis complex and their association with drug resistance (WHO, 2021).
Gabbassov, E., Moreno-Molina, M., Comas, I., Libbrecht, M. & Chindelevitch, L. SplitStrains, a tool to identify and separate mixed Mycobacterium tuberculosis infections from WGS data. Microb. Genom. 7, 000607 (2021).
Cohen, T. et al. Mixed-strain Mycobacterium tuberculosis infections and the implications for tuberculosis treatment and control. Clin. Microbiol. Rev. 25, 708–719 (2012).
Band, V. I. & Weiss, D. S. Heteroresistance: a cause of unexplained antibiotic treatment failure? PLoS Pathog. 15, e1007726 (2019).
Ernst, J. D. Mechanisms of M. tuberculosis immune evasion as challenges to TB vaccine design. Cell Host Microbe 24, 34–42 (2018).
Chai, Q., Wang, L., Liu, C. H. & Ge, B. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol. Immunol. 17, 901–913 (2020).
Prideaux, B. et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med. 21, 1223–1227 (2015).
Sarathy, J. P. & Dartois, V. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin. Microbiol. Rev. 33, e00159-19 (2020).
Mashabela, G. T., de Wet, T. J. & Warner, D. F. Mycobacterium tuberculosis metabolism. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.GPP3-0067-2019 (2019).
Hawn, T. R., Shah, J. A. & Kalman, D. New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics. Immunol. Rev. 264, 344–362 (2015).
Lai, R. P. et al. Transcriptomic characterization of tuberculous sputum reveals a host Warburg effect and microbial cholesterol catabolism. mBio 12, e0176621 (2021).
Larkins-Ford, J. et al. Systematic measurement of combination-drug landscapes to predict in vivo treatment outcomes for tuberculosis. Cell Syst. 12, 1046–1063.e7 (2021).
Bosch, B. et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 184, 4579–4592.e24 (2021).
Rock, J. M. et al. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2, 16274 (2017).
Cokol, M., Kuru, N., Bicak, E., Larkins-Ford, J. & Aldridge, B. B. Efficient measurement and factorization of high-order drug interactions in Mycobacterium tuberculosis. Sci. Adv. 3, e1701881 (2017).
Ma, S. et al. Transcriptomic signatures predict regulators of drug synergy and clinical regimen efficacy against tuberculosis. mBio 10, e02627-19 (2019).
Clemens, D. L. et al. Artificial intelligence enabled parabolic response surface platform identifies ultra-rapid near-universal TB drug treatment regimens comprising approved drugs. PLoS ONE 14, e0215607 (2019).
Lee, B. Y. et al. Ultra-rapid near universal TB drug regimen identified via parabolic response surface platform cures mice of both conventional and high susceptibility. PLoS ONE 13, e0207469 (2018).
Band, V. I. et al. Antibiotic combinations that exploit heteroresistance to multiple drugs effectively control infection. Nat. Microbiol. 4, 1627–1635 (2019).
Peterson, E. J. R., Ma, S., Sherman, D. R. & Baliga, N. S. Network analysis identifies Rv0324 and Rv0880 as regulators of bedaquiline tolerance in Mycobacterium tuberculosis. Nat. Microbiol. 1, 16078 (2016).
Walter, N. D. et al. Mycobacterium tuberculosis precursor rRNA as a measure of treatment-shortening activity of drugs and regimens. Nat. Commun. 12, 2899 (2021).
Gideon, H. P. et al. Multimodal profiling of lung granulomas reveals cellular correlates of tuberculosis control. bioRxiv https://doi.org/10.1101/2020.10.24.352492 (2021). Preprint at.
Dinkele, R. et al. Capture and visualization of live Mycobacterium tuberculosis bacilli from tuberculosis patient bioaerosols. PLoS Pathog. 17, e1009262 (2021).
Berry, M. P. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010).
Wells, G. et al. μCT analysis of the human tuberculous lung reveals remarkable heterogeneity in 3D granuloma morphology. Am. J. Respir. Crit. Care Med. 204, 583–595 (2021).
Chen, R. Y. et al. Radiological and functional evidence of the bronchial spread of tuberculosis: an observational analysis. Lancet Microbe 2, e518–e526 (2021).
Dartois, V. & Dick, T. A ginger root or plum model for the TB “granuloma”? Am. J. Respir. Crit. Care Med. 204, 505–507 (2021).
Hunter, R. L. The pathogenesis of tuberculosis — the Koch phenomenon reinstated. Pathogens 9, 813 (2020).
Libardo, J., Boshoff, H. I. & Barry, C. E. 3rd The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 42, 81–94 (2018).
Bartelink, I. H. et al. New paradigm for translational modeling to predict long-term tuberculosis treatment response. Clin. Transl. Sci. 10, 366–379 (2017).
Pienaar, E. et al. A computational tool integrating host immunity with antibiotic dynamics to study tuberculosis treatment. J. Theor. Biol. 367, 166–179 (2015).
Ernest, J. et al. Development of new tuberculosis drugs: translation to regimen composition for drug-sensitive and multidrug-resistant tuberculosis. Annu. Rev. Pharmacol. Toxicol. 61, 495–516 (2020).
Mudde, S. E. et al. Predictive modeling to study the treatment-shortening potential of novel tuberculosis drug regimens, towards bundling of preclinical data. J. Infect. Dis. https://doi.org/10.1093/infdis/jiab101 (2021).
Dooley, K. E., Hanna, D., Mave, V., Eisenach, K. & Savic, R. M. Advancing the development of new tuberculosis treatment regimens: the essential role of translational and clinical pharmacology and microbiology. PLoS Med. 16, e1002842 (2019).
Lenaerts, A., Barry, C. E. 3rd & Dartois, V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol. Rev. 264, 288–307 (2015).
Nuermberger, E. L. Preclinical efficacy testing of new drug candidates. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.TBTB2-0034-2017 (2017).
Via, L. E. et al. A sterilizing tuberculosis treatment regimen is associated with faster clearance of bacteria in cavitary lesions in marmosets. Antimicrob. Agents Chemother. 59, 4181–4189 (2015).
White, A. G. et al. Analysis of 18FDG PET/CT imaging as a tool for studying Mycobacterium tuberculosis infection and treatment in non-human primates. J. Vis. Exp. 127, 56375 (2017).
Xie, Y. L. et al. Fourteen-day PET/CT imaging to monitor drug combination activity in treated individuals with tuberculosis. Sci. Transl Med. 13, eabd7618 (2021).
Lienhardt, C. et al. Advances in clinical trial design: weaving tomorrow’s TB treatments. PLoS Med. 17, e1003059 (2020).
Davies, G., Boeree, M., Hermann, D. & Hoelscher, M. Accelerating the transition of new tuberculosis drug combinations from phase II to phase III trials: new technologies and innovative designs. PLoS Med. 16, e1002851 (2019).
Strydom, N. et al. Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: a mechanistic model and tool for regimen and dose optimization. PLoS Med. 16, e1002773 (2019).
Lewis, K. & Shan, Y. Why tolerance invites resistance. Science 355, 796 (2017).
Schrader, S. M., Vaubourgeix, J. & Nathan, C. Biology of antimicrobial resistance and approaches to combat it. Sci. Transl Med. 12, eaaz6992 (2020).
Hicks, N. D. et al. Clinically prevalent mutations in Mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat. Microbiol. 3, 1032–1042 (2018).
Zhu, J. H. et al. Rifampicin can induce antibiotic tolerance in mycobacteria via paradoxical changes in rpoB transcription. Nat. Commun. 9, 4218 (2018).
Bellerose, M. M. et al. Common variants in the glycerol kinase gene reduce tuberculosis drug efficacy. mBio 10, e00663-19 (2019).
Safi, H. et al. Phase variation in Mycobacterium tuberculosis glpK produces transiently heritable drug tolerance. Proc. Natl Acad. Sci. USA 116, 19665–19674 (2019).
Lee, J. J. et al. Transient drug-tolerance and permanent drug-resistance rely on the trehalose-catalytic shift in Mycobacterium tuberculosis. Nat. Commun. 10, 2928 (2019).
Lim, J. et al. Phosphoenolpyruvate depletion mediates both growth arrest and drug tolerance of Mycobacterium tuberculosis in hypoxia. Proc. Natl Acad. Sci. USA 118, e2105800118 (2021).
Baek, S. H., Li, A. H. & Sassetti, C. M. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 9, e1001065 (2011).
Martini, M. C. et al. Loss of RNase J leads to multi-drug tolerance and accumulation of highly structured mRNA fragments in Mycobacterium tuberculosis. Preprint at bioRxiv https://doi.org/10.1101/2022.02.13.480260 (2022).
Zheng, H. et al. Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nat. Chem. Biol. 13, 218–225 (2017).
Chaudhuri, S. et al. Kasugamycin potentiates rifampicin and limits emergence of resistance in Mycobacterium tuberculosis by specifically decreasing mycobacterial mistranslation. eLife 7, e36782 (2018).
Liu, Y. et al. Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J. Exp. Med. 213, 809–825 (2016).
Prosser, G. et al. The bacillary and macrophage response to hypoxia in tuberculosis and the consequences for T cell antigen recognition. Microbes Infect. 19, 177–192 (2017).
Tan, S., Sukumar, N., Abramovitch, R. B., Parish, T. & Russell, D. G. Mycobacterium tuberculosis responds to chloride and pH as synergistic cues to the immune status of its host cell. PLoS Pathog. 9, e1003282 (2013).
Datta, M. et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc. Natl Acad. Sci. USA 112, 1827–1832 (2015).
Boldrin, F., Provvedi, R., Cioetto Mazzabo, L., Segafreddo, G. & Manganelli, R. Tolerance and persistence to drugs: a main challenge in the fight against Mycobacterium tuberculosis. Front. Microbiol. 11, 1924 (2020).
Gessner, S. et al. The mycobacterial mutasome: composition and recruitment in live cells. bioRxiv https://doi.org/10.1101/2021.11.16.468908 (2021). Preprint at.
Ragheb, M. N. et al. Inhibiting the evolution of antibiotic resistance. Mol. Cell 73, 157–165.e5 (2019).
Awasthi, D. & Freundlich, J. S. Antimycobacterial metabolism: illuminating Mycobacterium tuberculosis biology and drug discovery. Trends Microbiol. 25, 756–767 (2017).
Varaksa, T. et al. Metabolic fate of human immunoactive sterols in Mycobacterium tuberculosis. J. Mol. Biol. 433, 166763 (2021).
Remm, S., Earp, J. C., Dick, T., Dartois, V. & Seeger, M. A. Critical discussion on drug efflux in Mycobacterium tuberculosis. FEMS Microbiol. Rev. 46, fuab050 (2022).
Lee, R. E. et al. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat. Med. 20, 152–158 (2014).
Laws, M., Jin, P. & Rahman, K. M. Efflux pumps in Mycobacterium tuberculosis and their inhibition to tackle antimicrobial resistance. Trends Microbiol. 30, 57–68 (2022).
World Health Organization. The end TB strategy (WHO, 2015).
Franzblau, S. G. et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 92, 453–488 (2012).
Zhang, M. et al. Streptomycin-starved Mycobacterium tuberculosis 18b, a drug discovery tool for latent tuberculosis. Antimicrob. Agents Chemother. 56, 5782–5789 (2012).
Mitchison, D. A. & Coates, A. R. Predictive in vitro models of the sterilizing activity of anti-tuberculosis drugs. Curr. Pharm. Des. 10, 3285–3295 (2004).
Bassett, I. M. et al. Detection of inhibitors of phenotypically drug-tolerant Mycobacterium tuberculosis using an in vitro bactericidal screen. J. Microbiol. 51, 651–658 (2013).
Darby, C. M. et al. Whole cell screen for inhibitors of pH homeostasis in Mycobacterium tuberculosis. PLoS ONE 8, e68942 (2013).
Early, J. et al. Identification of compounds with pH-dependent bactericidal activity against Mycobacterium tuberculosis. ACS Infect. Dis. 5, 272–280 (2019).
Gouzy, A., Healy, C., Black, K. A., Rhee, K. Y. & Ehrt, S. Growth of Mycobacterium tuberculosis at acidic pH depends on lipid assimilation and is accompanied by reduced GAPDH activity. Proc. Natl Acad. Sci. USA 118, e2024571118 (2021).
Grant, S. S. et al. Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem. Biol. 8, 2224–2234 (2013).
Dahl, J. L. et al. The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc. Natl Acad. Sci. USA 100, 10026–10031 (2003).
Betts, J. C., Lukey, P. T., Robb, L. C., McAdam, R. A. & Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731 (2002).
Loebel, R. O., Shorr, E. & Richardson, H. B. The influence of foodstuffs upon the respiratory metabolism and growth of human tubercle bacilli. J. Bacteriol. 26, 139–166 (1933).
Cho, S. H. et al. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 51, 1380–1385 (2007).
Wayne, L. G. & Hayes, L. G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64, 2062–2069 (1996).
Boon, C. & Dick, T. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J. Bacteriol. 184, 6760–6767 (2002).
Via, L. E. et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and non-human primates. Infect. Immun. 76, 2333–2340 (2008).
Lovewell, R. R., Sassetti, C. M. & VanderVen, B. C. Chewing the fat: lipid metabolism and homeostasis during M. tuberculosis infection. Curr. Opin. Microbiol. 29, 30–36 (2016).
Pandey, A. K. & Sassetti, C. M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl Acad. Sci. USA 105, 4376–4380 (2008).
Garton, N. J. et al. Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 5, e75 (2008).
Kim, M. J. et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2, 258–274 (2010).
Gold, B., Warrier, T. & Nathan, C. A multistress model for high throughput screening against nonreplicating Mycobacterium tuberculosis. Methods Mol. Biol. 2314, 611–635 (2021).
Christophe, T., Ewann, F., Jeon, H. K., Cechetto, J. & Brodin, P. High-content imaging of Mycobacterium tuberculosis-infected macrophages: an in vitro model for tuberculosis drug discovery. Future Med. Chem. 2, 1283–1293 (2010).
Song, O. R. et al. Phenotypic assays for Mycobacterium tuberculosis infection. Cytometry A 91, 983–994 (2017).
Chakraborty, P., Bajeli, S., Kaushal, D., Radotra, B. D. & Kumar, A. Biofilm formation in the lung contributes to virulence and drug tolerance of Mycobacterium tuberculosis. Nat. Commun. 12, 1606 (2021).
Ackart, D. F. et al. Expression of antimicrobial drug tolerance by attached communities of Mycobacterium tuberculosis. Pathog. Dis. 70, 359–369 (2014).
Ojha, A. K. et al. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69, 164–174 (2008).
Mukamolova, G. V., Turapov, O., Malkin, J., Woltmann, G. & Barer, M. R. Resuscitation-promoting factors reveal an occult population of tubercle bacilli in sputum. Am. J. Respir. Crit. Care Med. 181, 174–180 (2010).
Lipworth, S. et al. Defining dormancy in mycobacterial disease. Tuberculosis 99, 131–142 (2016).
Shetye, G. S., Franzblau, S. G. & Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 220, 68–97 (2020).
Singh, R. et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395 (2008).
Robertson, G. T. et al. Comparative analysis of pharmacodynamics in the C3HeB/FeJ mouse tuberculosis model for DprE1 inhibitors TBA-7371, PBTZ169, and OPC-167832. Antimicrob. Agents Chemother. 65, e0058321 (2021).
Tenero, D. et al. First-time-in-human study and prediction of early bactericidal activity for GSK3036656, a potent leucyl-tRNA synthetase inhibitor for tuberculosis treatment. Antimicrob. Agents Chemother. 63, e00240-19 (2019).
Zumla, A., Nahid, P. & Cole, S. T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 12, 388–404 (2013).
Rawson, T. M. et al. Optimizing antimicrobial use: challenges, advances and opportunities. Nat. Rev. Microbiol. 19, 747–758 (2021).
Roemhild, R., Bollenbach, T. & Andersson, D. I. The physiology and genetics of bacterial responses to antibiotic combinations. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-022-00700-5 (2022).
Tyers, M. & Wright, G. D. Drug combinations: a strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 17, 141–155 (2019).
Robertson, B. D. et al. Detection and treatment of subclinical tuberculosis. Tuberculosis 92, 447–452 (2012).
Vernon, A. & Bishai, W. Modeling treatment of latent tuberculosis: shortening the leap of faith? Am. J. Respir. Crit. Care Med. 201, 405–406 (2020).
Bucsan, A. N., Mehra, S., Khader, S. A. & Kaushal, D. The current state of animal models and genomic approaches towards identifying and validating molecular determinants of Mycobacterium tuberculosis infection and tuberculosis disease. Pathog. Dis. 77, ftz037 (2019).
Oh, C. E. & Menzies, D. Four months of rifampicin monotherapy for latent tuberculosis infection in children. Clin. Exp. Pediatr. https://doi.org/10.3345/cep.2021.01186 (2021).
Fox, G. J. et al. Preventing the spread of multidrug-resistant tuberculosis and protecting contacts of infectious cases. Clin. Microbiol. Infect. 23, 147–153 (2017).
Acknowledgements
The authors thank B. B. Aldridge and T. Dick for stimulating discussions. Support for work in the authors’ laboratories was provided by grants INV-009173 and INV-004704 from the Bill and Melinda Gates Foundation and grant U19AI142735 from the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
V. A. D. researched data for the article. Both authors contributed substantially to discussion of the content, wrote the article, and edited and reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Microbiology thanks David Sherman, Mel Spigelman and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov: https://clinicaltrials.gov
The World Bank database: https://data.worldbank.org/indicator/SH.TBS.INCD?end=2020&locations=ZA-RU-CN&start=2000&view=chart
WHO tuberculosis: https://www.who.int/news-room/fact-sheets/detail/tuberculosis
Working Group on New TB Drugs: https://www.newtbdrugs.org/pipeline/clinical
Supplementary information
Glossary
- Active TB
-
Disease that occurs in individuals infected with Mycobacterium tuberculosis and present symptoms that include cough, weight loss, night sweats and fever.
- Cavitary disease
-
Active disease characterized by the presence of open pulmonary cavities in the lung parenchyma, composed of a wall filled with necrotic debris and high bacterial burden, leading to poor treatment outcome and increased person-to-person transmission.
- Drug tolerance
-
Increased survival of bacterial cells in the presence of drug, generally not driven by genetic mutations but due to physiological or metabolic adaptations that lead to decreased growth rate upon encountering specific environmental conditions.
- Persister subpopulation
-
The subpopulation of bacteria that survives the bactericidal action of antibiotics but is genetically identical to susceptible bacteria and appears to be non-replicating or slowly growing in response to various stresses.
- Minimum inhibitory concentration
-
(MIC). The lowest drug concentration that inhibits growth.
- Resistance breakpoints
-
The minimum inhibitory concentration (MIC) above which an antimicrobial agent is considered to have a low probability of treatment success in the clinic.
- Synergistic drug interactions
-
Interactions that occur when the combined effect of two drugs is greater than the sum of each drug’s individual activity.
- Subclinical tuberculosis
-
Mycobacterium tuberculosis infection presenting with mild or no symptoms, where low bacterial burden persists in a quiescent state, can be detected by conventional immune-based assays and can be intermittently culture-positive but sputum smear-negative due to the low bacillary load.
- Mutasome
-
The protein components that assemble at a DNA lesion in the presence of a stalled replication complex to mediate translesion DNA synthesis in a mutagenic manner.
Rights and permissions
About this article

Cite this article
Dartois, V.A., Rubin, E.J. Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nat Rev Microbiol 20, 685–701 (2022). https://doi.org/10.1038/s41579-022-00731-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-022-00731-y
