
Abstract
SARS-CoV-2 infection is benign in most individuals but, in around 10% of cases, it triggers hypoxaemic COVID-19 pneumonia, which leads to critical illness in around 3% of cases. The ensuing risk of death (approximately 1% across age and gender) doubles every five years from childhood onwards and is around 1.5 times greater in men than in women. Here we review the molecular and cellular determinants of critical COVID-19 pneumonia. Inborn errors of type I interferons (IFNs), including autosomal TLR3 and X-chromosome-linked TLR7 deficiencies, are found in around 1–5% of patients with critical pneumonia under 60 years old, and a lower proportion in older patients. Pre-existing auto-antibodies neutralizing IFNα, IFNβ and/or IFNω, which are more common in men than in women, are found in approximately 15–20% of patients with critical pneumonia over 70 years old, and a lower proportion in younger patients. Thus, at least 15% of cases of critical COVID-19 pneumonia can be explained. The TLR3- and TLR7-dependent production of type I IFNs by respiratory epithelial cells and plasmacytoid dendritic cells, respectively, is essential for host defence against SARS-CoV-2. In ways that can depend on age and sex, insufficient type I IFN immunity in the respiratory tract during the first few days of infection may account for the spread of the virus, leading to pulmonary and systemic inflammation.
Similar content being viewed by others


Main
More than 5 million people have died from COVID-19, and infection fatality rates in unvaccinated populations are around 1% (refs. 1,2). Indeed, infection with SARS-CoV-2 is silent in around 40% of cases, underlies a benign upper respiratory tract disease in another 40% and causes pneumonia in approximately 20% of cases3,4. Non-hypoxaemic, moderate pneumonia is seen in around 10% of cases, whereas the remaining 10% of cases present hypoxaemic pneumonia, typically requiring hospitalization for oxygen therapy. In about 3% of cases, the administration of O2 at a rate of <6 l min−1 (the cut-off for severe pneumonia) is not sufficient to alleviate hypoxaemia. In such cases, high-flow oxygen (O2 > 6 l min−1), mechanical ventilation (non-invasive or by intubation) or extracorporeal membrane oxygenation is required (any of these three options, typically provided in intensive care units, defines critical pneumonia)5,6. The infection fatality rate increases exponentially with age, doubling every five years, from 0.001% in individuals aged 5–9 years to 8.29% in those over the age of 80 years1,7,8,9,10. Ancestry, social status and several comorbid conditions have been associated with higher disease severity and death rates, but with modest odds ratios (typically <1.5, rarely >2)7,8,9. Men have a 1.5 times greater risk of death than women, after adjustment for other risk factors1,11. Overall, the notable epidemiological feature of life-threatening COVID-19 is its strong dependence on age, steadily increasing throughout life, with a 10,000 times greater risk at ages >80 years compared with in the first decade of life1,12,13. A similar pattern is observed with the more transmissible viral variants14,15. The same viruses are found in patients with silent and lethal infections, excluding the hypothesis that interindividual clinical variability is primarily a consequence of viral diversity.
The hypothesis that a large amount of viral inoculum is more life-threatening than a small inoculum is more plausible, consistent with the findings of 100 years of experimental inoculations of animals with pathogens16. However, it is difficult to test this hypothesis in humans. One alternative hypothesis is that humans with life-threatening COVID-19 were particularly prone to critical illness due to an underlying and hitherto silent immunodeficiency17,18. The traditional view of immunodeficiency, characterized by overt immunological abnormalities and broad vulnerability to infectious agents—as shown in patients with acquired immunodeficiency syndrome or severe combined immunodeficiency, who lack T cells owing to HIV infection and germline mutations, respectively—has turned out to be the tip of an iceberg. Since 1996, previously healthy patients with rare or common infectious diseases but normal resistance to other infectious agents have been found to carry inborn errors of immunity (IEIs), rendering them particularly susceptible to specific microorganisms. Rare IEIs have been implicated in at least 20 different types of viral, bacterial, fungal and parasitic infections17,18. These rare IEIs led to the discovery of a common IEI, accounting for about 1% of cases of tuberculosis in populations of European descent19,20. On the basis of all of these findings, we launched the COVID Human Genetic Effort (www.covidhge.com) with the aim of discovering the molecular, cellular and immunological determinants of the various SARS-CoV-2-related disease manifestations by searching for causal IEIs13. Here we review these and other studies that have clarified the human genetic and immunological determinants of life-threatening COVID-19 pneumonia12,13,21,22,23,24. We do not consider other phenotypes, such as resistance to infection25, pernio (COVID toes)26, multisystem inflammatory syndrome in children or adults27, neuro-COVID28 or long COVID10,29, for which genetic and immunological studies have only just begun.
Inborn errors underlying critical influenza
The first breakthrough emerged from a study of candidate inborn errors of TLR3-, IRF7- and IRF9-dependent type I IFN immunity that had previously been shown to underlie life-threatening influenza pneumonia5,17,18,24,30,31,32 (Fig. 1). Predispositions to critical COVID-19 and influenza were hypothesized to be allelic because both conditions are respiratory infections caused by RNA viruses12. The first influenza-susceptibility gene discovered encodes IRF7, the inducible transcription factor that is responsible for amplifying type I and III IFN production in virus-infected cells33. Plasmacytoid dendritic cells (pDCs) constitutively express high levels of IRF7 and are the most potent producers of type I IFN34,35. The second encodes IRF9, the DNA-binding component of the interferon-stimulated gene factor 3 (ISGF-3) complex that is activated by type I and III IFNs36. The third encodes TLR3, an endosomal double-stranded RNA sensor that regulates basal levels of type I IFN in various non-haematopoietic cells37, possibly including respiratory epithelial cells (RECs)24,32. Germline mutations at these three human loci are causal for critical influenza pneumonia30,31,32. We also considered ten other genes, including IFNAR1 and IFNAR2, the products of which are biochemically and immunologically connected to these three core genes (Fig. 1), and for which deleterious genotypes have been shown to underlie other severe viral diseases (suggesting incomplete penetrance for influenza)5. These 13 loci encode proteins of which a genetic deficiency can be considered to confer a high risk of critical influenza.

There are 17 human type I IFNs (red), each encoded by a specific, intronless gene: 13 subtypes of IFNα, IFNβ, IFNε, IFNκ and IFNω, and three human type III IFNs (IFNλ1–3). Auto-antibodies to IFNα, IFNβ and/or IFNω have been identified in about 15% of patients with critical COVID-19 pneumonia. Monogenic inborn errors of TLR3- and/or TLR7-dependent type I IFN immunity have been identified in about 1–5% of patients with critical COVID-19 pneumonia (genes shown in red). SARS-CoV-2 infection can induce type I IFN production in a TLR3-dependent manner in tissue-resident RECs (which express TLR3 but not TLR7) and in a TLR7-dependent manner in circulating plasmacytoid dendritic cells (pDCs, which express TLR7 but not TLR3)200. IRF7 is constitutively expressed in pDCs, at higher levels than in other cell types, whereas it is mostly induced by viral infection in RECs200. IRF7 activation is required to produce type I IFNs other than IFNβ33.
Autosomal inborn errors of type I IFNs
Biochemically deleterious germline mutations of 8 of the 13 genes were found in 23 out of 659 patients with critical COVID-19 (3.5%) aged 17 to 77 years, including 18 patients under 60 years old (3.8%). Notably, four unrelated previously healthy adults, aged 26 to 50 years, had autosomal recessive complete IRF7 or IFNAR1 deficiency. The other patients had known (n = 11) or previously unreported (n = 8) autosomal dominant partial deficiencies. None of these patients had ever been hospitalized for other viral infections, including influenza. The penetrance of these disorders for critical COVID-19 is also probably incomplete, but higher for the autosomal recessive than for the autosomal dominant disorders, and for the known than for the unreported autosomal dominant disorders (Table 1). A 13-year-old boy with autosomal recessive IFNAR1 deficiency38,39 a 3-year old girl with autosomal recessive IFNAR1 deficiency40 and a 3-year old girl with autosomal recessive TBK1 deficiency41 were independently reported to have critical COVID-19. Fibroblasts presenting autosomal dominant or recessive TLR3 deficiency, autosomal recessive IRF7 deficiency or autosomal recessive IFNAR1 deficiency displayed defective type-I-IFN-dependent control of SARS-CoV-2 in vitro5, suggesting that RECs may display the same phenotype32. Moreover, pDCs from an IRF7-deficient patient were unable to induce type I IFNs after stimulation with SARS-CoV-2 in vitro. This experimental approach provided a proof of concept that IEIs that affect type I IFNs—including disorders of TLR3-dependent type I IFN immunity in RECs, and even autosomal recessive defects that blunt type I IFN immunity across cell types—can underlie life-threatening COVID-19 pneumonia in previously healthy patients12,21 (Fig. 1).
X-linked recessive TLR7 deficiency
In parallel, an X-chromosome-wide approach resulted in the discovery of X-linked recessive TLR7 deficiency, a previously unknown IEI42. In a cohort of 1,202 unrelated male patients with critical pneumonia, 17 patients (1.4%) from 16 kindreds were hemizygous for biochemically deleterious TLR7 variants, whereas none of the 331 men with asymptomatic or mild COVID-19 carried such mutations42. Sixteen of the seventeen patients are below the age of 60 years (1.8%). One of these patients also had ataxia–telangiectasia (AT), which was not causal for critical COVID-19 in other patients with AT infected with SARS-CoV-2 (ref. 43). TLR7 deficiency was also found in 1% of patients with severe, but not critical COVID-19 (that is, with O2 < 6 l min−1). The penetrance of X-linked recessive TLR7 deficiency for severe or critical COVID-19 among relatives of index cases was high, but incomplete, especially in children (Table 1). We also found that the cumulative minor allele frequency (MAF) of deleterious alleles in men was <6.5 × 10−4. Moreover, 6 out of the 11 TLR7 variants previously reported in other patients were deleterious (carried by 9 out of 16 patients)44,45,46,47, whereas the variants in another study were not disclosed48. We further showed that the TLR7 genotype was deleterious in patients’ Epstein–Barr-virus-transformed B cell lines. Overall, these genetic and biochemical data implicated X-linked recessive TLR7 deficiency due to deleterious variants in at least 1% of critical cases of COVID-19 in male patients under the age of 60 years, with high penetrance.
Deficiency of plasmacytoid dendritic cells
TLR7-deficient pDCs did not respond to the TLR7-specific agonists tested. Moreover, when challenged with SARS-CoV-2 in vitro, they displayed severely impaired, but not entirely absent type I IFN induction42. TLR9 is probably responsible for the residual response, as UNC-93B- and IRAK4-deficient pDCs do not respond at all to the virus49 (Fig. 1). The discovery of X-linked recessive TLR7 deficiency through an unbiased approach therefore confirmed the key role of type I IFN immunity in protecting against SARS-CoV-2 in the respiratory tract42. It also suggested that pDCs are essential for this process. It has long been known that pDCs are the most potent discernible type-I-IFN-producing cell type34,50,51,52; this experiment of nature suggests that these cells are essential for antiviral immunity, as the other TLR7-expressing myeloid and lymphoid cells are poor producers of type I IFNs53. Human TLR7 is now firmly established as having an important role in host defence. The activation of TLR7 by viral RNA was long known54,55,56,57,58, with its gene shown to be subject to strong negative selection in the general population59, but its role in host defence had remained unclear, as patients with deficiencies of MYD88 or IRAK4 displayed no severe viral illnesses and the viral infections observed in UNC-93B-deficient patients had been attributed to their TLR3 pathway defects60. Overall, TLR3-dependent type I immunity in RECs and TLR7-dependent type I IFN immunity in pDCs seem to be strong determinants of protection against SARS-CoV-2 in the respiratory tract.
Other inborn errors of type I IFN immunity
Nine IEIs of type I IFN immunity were therefore found to underlie life-threatening COVID-19 with low (autosomal dominant disorders) or high (autosomal recessive, X-linked recessive) penetrance. Moreover, five young patients with related IEIs—MYD88 (ref. 61) IRAK4 (ref. 62) and GATA2 deficiencies63,64—were hospitalized for COVID-19 pneumonia, albeit of moderate severity. Severe influenza infections had been reported in patients with GATA2 deficiency, probably caused at least in part by low counts of circulating pDCs65, which do not require TLR7 to sense influenza virus30,49. Other patients with MYD88, IRAK4 or GATA2 deficiency are probably susceptible to hypoxaemic COVID-19 pneumonia49. Defects of other genes involved in type I IFN immunity may also increase susceptibility to COVID-19 (Fig. 1). Overall, the nine IEIs of type I IFN immunity identified may already account for about 1–5% of life-threatening cases of COVID-19, especially among patients under 60 years old, with X-linked recessive TLR7 deficiency alone accounting for over 1% of critical cases in men. This proportion is high, exceeding the 1% of cases of tuberculosis in Europeans for which a genetic explanation has been obtained19,20. Other causal IEIs affecting type I IFN will probably be discovered in the future. Indeed, autosomal recessive IFNAR1 and IRF7 deficiencies have already acted like a compass, pointing us in the right direction for the discovery of a more common cause of life-threatening COVID-19.
From inborn errors to their phenocopy
Auto-antibodies against type I IFNs were first detected in the 1980s, in patients treated with type I IFN or with systemic lupus erythematosus66,67,68. Their production can be genetically driven, as in patients with autoimmune polyendocrine syndrome type-1 (APS-1) due to germline mutations of AIRE, which controls the thymic expression of peripheral self-antigens and, therefore, central T cell tolerance69,70,71. They are also found in men with immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) due to mutations in FOXP3, which encodes a protein that governs the development of regulatory T cells and therefore peripheral T cell tolerance72,73, and in patients with combined T or B cell immunodeficiency due to hypomorphic mutations of RAG1 or RAG2 (ref. 74). Auto-antibodies against type I IFN may also be produced in two overlapping conditions75 of unclear aetiology—thymoma76 and myasthenia gravis77,78. Patients with APS-1 and thymoma have thymic epithelial-intrinsic defects, whereas patients with RAG1, RAG2 and FOXP3 mutations have T-cell-intrinsic defects71,79,80. These auto-antibodies have been widely recognized for 40 years, and were even reported in otherwise healthy patients with severe varicella zoster virus infection by Ion Gresser81 as early as 1984, but they were not thought to confer a predisposition to viral diseases. By contrast, autoimmune phenocopies of IEIs disrupting type II IFN (IFNγ), IL-6 and IL-17A/F have long been known to underlie mycobacterial disease, staphylococcal disease, and mucocutaneous candidiasis, respectively18,82,83,84,85,86,87,88.
Auto-antibodies neutralizing type I IFNs
We found that at least 10% of individuals with critical COVID-19 had auto-antibodies neutralizing supraphysiological concentrations (10 ng ml−1, in plasma diluted 1/10) of IFNα2 and/or IFNω6. These findings were widely replicated89,90,91,92,93,94,95,96,97,98,99,100,101,102. In our study and another, these auto-antibodies were not found in patients with silent or benign SARS-CoV-2 infections6,92. Alarmingly, auto-antibodies neutralizing type I IFN were found in therapeutic convalescent plasma from a few patients hospitalized for COVID-19 (ref. 99). In the few patients tested, the auto-antibodies existed before the SARS-CoV-2 infection. Moreover, APS-1 patients—who produce such auto-antibodies from early childhood—were at very high risk of developing severe or critical COVID-19 pneumonia, especially in patients over 20 years old103,104. An elegant unbiased study reported that a number of patients with hypoxaemic COVID-19 pneumonia displayed diverse auto-antibodies92, most of which were probably triggered by SARS-CoV-2 infection and may have influenced the course of disease. This and a longitudinal study of a small group of patients suggested that SARS-CoV-2 infection might boost the levels of pre-existing type I IFN auto-antibodies105. The auto-antibodies blocked the protective effect of IFNα2 against SARS-CoV-2 in vitro6. Furthermore, circulating IFNα concentrations were low or undetectable in vivo in patients with auto-antibodies against IFNα2, which also target the 13 forms of IFNα6. These auto-antibodies also impair type I IFN activity in peripheral blood mononuclear cells93. Impaired expression of IFN-stimulated genes (ISGs) was also observed in the respiratory tract in patients with auto-antibodies96,106 (Fig. 2). Indeed, these auto-antibodies were also detected in tracheal aspirates and nasal swabs106,107.

In a two-step model of pathogenesis of critical COVID-19 (ref. 12), inadequate type I IFN immunity during the first few hours and days of infection results in the spread of the virus to the lungs, blood and beyond. This results, one to two weeks later, in pulmonary and systemic hyperinflammation, largely due to the recruitment and activation of leukocytes, which produce excessive amounts of cytokines in a last-ditch attempt to eradicate the virus that should have been eradicated by type I IFN but was not. The two-step model suggests that early administration of type I IFN at the onset of SARS-CoV-2 infection, in ambulatory patients, or even before infection in exposed individuals at risk of severe disease, may halt disease progression in patients without auto-antibodies to the corresponding type I IFN and without IEIs downstream from type I IFN receptors.
Neutralization of lower concentrations
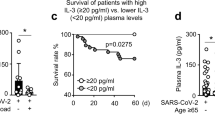
The physiological concentrations of IFNα in the blood during SARS-CoV-2 infection are much lower (between 1 and 100 pg ml−1 in undiluted plasma)108 compared with the concentrations used in our initial experiments (10 ng ml−1 in plasma diluted 1/10). We found that around 14% of patients with critical COVID-19 pneumonia had auto-antibodies neutralizing lower, more physiological, concentrations of IFNα and/or IFNω (100 pg ml−1 in plasma diluted 1/10)109. The proportion of such patients increased after the age of 65 years and was greater in men than in women. Moreover, another 1% of patients had auto-antibodies neutralizing 10 ng ml−1 IFNβ only. Globally, around 20% of patients with critical COVID-19 over 80 years of age, and about 20% of deceased patients across all ages, had these auto-antibodies. Moreover, approximately 7% of patients with severe, but not critical, COVID-19 also had these auto-antibodies. We estimated ORs by comparing the prevalence of auto-antibodies in patients with critical disease with the prevalence in patients with asymptomatic or mild infection109 (Table 1). For most categories of auto-antibodies against type I IFN, their prevalence was not null in patients with silent or mild infection, as previously documented for patients with APS-1 (ref. 103,104). The highest ORs were obtained for auto-antibodies neutralizing both IFNα and IFNω at concentrations of 10 ng ml−1 or 100 pg ml−1, followed by auto-antibodies against IFNα only, whereas the ORs for auto-antibodies against IFNω only were lower. For auto-antibodies against IFNβ only, the ORs for critical disease were even lower. However, auto-antibodies neutralizing only IFNβ can underlie life-threatening COVID-19, as can auto-antibodies against IFNα only or IFNω only6,109.
Auto-antibodies in the general population
We tested more than 34,000 individuals from the general population aged 18 to 100 years. We found that the prevalence of auto-antibodies neutralizing 10 ng ml−1 (or 100 pg ml−1) IFNα or IFNω was not only higher in men than in women, but also increased significantly with age in the general population, with 0.17% (1.1%) of individuals positive for these antibodies before the age of 70 years, and more than 1.4% (4.4%) positive after the age of 70 years109. This notable distribution probably contributes to the higher risk of death from COVID-19 in the ageing population. Interestingly, auto-antibodies neutralizing IFNα and/or IFNω are much more prevalent in the ageing population, whereas auto-antibodies neutralizing IFNβ seem to have a similar prevalence in all of the age groups tested. IFNω and the 13 forms of IFNα are very similar biochemically, closely related phylogenetically and found in the blood, whereas IFNβ, IFNε and IFNκ differ structurally and functionally. IFNβ is widely required to initiate the production of other type I IFNs, whereas IFNε and IFNκ are predominantly expressed in reproductive and cutaneous tissues (and not tested in our studies of auto-antibodies)110,111,112. Defective activity for all 13 forms of IFNα or IFNω or IFNβ, or a combination of these molecules may remain silent for long periods until a virus, such as SARS-CoV-2, reveals the deficiency112,113,114. Overall, auto-antibodies to type I IFNs appear to be strong determinants of critical COVID-19 pneumonia.
Clinical implications
Auto-antibodies neutralizing type I IFNs apparently underlie already almost 1 million deaths from COVID-19 worldwide (15–20%). Thus, these studies have clinical implications because (1) it is straightforward to test for these neutralizing auto-antibodies before infection; (2) individuals with these antibodies should be vaccinated early and given priority for booster injections; (3) it is also possible to test for these antibodies during the early stages of COVID-19; and (4) specific treatments—such as IFNβ, monoclonal antibodies neutralizing SARS-CoV-2 or plasma exchange—could then be considered and tested in unvaccinated individuals, and perhaps even in vaccinated individuals115,116. Finally, these auto-antibodies against type I IFNs also underlie severe adverse reactions to vaccination with the live attenuated virus vaccine against yellow fever and perhaps other viral infections81,117,118. Together with IEIs of type I IFN immunity, these findings may explain the pathogenesis of about 15–20% of cases of critical COVID-19 pneumonia, especially in patients over 70 years old (Table 1 and Fig. 3). We know from IPEX72, RAG1/2 deficiencies74, incontinentia pigmenti6,119 and APS-1 (refs. 103,104,105,120,121) that some IEIs can underlie the production of auto-antibodies against type I IFNs. It will be interesting to determine whether other IEIs also underlie the production of auto-antibodies against type I IFN64,122,123,124. It will also be interesting to elucidate the reasons for the sudden increase in these auto-antibodies after 65 years of age, especially in men.

a, Inborn errors of type I IFN immunity that confer predisposition to critical COVID-19 pneumonia are represented in a slightly declining proportion across age groups in the general population, as they may underlie critical influenza and related life-threatening viral illnesses. By contrast, the frequency of auto-antibodies against type I IFN increases exponentially after the age of 65 years, attesting to a breakdown of tolerance in the ageing population. b, Global type I IFN immunity in the respiratory tract mucosae (RECs) and in the blood (pDCs) is shown to decline with age, under the influence of ageing and environmental triggers190,191. This decline in global type I IFN immunity over time may increase the risk of life-threatening COVID-19 (referred to as penetrance, for both IEI and auto-antibodies) associated with genetic and immunological aetiologies in older patients. All three risk factors—IEIs, auto-antibodies and tonic levels of type I IFNs—may contribute to critical COVID-19 pneumonia. IEIs and auto-antibodies appear to affect different patients, while the gradual decrease in tonic levels of type I IFNs can aggravate the consequences of both IEIs and auto-antibodies. Overall, the cohort of patients with life-threatening COVID-19 is enriched with IEIs in young patients and with auto-antibodies in older patients. IFR, infection fatality rate.
Type I IFNs in unexplained COVID-19
Before the discovery that type I IFN deficiency may underlie critical COVID-19 in some patients, some observations suggested that type I IFN levels in the blood of a subset of patients with critical COVID-19 pneumonia were lower than for other forms of infection108,125,126,127. By contrast, other studies reported enhanced type I IFN activity in a subset of patients with critical COVID-19 (refs. 128,129,130). Studies on patients with no known determinant of critical disease are, by nature, inconclusive. At best, the abnormalities detected can be correlated with disease severity, but it remains unclear whether they are a cause or consequence of disease. In the infinite and multidimensional matrix of causes and consequences, involving countless viruses and cell types, in individual patients, each of whom is unique, from the first day of infection to the death of the patient or viral clearance, it is difficult to establish a causal relationship. This has always been a fundamental problem in the field of infectious diseases, and in medicine at large, and has resulted in observational studies in humans gradually being replaced by experimental studies of cells in vitro and of animals in vivo and, more recently, by the study of the human genetic determinants of infectious diseases17,18,24. The discovery of genetic lesions or pre-existing auto-antibodies has provided an anchor to which observations of COVID-19 or other infections can be fixed to establish causality.
Type I IFN biology in patients with deficiencies
Only one patient with a type I IFN IEI, autosomal recessive IRF9 deficiency, has been studied immunologically, early in the course of infection131. The impact of auto-antibodies on systemic and/or mucosal immunity has been studied using single-cell RNA sequencing in more patients93,96. These studies showed that critically ill patients had weaker ISG responses in myeloid cells, and this lack of responsiveness was particularly marked in patients with auto-antibodies against type I IFN93. Consistent with this, single-cell RNA-sequencing analysis of nasopharyngeal swabs showed that patients with critical COVID-19, including one patient with auto-antibodies against type I IFNs, had muted ISG responses96. Finally, auto-antibodies against type I IFN have been detected in nasal fluids, and nasal ISG responses have been shown to be correlated with nasal viral load, systematic ISG responses in leukocytes and blood type I IFNα levels106. The patients with auto-antibodies against type I IFN and critical COVID-19 tested also displayed increases in the levels of inflammatory cytokines in both the respiratory tract and the blood, suggesting a two-step model for the pathogenesis of critical COVID-19, with insufficient type I IFN in the first few days of infection leading to excessive inflammation from the second week onwards12. Overall, these extensive studies have suggested that patients with critical COVID-19 and auto-antibodies against type I IFN have insufficient systemic and nasal type I IFN activity early in the course of disease (Fig. 2).
Other inborn IEIs
Regarding what have we learned from the study of patients with IEIs that do not impair type I IFN immunity directly or through the production of auto-antibodies, in ten retrospective cohorts of patients with various IEIs, the natural history of SARS-CoV-2 infection seemed to resemble that in the general population, albeit apparently with higher mortality in some IEI subsets62,64,123,124,132,133,134,135,136,137. A prospective study of IEI patients reached similar conclusions61. Notably, patients with predominant antibody deficiencies are not prone to life-threatening COVID-19 pneumonia62,64,123,124,132,133,134,135,136,137. This is consistent with the findings for critical influenza pneumonia, which is specifically seen in patients with IEIs of type I IFN immunity, but not in other individuals, even those lacking T and/or B cells65. Patients with IEIs of T and/or B cells may suffer from chronic COVID-19 infection and prolonged viral shedding138,139,140,141, similar to patients with acquired adaptive immunodeficiencies142,143,144. Multimutated, potentially more pathogenic SARS-CoV-2 variants might arise in such cases of persistent infection138. No IEIs other than those impairing type I IFN immunity directly or through auto-antibodies have been genetically or mechanistically associated with life-threatening COVID-19, but their vast genetic and immunological heterogeneity, and their individual rarity suggest that targeted clinical surveys are warranted. In particular, type I and III IFNs both activate ISGF-3 and induce a largely overlapping range of ISGs65,112 (Fig. 1). It would be interesting to study the course of SARS-CoV-2 infection in patients with autosomal recessive IL-10RB deficiency, whose cells respond to type I but not type III IFNs (Fig. 1).
Genome-wide association studies
The key result of genome-wide association studies (GWAS) is the identification of common variants of chromosomal region 3p21.31 that are associated with critical COVID-19 (refs. 145,146,147,148). The risk haplotype, inherited from Neanderthals, confers an estimated odds ratio per copy of between 1.6 and 2.1, with higher values for individuals under 60 years old148,149,150. The region encompasses six genes, including CXCR6 and LZTFL1. Five other genome-wide regions have been shown to be significantly associated with critical COVID-19 (ref. 147). Three of these regions encompass genes that are involved in type I IFN immunity. The first, on chr12q24.13, containing protective variants inherited from Neanderthals, includes the OAS1, OAS2 and OAS3 cluster—ISGs that are required for the activation of anti-viral RNaseL151. The second, a region on chr21q22.1, includes IFNAR2. The third, a region on chr19p13.2, includes TYK2. In these regions, one copy of the risk allele increases the risk of critical COVID-19 slightly, with odds ratios below 1.5. An odds ratio of 1.5 is often presented as increasing the risk by 50% but, assuming that the odds ratio does not overestimate the relative risk, the mathematical and clinical reality is that, for a COVID-19 mortality risk of 0.006% at the age of 20 years, 0.2% at the age of 50 years and 8.3% at the age of 80 years1, individuals carrying the at-risk genotype have risks of 0.009%, 0.3% and 12.5%, respectively. Although modest at the individual level, the impact of these findings is important at the population level152 (Table 1). These studies may not only reveal genetic modifiers of stronger determinants of disease, but also mechanisms that are dependent or independent of type I IFN.
Genome-wide search for rare variants
A population-based exome-wide association study48 sought to identify rare genetic variants associated with COVID-19 outcomes. Although no individual variant or gene was detected at the stringent genome-wide significance threshold corrected for the number of variants and traits tested (P < 9.6 × 10−10), the authors identified eight genes at a less conservative significance threshold of P < 5 × 10−8, one of which, TLR7, displayed an enrichment in predicted loss of function and in-frame variants with a MAF < 10−5 in critically ill patients with COVID-19 relative to individuals of unknown or seronegative status for SARS-CoV-2 infection. By contrast, this study and a previous rare-variant candidate gene association study153 reported no enrichment in predicted loss of function variants of 13 type-I-IFN-related influenza susceptibility genes5 in patients with critical COVID-19 pneumonia. Two possible reasons for this apparent discrepancy are of particular importance154. First, age—the key epidemiological factor driving COVID-19 severity—was ignored. Our cohort was much younger (mean age of 52 versus 66 years) and these IEIs are more frequent in patients under the age of 60 years154. Second, no tests were performed for auto-antibodies against type I IFN, the most common known determinant of critical COVID-19, especially in patients over 60 years old154. Importantly, the proportions of patients with critical COVID-19 due to autosomal recessive, X-linked recessive and autosomal dominant IEIs at these (or other) loci may vary from population to population. Finally, their causal link to critical COVID-19 cannot be concluded or excluded from an enrichment analysis of untested variants—it should be based on biochemical, virological and immunological experiments mechanistically connecting germline genotypes with clinical phenotypes5,24,40,41,42.
SARS-CoV-2 interference with type I IFN
The discovery that insufficient type I IFN can underlie critical COVID-19 pneumonia in vivo is remarkably convergent with various elegant virological studies conducted in human cells in vitro. Indeed, SARS-CoV-2 induces type I IFN production less strongly than seasonal influenza A viruses (IAV)155 or Sendai virus (SeV)156. The ability of SARS-CoV-2 to evade type I IFN induction results not only from the non-specific inhibition of host cellular functions, such as transcription and translation157,158,159, but also from the specific suppression of type I IFN induction pathways. Despite the limitations of overexpression systems, numerous studies have shown that at least 14 of the 31 products of known open reading frames (ORFs) of SARS-CoV-2 (Nsp1, Nsp5, Nsp6, Nsp13, Nsp14, Nsp15, ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF9b, M and N) target host proteins that govern type I IFN induction, including IRF3, TBK1, MAVS, RIG-I and NEMO, or self-amplification, including IFNAR1, STAT1, STAT2 and TYK2 (refs. 160,161,162,163,164,165,166,167,168). Moreover, an Nsp1 mutation (ΔD500–532) that is frequent in viral variants is associated with even lower levels of type I IFN production169. It remains to be tested whether the ability of SARS-CoV-2 to resist type I IFN is also increasing in emerging variants, such as B.1, B.1.1.7/Alpha, B.1.1351/Beta, B.1.617.2/Delta and B.1.1.529/Omicron. Current findings suggest that being able to evade type I IFN immunity is essential for viral fitness160,170.
Viral and human fitness depends on type I IFNs
Notably, three targets of the virus, IFNAR1 (ref. 167) IRF3 (refs. 164,168) and TBK1 (ref. 165), are encoded by COVID-19-susceptibility genes (Fig. 1). We expect that a greater convergence of viral targets and susceptibility genes will emerge with the genetic testing of viral targets in vivo, and the virological testing of susceptibility genes in vitro158,159,171,172,173,174,175,176,177,178,179. Suppression of the type I IFN response is essential for viral fitness, whereas the maintenance of type I IFN immunity is essential for human fitness. The type I IFN-blocking proteins of SARS-CoV-2 make the small amounts of type I IFN produced by infected cells in individual patients even more consequential, as attested by the catastrophic outcome of genetic or autoimmune deficiencies of type I IFN in vivo. Any further decrease in type I IFN levels due to the selection of new viral variants would tip the balance further in favour of the virus. Encouragingly, despite the ability of SARS-CoV-2 and its variants to evade type I IFN induction, these viral variants remain highly sensitive to type I IFN pretreatment in vitro161,180. However, the immense numbers of viral variants worldwide raise concerns about the emergence of new variants that are capable of impairing type I IFN immunity to an even greater extent.
Concluding remarks
IEIs of type I IFN immunity, and pre-existing auto-antibodies neutralizing type I IFNs appear to be strong determinants of critical COVID-19 pneumonia in about 15–20% of patients. This is unprecedented among common infectious diseases, as this proportion is much higher than the next best example—the possible explanation of only 1% of European cases of tuberculosis19,20. As these findings are consistent with those of in vitro virological studies and in vivo animal models156,181,182,183,184,185,186,187, they may reflect a general mechanism of disease. Individuals with insufficient type I IFN in the respiratory epithelium, whatever the underlying determinants, may be unable to prevent the spread of the virus to the lungs, blood and other organs during the first few days of infection. Inflammation may then develop when activated leukocytes, including myeloid and lymphoid cells of an innate or adaptive nature are attracted to the site of infection and attempt to resolve the pulmonary and systemic infection that became established because of the lack of control by type I IFN10,24,188 (Fig. 2). Understandably, at such a late inflammatory stage, therapeutic type I IFN did not help hospitalized patients189; clinical trials of early administration in ambulatory patients are ongoing115. The penetrance of known IEIs of type I IFN immunity and of auto-antibodies varies, with a higher penetrance for autosomal recessive and X-linked recessive than for autosomal dominant disorders, and for auto-antibodies neutralizing high concentrations of most type I IFNs relative to those neutralizing low concentrations of a single type I IFN (Table 1). Penetrance may be influenced by the size of the viral inoculum, by previous infection with other viruses that trigger type I IFN, especially in children190, or by human determinants, such as the age-dependent decline of pDCs163,191,192,193,194 and local respiratory type I IFN activity36,195, or common genetic variants, including those discovered by GWAS145,146,147 (Fig. 3).
In terms of what underlies critical COVID-19 pneumonia in the remaining 80% of cases, it would not be surprising to discover other IEIs of type I IFN immunity, including some affecting genes encoding proteins acting upstream or downstream from type I IFNs. These findings would further clarify the pathogenesis of critical COVID-19, while revealing the corresponding redundancy of these loci against other viral infections. The considerable redundancy of type I IFN in host defence against viruses is already a major surprise. Indeed, most patients with critical COVID-19 pneumonia due to an IEI or auto-antibody production had never before been hospitalized for another severe viral illness, including patients with autosomal recessive (IRF7, IFNAR1) or X-linked recessive (TLR7) inborn errors of type I IFN immunity. These findings suggest that there are type-I-IFN-independent mechanisms of cell-intrinsic immunity that provide protection against a wide range of viruses16,24. Another important question is whether adaptive immunity to the vaccine can compensate for a constitutive deficiency of type I IFN. Encouragingly, monoclonal antibodies neutralizing SARS-CoV-2 protected an unvaccinated but infected child with inherited IRF9 deficiency131. Despite their current success, it is unclear whether vaccines will remain effective in the long term and against new viral variants196,197,198,199. The recent spread of the Omicron variant—which not only is more contagious, but its S protein is also structurally distant from that encoded by existing vaccines—is particularly worrisome. Even before the emergence of Omicron, an alarming increase has been reported in the number of breakthrough cases, defined as infection in fully vaccinated individuals, including cases of hypoxaemic pneumonia and even death. It is tempting to hypothesize that some IEIs or auto-antibodies against type I IFN may underlie some life-threatening breakthrough cases. The search for human genetic and immunological determinants of life-threatening COVID-19 pneumonia must now encompass not only various viral variants, but also both unvaccinated and vaccinated patients.
References
O’Driscoll, M. et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 590, 140–145 (2021). Evidence that the mortality of COVID-19 doubles every 5 years from childhood onwards, accounting for a 10,000-fold greater risk at 85 years of age (10%) than at 5 years of age (0.001%).
Sen, P., Yamana, T. K., Kandula, S., Galanti, M. & Shaman, J. Burden and characteristics of COVID-19 in the United States during 2020. Nature 598, 338–341 (2021).
Sah, P. et al. Asymptomatic SARS-CoV-2 infection: a systematic review and meta-analysis. Proc. Natl Acad. Sci. USA 118, e2109229118 (2021).
Oran, D. P. & Topol, E. J. Prevalence of asymptomatic SARS-CoV-2 infection: a narrative review. Ann. Intern. Med. 173, 362–367 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020). Report of autosomal inborn errors of type I IFN, including autosomal dominant TLR3, autosomal recessive IRF7 and IFNAR1 deficiencies, as human genetic and immunological determinants of life-threatening COVID-19 pneumonia.
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020). Report of auto-antibodies against type I IFNs as immunological determinants of life-threatening COVID-19 pneumonia, with auto-antibodies neutralizing high concentrations of IFNα and/or IFNω.
Navaratnam, A. V., Gray, W. K., Day, J., Wendon, J. & Briggs, T. W. R. Patient factors and temporal trends associated with COVID-19 in-hospital mortality in England: an observational study using administrative data. Lancet Respir. Med. 9, 397–406 (2021).
Bennett, T. D. et al. Clinical characterization and prediction of clinical severity of SARS-CoV-2 infection among US adults using data from the US National COVID Cohort Collaborative. JAMA Netw. Open 4, e2116901 (2021).
Ricoca Peixoto, V. et al. Determinants for hospitalisations, intensive care unit admission and death among 20,293 reported COVID-19 cases in Portugal, March to April 2020. Euro Surveill. 26, 2001059 (2021).
Brodin, P. Immune determinants of COVID-19 disease presentation and severity. Nat. Med. 27, 28–33 (2021). Review of the immunological underpinnings, correlates and consequences of COVID-19, covering intrinsic, innate and adaptive immunity.
Takahashi, T. et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 588, 315–320 (2020).
Zhang, Q. et al. Life-threatening COVID-19: defective interferons unleash excessive inflammation. Med 1, 14–20 (2020).
Casanova, J. L., Su, H. C. & the COVID Human Genetic Effort. A global effort to define the human genetics of protective immunity to SARS-CoV-2 infection. Cell 181, 1194–1199 (2020).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Telenti, A. et al. After the pandemic: perspectives on the future trajectory of COVID-19. Nature 596, 495–504 (2021).
Meyts, I. & Casanova, J. L. Viral infections in humans and mice with genetic deficiencies of the type I IFN response pathway. Eur. J. Immunol. 51, 1039–1061 (2021).
Casanova, J. L. & Abel, L. The human genetic determinism of life-threatening infectious diseases: genetic heterogeneity and physiological homogeneity? Hum. Genet. 139, 681–694 (2020). Review of current concepts and approaches in the study of the human genetic determinants of life-threatening infectious diseases.
Casanova, J. L. & Abel, L. Lethal infectious diseases as inborn errors of immunity: toward a synthesis of the germ and genetic theories. Annu. Rev. Pathol. 16, 23–50 (2021). Review of the history of concepts and findings in the field of human genetics of infectious diseases.
Kerner, G. et al. Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc. Natl Acad. Sci. USA 116, 10430–10434 (2019).
Kerner, G. et al. Human ancient DNA analyses reveal the high burden of tuberculosis in Europeans over the last 2,000 years. Am. J. Hum. Genet. 108, 517–524 (2021).
Zhang, S. Y., Zhang, Q., Casanova, J. L. & Su, H. C., COVID Team. Severe COVID-19 in the young and healthy: monogenic inborn errors of immunity? Nat. Rev. Immunol. 20, 455–456 (2020).
Stertz, S. & Hale, B. G. Interferon system deficiencies exacerbating severe pandemic virus infections. Trends Microbiol. 29, 973–982 (2021).
Carvalho, T., Krammer, F. & Iwasaki, A. The first 12 months of COVID-19: a timeline of immunological insights. Nat. Rev. Immunol. 21, 245–256 (2021).
Casanova, J. L. & Abel, L. Mechanisms of viral inflammation and disease in humans. Science 374, 1080–1086 (2021). Review of the human genetic and immunological determinants of viral diseases of the skin, brain, and lungs.
Andreakos, E. et al. A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat. Immunol. 23, 159–164 (2022).
Arkin, L. M. et al. From your nose to your toes: a review of SARS-CoV-2 pandemic-associated pernio. J. Invest. Dermatol. 141, 2791–2796 (2021).
Sancho-Shimizu, V. et al. SARS-CoV-2-related MIS-C: a key to the viral and genetic causes of Kawasaki disease? J. Exp. Med. 218, e20210446 (2021).
Helms, J. et al. Neurologic features in severe SARS-CoV-2 infection. N. Engl. J. Med. 382, 2268–2270 (2020).
Huang, L. et al. 1-year outcomes in hospital survivors with COVID-19: a longitudinal cohort study. Lancet 398, 747–758 (2021).
Ciancanelli, M. J. et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348, 448–453 (2015). Earliest report of an inborn error of immunity underlying life-threatening influenza pneumonia in an otherwise healthy child.
Hernandez, N. et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med. 215, 2567–2585 (2018).
Lim, H. K. et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 216, 2038–2056 (2019).
Honda, K. et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777 (2005).
Reizis, B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity 50, 37–50 (2019).
Honda, K. & Taniguchi, T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658 (2006).
Stark, G. R. & Darnell, J. E. Jr The JAK-STAT pathway at twenty. Immunity 36, 503–514 (2012).
Gao, D. et al. TLR3 controls constitutive IFN-beta antiviral immunity in human fibroblasts and cortical neurons. J. Clin. Invest. 131, e134529 (2021).
Hernandez, N. et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J. Exp. Med. 216, 2057–2070 (2019).
Khanmohammadi, S., Rezaei, N., Khazaei, M. & Shirkani, A. A case of autosomal recessive interferon alpha/beta receptor alpha chain (IFNAR1) deficiency with severe COVID-19. J. Clin. Immunol. 42, 19–24 (2022).
Hassan, A. N. et al. Inherited IFNAR1 deficiency in a child with both critical COVID-19 pneumonia and multisystem inflammatory syndrome. J. Clin. Immunol. https://doi.org/10.1007/s10875-022-01215-7 (2022).
Schmidt, A. et al. TBK1 and TNFRSF13B mutations and an autoinflammatory disease in a child with lethal COVID-19. NPJ Genom. Med. 6, 55 (2021).
Asano, T. et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 6, eabl4348 (2021). Report of X-linked recessive TLR7 deficiency as a human genetic and immunological determinant of life-threatening COVID-19 pneumonia in male patients.
Abolhassani, H. et al. X-Linked TLR7 deficiency underlies critical COVID-19 pneumonia in a male patient with ataxia-telangiectasia. J. Clin. Immunol. 42, 1–9 (2022).
van der Made, C. I. et al. Presence of genetic variants among young men with severe COVID-19. JAMA 324, 663–673 (2020).
Fallerini, C. et al. Association of Toll-like receptor 7 variants with life-threatening COVID-19 disease in males: findings from a nested case-control study. eLife 10, e67569 (2021).
Pessoa, N. L. et al. Case report: hepatitis in a child infected with SARS-CoV-2 presenting toll-like receptor 7 Gln11Leu single nucleotide polymorphism. Virol. J. 18, 180 (2021).
Solanich, X. et al. Genetic screening for TLR7 variants in young and previously healthy men with severe COVID-19. Front. Immunol. 12, 719115 (2021).
Kosmicki, J. A. et al. Pan-ancestry exome-wide association analyses of COVID-19 outcomes in 586,157 individuals. Am. J. Hum. Genet. 108, 1350–1355 (2021).
Onodi, F. et al. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J. Exp. Med. 218, e20201387 (2021). Evidence that human pDCs sense SARS-CoV-2 through UNC93B and IRAK4 and, by inference, through TLR7 and/or TLR9.
Swiecki, M. & Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 15, 471–485 (2015).
Colonna, M., Trinchieri, G. & Liu, Y. J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5, 1219–1226 (2004).
Liu, Y.-J. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 106, 259–262 (2001).
Severa, M. et al. Differential plasmacytoid dendritic cell phenotype and type I Interferon response in asymptomatic and severe COVID-19 infection. PLoS Pathog. 17, e1009878 (2021).
Beutler, B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430, 257–263 (2004).
Hemmi, H. et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3, 196–200 (2202).
Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. & Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004).
Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529 (2204).
Lund, J. M. et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl Acad. Sci. USA 101, 5598–5603 (2004).
Barreiro, L. B. et al. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 5, e1000562 (2209).
Zhang, S. Y. et al. Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev 220, 225–236 (2007).
Deya-Martinez, A. et al. COVID-19 in children and young adults with moderate/severe inborn errors of immunity in a high burden area in pre-vaccine era. Clin. Immunol. 230, 108821 (2021).
Goudouris, E. S. et al. Outcome of SARS-CoV-2 infection in 121 patients with inborn errors of immunity: a cross-sectional study. J. Clin. Immunol. 41, 1479–1489 (2021).
Shields, A. M., Burns, S. O., Savic, S. & Richter, A. G., UK PIN COVID-19 Consortium. COVID-19 in patients with primary and secondary immunodeficiency: the United Kingdom experience. J. Allergy Clin Immunol 147, 870–875 (2021).
Meyts, I. et al. Coronavirus disease 2019 in patients with inborn errors of immunity: an international study. J. Allergy Clin. Immunol. 147, 520–531 (2021).
Zhang, Q. Human genetics of life-threatening influenza pneumonitis. Hum. Genet. 139, 941–948 (2020).
Vallbracht, A., Treuner, J., Flehmig, B., Joester, K. E. & Niethammer, D. Interferon-neutralizing antibodies in a patient treated with human fibroblast interferon. Nature 289, 496–497 (1981).
Panem, S., Check, I. J., Henriksen, D. & Vilcek, J. Antibodies to alpha-interferon in a patient with systemic lupus erythematosus. J. Immunol. 129, 1–3 (1982).
Gupta, S. et al. Distinct functions of autoantibodies against interferon in systemic lupus erythematosus: a comprehensive analysis of anticytokine autoantibodies in common rheumatic diseases. Arthritis Rheumatol. 68, 1677–1687 (2016).
Levin, M. Anti-interferon auto-antibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 3, e292 (2006).
Meager, A. et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 3, e289 (2006). Initial report that patients with APS-1 carry auto-antibodies against type I IFNs.
Meyer, S. et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell 166, 582–595 (2016).
Rosenberg, J. M. et al. Neutralizing anti-cytokine autoantibodies against interferon-α in immunodysregulation polyendocrinopathy enteropathy X-Linked. Front. Immunol. 9, 544 (2018).
Eriksson, D. et al. The autoimmune targets in IPEX are dominated by gut epithelial proteins. J. Allergy Clin. Immunol. 144, 327–330 (2019).
Walter, J. E. et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J. Clin. Invest. 125, 4135–4148 (2015).
Romi, F. Thymoma in myasthenia gravis: from diagnosis to treatment. Autoimmune Dis. 2011, 474512 (2011).
Shiono, H. et al. Spontaneous production of anti-IFN-alpha and anti-IL-12 autoantibodies by thymoma cells from myasthenia gravis patients suggests autoimmunization in the tumor. Int. Immunol. 15, 903–913 (2003).
Bello-Rivero, I. et al. Characterization of the immunoreactivity of anti-interferon alpha antibodies in myasthenia gravis patients. Epitope mapping. J. Autoimmun. 23, 63–73 (2004).
Meager, A. et al. Anti-cytokine autoantibodies in autoimmunity: preponderance of neutralizing autoantibodies against interferon-alpha, interferon-omega and interleukin-12 in patients with thymoma and/or myasthenia gravis. Clin. Exp. Immunol. 132, 128–136 (2003).
Anderson, M. S. et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002).
Cheng, M. H. et al. Acquired autoimmune polyglandular syndrome, thymoma, and an AIRE defect. N. Engl. J. Med. 362, 764–766 (2010).
Pozzetto, B., Mogensen, K. E., Tovey, M. G. & Gresser, I. Characteristics of autoantibodies to human interferon in a patient with varicella-zoster disease. J. Infect. Dis. 150, 707–713 (1984). Seminal report of a single patient with severe varicella zoster virus disease due to auto-antibodies neutralizing type I IFNs, which is also the first infectious disease causally attributed to an auto-antibody directed against a cytokine.
Doffinger, R. et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin. Infect. Dis. 38, e10–e14 (2004).
Hoflich, C. et al. Naturally occurring anti-IFN-gamma autoantibody and severe infections with Mycobacterium cheloneae and Burkholderia cocovenenans. Blood 103, 673–675 (2004).
Kampmann, B. et al. Acquired predisposition to mycobacterial disease due to autoantibodies to IFN-γ. J. Clin. Invest. 115, 2480–2488 (2005).
Puel, A. et al. Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J. Immunol. 180, 647–654 (2008).
Puel, A. et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 207, 291–297 (2010).
Ku, C. L., Chi, C. Y., von Bernuth, H. & Doffinger, R. Autoantibodies against cytokines: phenocopies of primary immunodeficiencies? Hum. Genet. 139, 783–794 (2020).
Kisand, K. et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 207, 299–308 (2010).
Koning, R. et al. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med. 47, 704–706 (2021).
Troya, J. et al. Neutralizing autoantibodies to type I IFNs in >10% of patients with severe COVID-19 pneumonia hospitalized in Madrid, Spain. J. Clin. Immunol. 41, 914–922 (2021).
Goncalves, D. et al. Antibodies against type I interferon: detection and association with severe clinical outcome in COVID-19 patients. Clin. Transl. Immunol. 10, e1327 (2021).
Wang, E. Y. et al. Diverse functional autoantibodies in patients with COVID-19. Nature 595, 283–288 (2021). Replication of the enrichment of auto-antibodies against type I IFNs in patients with severe COVID-19 and evidence that SARS-CoV-2 infection can trigger other auto-antibodies.
van der Wijst, M. G. P. et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 13, eabh2624 (2021). Single-cell study of blood leukocytes showing that patients with auto-antibodies against type I IFNs are immunologically similar to other patients with critical COVID-19 pneumonia.
Acosta-Ampudia, Y. et al. COVID-19 convalescent plasma composition and immunological effects in severe patients. J. Autoimmun. 118, 102598 (2021).
Chang, S. E. et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat. Commun. 12, 5417 (2021).
Ziegler, C. G. K. et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 184, 4713–4733 (2021). A comprehensive and in-depth study of intrinsic immunity in the respiratory tract of patients with COVID-19.
Solanich, X. et al. Pre-existing autoantibodies neutralizing high concentrations of type i interferons in almost 10% of COVID-19 patients admitted to intensive care in Barcelona. J. Clin. Immunol. 41, 1733–1744 (2021).
Abers, M. S. et al. Neutralizing type-I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID-19. Immunol. Cell Biol. 99, 917–921 (2021).
Vazquez, S. E. et al. Neutralizing autoantibodies to type I interferons in COVID-19 convalescent donor plasma. J. Clin. Immunol. 41, 1169–1171 (2021).
Chauvineau-Grenier, A. et al. Autoantibodies neutralizing type I interferons in 20% of COVID-19 deaths in a French hospital. Res. Sq. https://doi.org/10.21203/rs.3.rs-915062/v1 (2021).
Carapito, R. et al. Identification of driver genes for critical forms of COVID-19 in a deeply phenotyped young patient cohort. Sci. Transl. Med. 14, eabj7521 (2021).
Raadsen, M. P. et al. Interferon-α2 auto-antibodies in convalescent plasma therapy for COVID-19. J. Clin. Immunol. 42, 232–239 (2022).
Bastard, P. et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J. Exp. Med. 218, e20210554 (2021). Evidence that patients with APS-1 are at very high risk of critical COVID-19 due to their auto-antibodies against type I IFNs.
Meisel, C. et al. Mild COVID-19 despite autoantibodies against type I IFNs in autoimmune polyendocrine syndrome type 1. J. Clin. Invest. 131, e150867 (2021).
Shaw, E. R. et al. Temporal dynamics of anti-type 1 interferon autoantibodies in COVID-19 patients. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciab1002 (2021).
Lopez, J. et al. Early nasal type I IFN immunity against SARS-CoV-2 is compromised in patients with autoantibodies against type I IFNs. J. Exp. Med. 218, e20211211 (2021). Evidence that auto-antibodies against type I IFNs are found in nasal secretions and impair type I IFN immunity in the corresponding mucosa.
de Prost, N. et al. Plasma exchange to rescue patients with autoantibodies against type i interferons and life-threatening COVID-19 pneumonia. J. Clin. Immunol. 41, 536–544 (2021).
Galani, I. E. et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 22, 32–40 (2021). Longitudinal study of antiviral immunity, including type I and III IFNs, in patients with diverse forms of COVID-19 pneumonia, in comparison with influenza pneumonia.
Bastard, P. et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci. Immunol. 6, eabl4340 (2021). Discovery of auto-antibodies neutralizing low concentrations of type I IFNs in the general population and as immunological determinants of life-threatening COVID-19 pneumonia, especially in the aging population.
LaFleur, D. W. et al. Interferon-κ, a novel type I interferon expressed in human keratinocytes. J. Biol. Chem. 276, 39765–39771 (2001).
Marks, Z. R. C. et al. Properties and functions of the novel type I interferon epsilon. Semin. Immunol. 43, 101328 (2019).
Lazear, H. M., Schoggins, J. W. & Diamond, M. S. Shared and distinct functions of type I and type III interferons. Immunity 50, 907–923 (2019).
Manry, J. et al. Evolutionary genetic dissection of human interferons. J. Exp. Med. 208, 2747–2759 (2011).
Park, A. & Iwasaki, A. Type I and Type III interferons—induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27, 870–878 (2020).
Vinh, D. C. et al. Harnessing type I IFN immunity against SARS-CoV-2 with early administration of IFN-β. J. Clin. Immunol. 41, 1425–1442 (2021).
de Prost, N. et al. Plasma exchange to rescue patients with autoantibodies against type I interferons and life-threatening COVID-19 pneumonia. J. Clin. Immunol. 41, 536–544 (2021).
Bastard, P. et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J. Exp. Med. 218, e20202486 (2021). Evidence that auto-antibodies neutralizing type I IFNs can underlie life-threatening adverse reactions to live-attenuated yellow fever virus vaccine in previously healthy patients.
Hetemaki, I. et al. Patients with autoimmune polyendocrine syndrome type 1 have an increased susceptibility to severe herpesvirus infections. Clin. Immunol. 231, 108851 (2021).
Bastard, P. et al. Interferon-β therapy in a patient with incontinentia pigmenti and autoantibodies against type I IFNs infected with SARS-CoV-2. J. Clin. Immunol. 41, 931–933 (2021).
Beccuti, G. et al. A COVID-19 pneumonia case report of autoimmune polyendocrine syndrome type 1 in Lombardy, Italy: letter to the editor. J. Endocrinol. Invest. 43, 1175–1177 (2020).
Lemarquis, A. et al. Severe COVID-19 in an APS1 patient with interferon autoantibodies treated with plasmapheresis. J. Allergy Clin. Immunol. 148, 96–98 (2021).
Abraham, R. S. et al. Severe SARS-CoV-2 disease in the context of a NF-κB2 loss-of-function pathogenic variant. J. Allergy Clin. Immunol. 147, 532–544 (2021).
Drabe, C. H. et al. Low morbidity in Danish patients with common variable immunodeficiency disorder infected with severe acute respiratory syndrome coronavirus 2. Infect. Dis. 53, 953–958 (2021).
Marcus, N. et al. Minor clinical impact of COVID-19 pandemic on patients with primary immunodeficiency in Israel. Front. Immunol. 11, 614086 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Trouillet-Assant, S. et al. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 146, 206–208 (2020).
Liu, C. et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 184, 1836–1857 (2021).
Lucas, C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020).
Lee, J. S. et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 5, eabd1554 (2020).
Sposito, B. et al. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell 184, 4953–4968 (2021). Comprehensive and in-depth study of intrinsic and innate immunity along the respiratory tract in patients with severe COVID-19.
Lévy, R. et al. Monoclonal antibody-mediated neutralization of SARS-CoV-2 in an IRF9-deficient child. Proc. Natl Acad. Sci. USA 118, e2114390118 (2021). Evidence that monoclonal antibodies neutralizing SARS-CoV-2 were able to overcome a complete deficiency of both type I and III IFNs in a child with autosomal recesive IRF9 deficiency.
Delavari, S. et al. Impact of SARS-CoV-2 pandemic on patients with primary immunodeficiency. J. Clin. Immunol. 41, 345–355 (2021).
Esenboga, S. et al. COVID-19 in patients with primary immunodeficiency. J. Clin. Immunol. 41, 1515–1522 (2021).
Ho, H. E., Mathew, S., Peluso, M. J. & Cunningham-Rundles, C. Clinical outcomes and features of COVID-19 in patients with primary immunodeficiencies in New York City. J. Allergy Clin. Immunol. Pract. 9, 490–493 (2021).
Milito, C. et al. Clinical outcome, incidence, and SARS-CoV-2 infection-fatality rates in Italian patients with inborn errors of immunity. J. Allergy Clin. Immunol. Pract. 9, 2904–2906 e2902 (2021).
Castano-Jaramillo, L. M. et al. COVID-19 in the context of inborn errors of immunity: a case series of 31 patients from Mexico. J. Clin. Immunol. 41, 1463–1478 (2021).
Karakoc Aydiner, E. et al. Adverse COVID-19 outcomes in immune deficiencies: inequality exists between subclasses. Allergy 77, 282–295 (2021).
Corey, L. et al. SARS-CoV-2 variants in patients with immunosuppression. N. Engl. J. Med. 385, 562–566 (2021).
Giardino, G. et al. SARS-CoV-2 infection in the immunodeficient host: necessary and dispensable immune pathways. J. Allergy Clin. Immunol. Pract. 9, 3237–3248 (2021).
Hariharan, S. V., Muthusamy, S. & Asokan, S. K. Persistent viral shedding after SARS-CoV-2 infection in an infant with severe combined immunodeficiency. Indian J. Pediatr. 89, 94 (2021).
Mohanty, M. C., Taur, P. D., Sawant, U. P., Yadav, R. M. & Potdar, V. Prolonged fecal shedding of SARS-CoV-2 in asymptomatic children with inborn errors of immunity. J. Clin. Immunol. 41, 1748–1753 (2021).
Ambrosioni, J. et al. Overview of SARS-CoV-2 infection in adults living with HIV. Lancet HIV 8, e294–e305 (2021).
Kemp, S. A. et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 592, 277–282 (2021).
Choi, B. et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N. Engl. J. Med. 383, 2291–2293 (2020).
The Severe Covid-19 GWAS Group. Genomewide association study of severe COVID-19 with respiratory failure. N. Engl. J. Med. 383, 1522–1534 (2020).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in COVID-19. Nature 591, 92–98 (2021).
COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 600, 472–477 (2021). Genome-wide association meta-analysis of COVID-19, including genome-wide significant loci that are weakly associated with SARS-CoV-2 infection or severe COVID-19.
Nakanishi, T. et al. Age-dependent impact of the major common genetic risk factor for COVID-19 on severity and mortality. J. Clin. Invest. 131, e152386 (2021).
Zeberg, H. & Paabo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020).
Kerner, G., Patin, E. & Quintana-Murci, L. New insights into human immunity from ancient genomics. Curr. Opin. Immunol. 72, 116–125 (2021).
Zeberg, H. & Paabo, S. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc. Natl Acad. Sci. USA 118, e2026309118 (2021).
Colona, V. L., Biancolella, M., Novelli, A. & Novelli, G. Will GWAS eventually allow the identification of genomic biomarkers for COVID-19 severity and mortality? J. Clin. Invest. 131, e155011 (2021).
Povysil, G. et al. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J. Clin. Invest. 131, e147834 (2021).
Zhang, Q. et al. Association of rare predicted loss-of-function variants of influenza-related type I IFN genes with critical COVID-19 pneumonia. J. Clin. Invest. 131, e152474 (2021).
Hatton, C. F. et al. Delayed induction of type I and III interferons mediates nasal epithelial cell permissiveness to SARS-CoV-2. Nat. Commun. 12, 7092 (2021).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045 (2020). A comprehensive study of the virus–host dynamics that underlie COVID-19, with low levels of type I and III IFNs and high levels of other chemokines and cytokines.
Banerjee, A. K. et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339 (2020).
Flynn, R. A. et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell 184, 2394–2411 (2021).
Schneider, W. M. et al. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell 184, 120–132 (2021). Identification of human cell factors that restrict SARS-CoV-2 proliferation and spreading.
Palermo, E., Di Carlo, D., Sgarbanti, M. & Hiscott, J. Type I interferons in COVID-19 pathogenesis. Biology 10, 829 (2021).
Lowery, S. A., Sariol, A. & Perlman, S. Innate immune and inflammatory responses to SARS-CoV-2: implications for COVID-19. Cell Host Microbe 29, 1052–1062 (2021). A review of the mechanisms by which SARS-CoV-2 can induce or antagonize type I and III IFN activity.
Mariano, G., Farthing, R. J., Lale-Farjat, S. L. M. & Bergeron, J. R. C. Structural characterization of SARS-CoV-2: where we are, and where we need to be. Front. Mol. Biosci. 7, 605236 (2020).
Schultze, J. L. & Aschenbrenner, A. C. COVID-19 and the human innate immune system. Cell 184, 1671–1692 (2021).
Konno, Y. et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 32, 108185 (2020).
Sui, L. et al. SARS-CoV-2 membrane protein inhibits type I interferon production through ubiquitin-mediated degradation of TBK1. Front. Immunol. 12, 662989 (2021).
Miorin, L. et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl Acad. Sci. USA 117, 28344–28354 (2020).
Hayn, M. et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 35, 109126 (2021).
Yuen, C. K. et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 9, 1418–1428 (2020).
Lin, J. W. et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe 29, 489–502 (2021).
Mlcochova, P. et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 599, 114–119 (2021).
Daniloski, Z. et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 184, 92–105 (2021). Identification of human cell factors that restrict SARS-CoV-2 proliferation and spreading.
Hoffmann, H. H. et al. TMEM41B is a pan-flavivirus host factor. Cell 184, 133–148 (2021). Identification of human cell factors that restrict SARS-CoV-2 proliferation and spreading.
Wang, R. et al. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell 184, 106–119 (2021). Identification of human cell factors that restrict SARS-CoV-2 proliferation and spreading.
Wei, J. et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell 184, 76–91 (2021). Identification of human cell factors that restrict SARS-CoV-2 proliferation and spreading.
Hoffmann, H. H. et al. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 29, 267–280 (2021). Identification of human cell factors that restrict SARS-CoV-2 proliferation and spreading.
Katsura, H. et al. Human lung stem cell-based alveolospheres provide insights into SARS-CoV-2-mediated interferon responses and pneumocyte dysfunction. Cell Stem Cell 27, 890–904 (2020).
Martin-Sancho, L. et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 81, 2656–2668 (2021).
Pfaender, S. et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 5, 1330–1339 (2020).
Gordon, D. E. et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370, eabe9403 (2020).
Lokugamage, K. G. et al. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 94, e01410-20 (2020).
Munoz-Fontela, C. et al. Animal models for COVID-19. Nature 586, 509–515 (2020). Review of animal models for SARS-CoV-2 infection and their applications.
Bessiere, P. et al. Intranasal type I interferon treatment is beneficial only when administered before clinical signs onset in the SARS-CoV-2 hamster model. PLoS Pathog. 17, e1009427 (2021).
Hoagland, D. A. et al. Leveraging the antiviral type I interferon system as a first line of defense against SARS-CoV-2 pathogenicity. Immunity 54, 557–570 (2021).
Hassan, A. O. et al. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell 182, 744–753 (2020).
Israelow, B. et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J. Exp. Med. 217, e20201241 (2020).
Leist, S. R. et al. A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard laboratory mice. Cell 183, 1070–1085 (2020).
Dinnon, K. H. III et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 586, 560–566 (2020).
Sette, A. & Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184, 861–880 (2021).
WHO Solidarity Trial Consortium. Repurposed antiviral drugs for COVID-19—interim WHO solidarity trial results. N. Engl. J. Med. 384, 497–511 (2021).
Loske J. et al. Pre-activated antiviral innate immunity in the upper airways controls early SARS-CoV-2 infection in children. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01037-9 (2021). Report of stronger type I IFN activity in the respiratory tract in SARS-CoV-2-infected children than in adults.
Splunter, M. V. et al. Plasmacytoid dendritic cell and myeloid dendritic cell function in ageing: a comparison between elderly and young adult women. PLoS ONE 14, e0225825 (2019). Report of weaker TLR7- and TLR9-dependent responses of plasmacytoid dendritic cells in older adults compared with in younger adults.
Pierce, C. A. et al. Immune responses to SARS-CoV-2 infection in hospitalized pediatric and adult patients. Sci. Transl. Med. 12, eabd5487 (2020).
Pierce, C. A. et al. Natural mucosal barriers and COVID-19 in children. JCI Insight 6, e148694 (2021).
Shaw, A. C., Goldstein, D. R. & Montgomery, R. R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 13, 875–887 (2013).
Bartleson, J. M. et al. SARS-CoV-2, COVID-19 and the aging immune system. Nat. Aging 1, 769–782 (2021).
Krause, P. R. et al. SARS-CoV-2 variants and vaccines. N. Engl. J. Med. 385, 179–186 (2021).
Hacisuleyman, E. et al. Vaccine breakthrough infections with SARS-CoV-2 variants. N. Engl. J. Med. 384, 2212–2218 (2021).
Kustin, T. et al. Evidence for increased breakthrough rates of SARS-CoV-2 variants of concern in BNT162b2-mRNA-vaccinated individuals. Nat. Med. 27, 1379–1384 (2021).
Pegu, A. et al. Durability of mRNA-1273 vaccine-induced antibodies against SARS-CoV-2 variants. Science 373, 1372–1377 (2021).
Gilliet, M., Cao, W. & Liu, Y. J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606 (2008).
Acknowledgements
The Laboratory of Human Genetics of Infectious Diseases is supported by the Howard Hughes Medical Institute, the Rockefeller University, the St Giles Foundation, the National Institutes of Health (NIH) (R01AI088364 and R01AI163029), the National Center for Advancing Translational Sciences (NCATS), the NIH Clinical and Translational Science Award (CTSA) program (UL1 TR001866), a Fast Grant from Emergent Ventures, the Mercatus Center at George Mason University, the Yale Center for Mendelian Genomics and the GSP Coordinating Center funded by the National Human Genome Research Institute (NHGRI) (UM1HG006504 and U24HG008956), the Yale High Performance Computing Center (S10OD018521), the Fisher Center for Alzheimer’s Research Foundation, the Meyer Foundation, the JPB Foundation, the French National Research Agency (ANR) under the ‘Investments for the Future’ program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the French Foundation for Medical Research (FRM) (EQU201903007798), the FRM and ANR GENCOVID project (ANR-20-COVI-0003), ANRS Nord-Sud (ANRS-COV05), ANR grant GENVIR (ANR-20-CE93-003), ANR AABIFNCOV (ANR-20-CO11-0001) and ANR MIS-C (ANR 21-COVR-0039, GenMIS-C) projects, the European Union’s Horizon 2020 research and innovation program under grant agreement no. 824110 (EASI-Genomics), the Square Foundation, Grandir—Fonds de solidarité pour l’enfance, the SCOR Corporate Foundation for Science, Fondation du Souffle, The French Ministry of Higher Education, Research, and Innovation (MESRI-COVID-19), Institut National de la Santé et de la Recherche Médicale (INSERM), REACTing-INSERM, and the University of Paris. P.B. was supported by the FRM (EA20170638020) and the MD-PhD programme of the Imagine Institute (with the support of the Fondation Bettencourt Schueller). G.N. is supported by Regione Lazio (Research Group Projects 2020) no. A0375-2020-36663, GecoBiomark. H.C.S. and L.D.N. are supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Author information
Authors and Affiliations
Consortia
Contributions
Q.Z., P.B. and A.C. contributed to writing the main text, auto-antibody and GWAS sections, respectively. J.-L.C. contributed to writing the abstract, main text and conclusion. Q.Z., P.B., A.C., J.-L.C. and the COVID Human Genetic Effort consortium members contributed to conceptualizing and editing the Review, either by directly editing the manuscript or figures or by providing key references and clinical and scientific expertise.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, Q., Bastard, P., COVID Human Genetic Effort. et al. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 603, 587–598 (2022). https://doi.org/10.1038/s41586-022-04447-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04447-0
This article is cited by
-
Kinome and phosphoproteome reprogramming underlies the aberrant immune responses in critically ill COVID-19 patients
Clinical Proteomics (2024)
-
Metacell-based differential expression analysis identifies cell type specific temporal gene response programs in COVID-19 patient PBMCs
npj Systems Biology and Applications (2024)
-
Recent advances and evolving concepts in Still’s disease
Nature Reviews Rheumatology (2024)
-
Evolution of enhanced innate immune suppression by SARS-CoV-2 Omicron subvariants
Nature Microbiology (2024)
-
Chest Computed Tomography Characteristics of Critically Ill COVID-19 Patients with Auto-antibodies Against Type I Interferons
Journal of Clinical Immunology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
