
Abstract
The animal reservoir of SARS-CoV-2 is unknown despite reports of SARS-CoV-2-related viruses in Asian Rhinolophus bats1,2,3,4, including the closest virus from R. affinis, RaTG13 (refs. 5,6), and pangolins7,8,9. SARS-CoV-2 has a mosaic genome, to which different progenitors contribute. The spike sequence determines the binding affinity and accessibility of its receptor-binding domain to the cellular angiotensin-converting enzyme 2 (ACE2) receptor and is responsible for host range10,11,12. SARS-CoV-2 progenitor bat viruses genetically close to SARS-CoV-2 and able to enter human cells through a human ACE2 (hACE2) pathway have not yet been identified, although they would be key in understanding the origin of the epidemic. Here we show that such viruses circulate in cave bats living in the limestone karstic terrain in northern Laos, in the Indochinese peninsula. We found that the receptor-binding domains of these viruses differ from that of SARS-CoV-2 by only one or two residues at the interface with ACE2, bind more efficiently to the hACE2 protein than that of the SARS-CoV-2 strain isolated in Wuhan from early human cases, and mediate hACE2-dependent entry and replication in human cells, which is inhibited by antibodies that neutralize SARS-CoV-2. None of these bat viruses contains a furin cleavage site in the spike protein. Our findings therefore indicate that bat-borne SARS-CoV-2-like viruses that are potentially infectious for humans circulate in Rhinolophus spp. in the Indochinese peninsula.
Similar content being viewed by others


Main
The origin of SARS-CoV-2, as well as its mode of introduction into the human population, are unknown at present. Since its emergence, numerous animal species have been studied to identify possible reservoirs and/or intermediate hosts of the virus, including a large diversity of insectivorous bats of the genus Rhinolophus. Despite the recent report of various SARS-CoV-2-related viruses in R. shameli (isolated in Cambodia in 201013), R. pusillus and R. malayanus (China, 2020 and 2019, respectively2), R. acuminatus (Thailand, 20203) and R. cornutus (Japan, 20134), the closest SARS-CoV-2 bat-borne genome still remains the one from R. affinis, RaTG13 (China, 20135,6), with 96.1% identity at the whole-genome level. Several studies also suggested the involvement of pangolin coronaviruses in the emergence of SARS-CoV-2 (refs. 7,8,9). Since its appearance in humans, SARS-CoV-2 has evolved through sporadic mutations and recombination events14, some of which correspond to gains in fitness allowing the virus to spread more widely, or to escape neutralizing antibodies15.
To decipher the origin of SARS-CoV-2, it is therefore essential to ascertain the diversity of animal coronaviruses, and more specifically, that of bat coronaviruses. Although the identification of SARS-CoV-2 in bats is a main goal, a more realistic objective is to identify the sequences that contribute to its mosaicism. The spike sequence seems essential, as it determines the binding affinity and accessibility of the receptor-binding domain (RBD) to the cellular ACE2 receptor and is therefore responsible for host range10,11,12. The closest related bat strain identified so far (RaTG13) has a low RBD sequence similarity to SARS-CoV-2, and with only 11/17 hACE2 contact amino acid residues conserved with SARS-CoV-2, its affinity for hACE2 is very limited16. Moreover, SARS-CoV-2 poorly infects bats and bat cells tested so far17. In addition, no bat SARS-CoV-2-like virus has been shown to use hACE2 to efficiently enter human cells, and none has the furin cleavage site that is associated with an increased pathogenicity in humans18. The SARS-CoV-2 RBD binds to R. macrotis ACE2 with a lower affinity than to hACE2 (ref. 19). An essential piece of information—finding bat viruses with an RBD motif genetically close to that of SARS-CoV-2 and capable of binding to hACE2 with high affinity—is therefore missing.
We speculated that this type of virus could be identified in bats living in the limestone karstic terrain common to China, Laos and Vietnam in the Indochinese peninsula. Here we report the presence of sarbecoviruses close to SARS-CoV-2 whose RBDs differ from that of SARS-CoV-2 by only one or two contact residues, strongly bind to the hACE2 protein and mediate hACE2-dependent entry and replication into human cells. Despite the absence of the furin cleavage site, these viruses may have contributed to the origin of SARS-CoV-2 and may intrinsically pose a future risk of direct transmission to humans.
Diversity of bat and coronavirus species
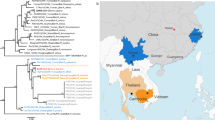
A total of 645 bats belonging to 6 families and 46 species were captured (Supplementary Table 1). Two hundred and forty-seven blood samples, 608 saliva, 539 anal/faecal and 157 urine swabs were collected from bats in the northern part of Laos (Supplementary Table 2). We first screened all 539 faecal samples through a pan-coronavirus nested RT-PCR analysis20. Overall, 24 individuals of 10 species were positive, and 1 individual (BANAL-27) was concomitantly infected by an alphacoronavirus and a betacoronavirus (Supplementary Table 3). BLAST analysis of amplicons identified alphacoronavirus sequences of the Decacovirus, Pedacovirus and Rhinacovirus subgenera and betacoronavirus sequences of the Nobecovirus and Sarbecovirus subgenera. Sequences of the Sarbecovirus subgenus were all identified from Rhinolophus individuals belonging to three different species (R. malayanus, R. marshalli and R. pusillus). Positive individuals were trapped in three different districts, and those infected with a sarbecovirus were all from the Fueng district in Vientiane province (Fig. 1a, site 1).

a, Map of sampling sites. All BANAL isolates were collected from the same site (site 1). The map was downloaded from DIVA-GIS (https://www.diva-gis.org/gdata). b, Phylogenetic analysis of the protein sequence of the RBD of Laotian and representative human, bat, and pangolin sarbecoviruses. Sequences were aligned with MAFFT (ref. 49) in auto mode, and maximum-likelihood phylogenetic reconstruction was performed with PhyML implemented through the NGPhylogeny portal50 with the LG + G substitution model. Branch support was evaluated with the aBayes parameter. Bat species are specified in the name of the sequences. Sequences are coloured according to Fig. 1c. c, Similarity plot analysis of Laotian and representative bat and pangolin sarbecoviruses based on the full-length genome sequence of the SARS-CoV-2 human prototype strain (NC_045512, Wuhan-Hu-1) used as a reference. The analysis was performed with the SimPlot program version 3.5.1 (ref. 51) with the Kimura two-parameter model, a window size of 1,000 base pairs, a step size of 100 base pairs, a transition/transversion rate (T/t) of 2.0, and a Gap/Strip parameter: on51. nsp, non-structural protein; RdRP, RNA-dependent RNA polymerase. d, Heat map of identities at the protein level of representative human, bat and pangolin sarbecoviruses compared to human SARS-CoV-2 lineage B (NC_045512). Spike protein has been divided into functional domains, and the sequences are ordered according to percentage of identity of the RBD domain. The asterisk marks the absence of a functional ORF10 in Thai bat RacCS203 (accession number MW251308). The heat map was created using the gplots package in R (version 3.6.3). AA, amino acid.
Next-generation sequencing (NGS) and Sanger sequencing were used to obtain a complete genomic sequence of five of the seven sarbecoviruses (Fig. 1 and Supplementary Table 4). The coverage of the genome of the remaining two sarbecoviruses (BANAL-27 and BANAL-242 sampled from R. pusillus and R. malayanus bats, respectively) was 90%; therefore, they were not included in the final analyses. Phylogenetic analyses performed on the receptor-binding protein domain of lineages A and B human SARS-CoV-2 (ref. 21), and on representative bat and pangolin sarbecoviruses, placed the Laotian R. malayanus BANAL-52, R. pusillus BANAL-103 and R. marshalli BANAL-236 coronaviruses close to human SARS-CoV-2 and pangolin coronaviruses collected in 2019, whereas R. malayanus BANAL-116 and BANAL-247 coronaviruses belonged to a sister clade with other bat coronaviruses (RmYN02, RacCS203, RpYN06 and PrC31) from different Rhinolophus species. Pangolin coronaviruses sampled in 2017 exhibited a basal position relative to these strains (Fig. 1b). Very similar SARS-CoV-2-like viruses are shared by different bat species, suggesting possible circulation of viruses between different species living sympatrically in the same caves. These results are consistent with the similarity plot analysis showing that RaTG13 and BANAL-52 bat coronaviruses exhibit high nucleotide identity with SARS-CoV-2 throughout the length of the genome (96.8% for BANAL-52 and 96.1% for RaTG13). Notably, BANAL-52 has a higher level of nucleotide conservation than RaTG13 in the S1 domain of the spike protein, and especially in the amino-terminal domain (NTD) and RBD of the spike protein (Fig. 1c). These observations are congruent with amino acid identities between human SARS-CoV-2 and representative bat and pangolin coronaviruses, which have a high level of conservation, except for the open reading frame 8 (ORF8) of bat BANAL-116, BANAL-247, Rc-o319 and RmYN02 (Supplementary Fig. 1). The S1 domain of the spike protein (and especially the NTD) has a lower degree of conservation in several bat coronaviruses, suggesting that this domain may reflect a relative degree of adaptation of the virus to its mammalian host (Fig. 1d and Extended Data Fig. 1).
Bat Sarbecovirus evolutionary history
Following analysis using a genetic algorithm for recombination detection (GARD), we identified 14 recombinant breakpoints during the evolutionary history of sarbecoviruses, which were further confirmed by phylogenetic analyses performed on the 15 fragments of sequences defined by the breakpoints (Fig. 2 and Supplementary Fig. 2). No specific signature was identified in the breakpoints (Supplementary Table 5). SARS-CoV-2 has a mosaic genome, to which more than five sequences close to sequences published or determined during this study contributed: R. malayanus RmYN02 and R. pusillus RpYN06 viruses found in China in 2019, R. affinis RaTG13 coronavirus found in China in 2013, and R. malayanus BANAL-52 and R. pusillus BANAL-103 found in northern Laos in 2020 (this study). No pangolin coronavirus sequence was immediately associated with a recombination event at the origin of SARS-CoV-2. Laotian Rhinolophus bat coronaviruses had a lower degree of recombination compared to SARS-CoV-2. Such recombination events occurred between other BANAL viruses isolated from bats living sympatrically in caves in the same area.

Representation of the 15 recombinant fragments of relevant Sarbecovirus genomes compared to the SARS-CoV-2 human prototype strain (NC_045512). The coordinates of the breakpoints refer to the nucleotide position in the alignment. Where possible, the closest viral sequence is indicated for each fragment. In other cases, MULT indicates a group of multiple sequences. The asterisk marks unresolved fragment phylogeny (fragment 13, from positions 27,344 to 27,800 in the alignment). Sequences are coloured as in Fig. 1. The complete phylogenetic analyses are presented in Supplementary Fig. 2.
Notably, the origin of several fragments of SARS-CoV-2 genomes could be assigned to several donor strains and not a unique donor sequence. For example, a breakpoint was identified seven nucleotides upstream of the RBD region of S1: the downstream fragment of SARS-CoV-2, which comprises the RBD and the beginning of S2, could involve the R. malayanus BANAL-52, R. pusillus BANAL-103 and R. marshalli BANAL-236 viruses, which formed a highly supported sister clade of SARS-CoV-2 (fragment 11, Supplementary Fig. 2). In a more basal position are R. shameli bat coronaviruses and pangolin-2019 coronaviruses, consistent with the conservation of RBD amino acid sequences among SARS-CoV-2 and representative bat and pangolin coronaviruses (Extended Data Fig. 2). Among the 17 residues that interact with hACE2, 16 are conserved between SARS-CoV-2 and BANAL-52 or BANAL-103 (one mismatch, H498Q), and 15/17 are conserved for BANAL-236 (two mismatches, K493Q and H498Q) whereas only 13/17 residues are conserved for the Cambodian bat R. shameli virus and 11/17 for the Chinese bat R. affinis RaTG13 virus. At the full spike protein level, bat R. affinis RaTG13 and pangolin-2017 P4L viruses seemed closer to SARS-CoV-2 than bat R. malayanus BANAL-52, but this effect is due to a higher degree of conservation in S2. All of these viruses shared the absence of a furin cleavage site and the conservation of the internal fusion peptide (Supplementary Figs. 1 and 3 and Extended Data Fig. 3).
Interaction of BANAL RBDs with ACE2
Biolayer interferometry experiments to measure the interaction between hACE2 and the RBDs of BANAL-52/103 (which have identical residues in the receptor-binding motif; Extended Data Fig. 2), BANAL-236 and SARS-CoV-2 (residues 233–524) resulted in a dissociation constant Kd three times lower for the BANAL RBDs compared to SARS-CoV-2 (Fig. 3a). This higher affinity can be attributed to the Q498H mismatch that has been reported to increase the affinity of the SARS-CoV-2 RBD for hACE2, and also to be involved in the host range expansion of SARS-CoV-2 and SARS-CoV-2-like viruses22,23,24,25,26,27,28,29.

a, Biolayer interferometry (BLI) binding analysis of the hACE2 peptidase domain to immobilized BANAL-52/103, BANAL-236 or SARS-CoV-2 RBDs. Black lines correspond to a global fit of the data using a 1:1 binding model. b, Frequency of formation of salt bridges close to the RBD–ACE2 interface (from left to right: D30/K417, E35/K493, D38/K493, K31/E35 and D38/K353) during the course of the MD simulations. The analysis was performed for nine different MD simulations (three replicates for each complex) of hACE2 in complex with RBDs from SARS-CoV-2 (SARS-CoV-2.1, SARS-CoV-2.2 and SARS-CoV-2.3, shades of green), BANAL-236 (BANAL 236-CoV.1, BANAL 236-CoV.2 and BANAL 236-CoV.3, shades of red) and BANAL-52/103 (BANAL 52/103-CoV.1, BANAL 52/103-CoV.2 and BANAL 52/103-CoV.3, shades of blue). c, Ribbon representations of the crystal structures of the hACE2 peptidase domain (cyan) in complex with SARS-CoV-2 (PDB 6M0J) or BANAL-236 (this study, PDB 7PKI) RBDs (pink). Black arrows in the overall structures indicate the structural difference between the two complexes at the level of helix H4. The magnifications show the main interactions in the ACE2–RBD interfaces. Residues in the receptor-binding motif altered between SARS-CoV-2 and BANAL-236 are highlighted with coloured outlines. Numbers 1, 2 and 3 indicate the three main clusters of interactions between hACE2 and RBDs.
To study the effect of the alterations at the interface between these RBDs and hACE2, we performed molecular dynamics (MD) simulations of the complexes between the RBDs of SARS-CoV-2, BANAL-236 and BANAL-52/103 and hACE2 initiated from the crystal structure and homology models of these systems, respectively (Supplementary Table 6). Cluster analysis of the MD trajectories showed that, at the RBD–hACE2 interface, both BANAL complexes were identical to the complex between the SARS-CoV-2 RBD and hACE2 within 2 Å backbone root mean square deviation (RMSD; Extended Data Fig. 4), except for one of the BANAL-52/103 simulations that exhibited larger conformational variability of the RBD residues S443–Y449 (Supplementary Methods and Extended Data Fig. 5). Empirical scoring functions predicted a similar RBD–hACE2 binding energy in all three complexes (Extended Data Fig. 6).
The analysis of the persistence of hydrogen bonds and salt bridges provided further insights into the effect of the substitutions at the RBD–hACE2 interface (Fig. 3b and Extended Data Fig. 7). The H498Q mismatch present in both BANAL-52/103 and BANAL-236 RBDs disrupted the hydrogen bonds between RBD Q498 and both hACE2 K353 and Q42. However, these hydrogen bonds were only transiently formed in the SARS-CoV-2 complex: more persistent hydrogen bonds in this region (RBD T500 to hACE2 D355; RBD G502 to hACE2 K353; and RBD Y505 to hACE2 E37) were not affected. The K493Q mismatch enabled the formation of two salt bridges between RBD and hACE2 that were not present in the SARS-CoV-2 complex (RBD K493 to hACE2 E35 and RBD K493 to hACE2 D38).
For further insight into the molecular details of these interactions, we determined the crystal structure of the complex between the BANAL-236 RBD and the hACE2 peptidase domain to 2.9 Å resolution (Supplementary Table 7). The overall structure of this RBD is identical to that of SARS-CoV-2 (RMSD 0.360 Å, 150 Cα). The only significant difference is in the region between amino acids D363 and S375 (Fig. 3c, arrow). In this region, BANAL spikes have the A372T mismatch, which converts the sequence 370NSA372to 370NST372, an N-linked glycosylation sequon. The crystals indeed showed clear electron density for the first N-acetylglucosamine residue of glycan attached to N370. The main chain at residue T372 makes a hydrogen bond with the glycan residue, altering the conformation of the main chain downstream, which results in partial unwinding of helix H4 in RBD 236. The calculation of a simulated annealing composite omit map for the segment D363–S375 confirms the correct assignment of the structure for this polypeptide segment and the N-glycosylation at residue N370 (Extended Data Fig. 8).
As expected, most of the interactions observed in the SARS-CoV-2 RBD–hACE2 complex30 are also present in the structure of the complex between the BANAL-236 RBD and hACE2. In these interfaces, there are three main clusters of interactions as indicated in Fig. 3c (insets). The sequence mismatches are in clusters 2 (making the salt bridge between RBD K493 and hACE2 E35) and 3 (hydrogen bond between RBD H498 and D38). Although the interaction K493–E35 contributes to stabilizing the complex, it does not seem to markedly affect the binding to hACE2 because both BANAL RBDs have similar Kd values.
Virus replication in human cells
To assess whether the BANAL-236 spike protein could mediate entry into cells expressing hACE2, we generated lentiviral particles pseudotyped with spike from the SARS-CoV-2 strain first detected in Wuhan or BANAL-236 (Supplementary Fig. 4). We detected spike-mediated entry of the BANAL-236 spike-pseudotyped lentivirus into hACE2-expressing HEK-293T cells, but no such entry into control cells not expressing hACE2 (Fig. 4a). Entry was blocked by human sera neutralizing SARS-CoV-2, but not by control non-neutralizing sera, demonstrating that neutralization of BANAL-236 was specific for epitopes shared with the spike protein of SARS-CoV-2 (Fig. 4b).

a, Results of spike-pseudotyped BANAL-236 (squares) and SARS-CoV-2 (strain BetaCoV/France/IDF0372/2020, GISAID accession number EPI_ISL_406596, diamonds) pseudovirus entry assay in HEK-293T cells expressing (purple lines) or not (grey lines) the hACE2 receptor, shown in relative luminescence units (RLUs) produced by the firefly luciferase present in the lentiviral backbone and the Bright-Glo luciferase substrate. A single experiment performed in triplicate representative of two experiments is shown. Centre values represent the average of the three replicates and error bars indicate s.d. b, Results of spike-pseudotyped BANAL-236 (black) and SARS-CoV-2 (strain BetaCoV/France/IDF0372/2020, grey) neutralization assay expressed as a percentage of neutralization of luciferase activity in the absence of serum. Sera neutralizing SARS-CoV-2 were from patients with confirmed infections whereas non-neutralizing sera samples were collected before the spread of SARS-CoV-2 Laos. The dashed line marks the neutralization threshold. A single experiment representative of three independent experiments is shown. c, Human cell lines expressing endogenous ACE2, Calu-3 (blue lines) and Caco-2 (green lines), were infected at an MOI of 0.01 with the BANAL-236 virus (squares) and the virus first detected in Wuhan (diamonds). VeroE6 cells were infected at an MOI of 0.0001 with BANAL-236 virus (squares) and SARS-CoV-2 (strain BetaCoV/France/IDF0372/2020, diamonds) pre-incubated with (grey lines) or without (pink lines) soluble hACE2 (sACE2) at 25 µg ml−1 for 30 min. Genome copy number was determined by quantitative RT-PCR in the supernatants recovered 3 and 4 days post-infection. A single experiment performed in triplicate is shown. Centre values represent the average of the three replicates and error bars indicate s.d.
To isolate infectious viruses, rectal swabs were inoculated on VeroE6 cells. No cytopathic effect (CPE) was observed 3 and 4 days after infection, but viral RNAs were detected for one of the two wells inoculated with the BANAL-236 sample (cycle threshold (Ct) = 25.1 at day 3, Ct = 21.7 at day 4). The culture supernatant (C1) formed plaques on VeroE6 and the titre was 3,800 pfu ml−1. A C2 viral stock was prepared by amplification on VeroE6 at a multiplicity of infection (MOI) of 10−4. The culture supernatant was collected on day 4 when CPE was observed and titrated on VeroE6 (Extended Data Fig. 9). The plaques’ phenotype was small, but the titre reached 2.6.106 pfu ml−1. The random NGS performed on the RNA extracted from this stock confirmed that the culture was pure and corresponded to the BANAL-236 virus, without any non-synonymous mutations between the original BANAL-236 genome and the C2 viral stock. Replication of BANAL-236 in VeroE6 was efficiently inhibited by soluble hACE2, thus showing that entry and propagation was largely ACE2 dependent (Fig. 4c and Extended Data Fig. 9). Furthermore, BANAL-236 replicated in human cell lines expressing endogenous levels of ACE2, Calu-3 and Caco-2 (Fig. 4c). The kinetics of RNA synthesis were slower compared to those for SARS-CoV-2. Infectious viral particles were produced at day 4 in the supernatant of Caco-2 and Calu-3 cells (respectively 104.7 and 102.9 pfu ml−1).
Discussion
Many sarbecoviruses circulate in Rhinolophus colonies living in caves in China and probably also in neighbouring countries further south31,32,33. During the course of a prospective study in northern Laos, we have identified, among other coronaviruses, five sarbecoviruses for which we obtained full-length sequences. Among these, three (BANAL-52, BANAL-103 and BANAL-236) were considered to be close to SARS-CoV-2 because of the similarity of one of the S1 domains (NTD or RBD) or S2 to that of SARS-CoV-2.
As genomic regions subject to recombination are probably contributing to host–virus interactions, we compared SARS-CoV-2 strains from the two lineages identified at the onset of the coronavirus disease 2019 (COVID-19) outbreak21 to these new bat sarbecoviruses and to pangolin strains in the SARS-CoV-2 clade. Strains close to R. pusillus RpYN06, R. malayanus RmYN02 and Rhinolophus sp. PrC31 isolated in China in 2018–2019, along with R. malayanus BANAL-52, R. pusillus BANAL-103 and R. marshalli BANAL-236 isolated in Laos in 2020, contributed to the appearance of SARS-CoV-2 in different regions of the genome. No closer viral genome has yet been identified as a possible contributor, and pangolin coronaviruses seem more distantly related than bat coronaviruses. We identified potential recombination sites, allowing for the reconstruction of the phylogenetic history of early isolated SARS-CoV-2 strains between homologous regions defined by recombination points. We identified a breakpoint at the beginning of the SARS-CoV-2 RBD, resulting in a downstream fragment key for the virus tropism and host spectrum composed of the RBD and the furin cleavage site, and ending in the N-terminal region of S2. Despite the absence of the furin site, phylogenetic reconstruction of this fragment showed that the Laotian R. malayanus BANAL-52, R. pusillus BANAL-103 and R. marshalli BANAL-236 coronaviruses are the closest ancestors of SARS-CoV-2 known so far. ORF8 was highly divergent between SARS-CoV-2 related genomes. The ORF8 genes from the strains BANAL-52, BANAL-103 and BANAL-236, like that of RaTG13, were closer to that of SARS-CoV-2 than to those of pangolin strains. ORF8 encodes a protein that has been proposed to participate in immune evasion34 and is deleted in many human SARS-CoV-2 strains that appeared after March 202035, which is reminiscent of the deletions identified during the 2003 severe acute respiratory syndrome epidemic36. Therefore, the presence of ORF8 is consistent with bats acting as a natural reservoir of early strains of SARS-CoV-2.
Structural and functional biology studies have identified the RBD domain that mediates the interaction with hACE2 and host range, as well as the main amino acids that are involved30,37,38. The RBDs (BANAL-52, BANAL-103 and BANAL-236) are closer to that of SARS-CoV-2 than that of any other bat strain described so far, including that of RaTG13, the virus identified in R. affinis from the Mojiang mineshaft where pneumonia cases with clinical characteristics a posteriori interpreted as similar to COVID-19 (ref. 6) were recorded in 201239,40. Overall, one (H498Q (BANAL-103 and BANAL-52)) or two (K493Q and H498Q (BANAL-236)) amino acids interacting with hACE2 are substituted in these strains in comparison to SARS-CoV-2. These substitutions did not destabilize the interface between BANAL-236 and hACE2, as shown by the biolayer interferometry experiments (Fig. 3a) and analysed by MD simulations.
Our results therefore support the hypothesis that SARS-CoV-2 could originally result from a recombination of sequences pre-existing in Rhinolophus bats living in the extensive limestone cave systems of Southeast Asia and South China41,42. Many species forage in the same cave areas, including R. malayanus and R. pusillus43. In addition, the distributions of R. marshalli, R. malayanus and R. pusillus overlap in the Indochinese subregion (Supplementary Fig. 5), which means that they may share caves as roost sites and foraging habitats44. With the new viruses described here, understanding the emergence of SARS-CoV-2 does not require speculation of recombination or natural selection for increased RBD affinity for hACE2 in an intermediate host such as the pangolin before spillover45, nor natural selection in humans following spillover46. However, we found no furin cleavage site in any of these viruses on sequences determined directly from original faecal swab samples, which prevent from any risk of counterselection of the furin site by amplification in Vero cells18. The lack of the furin cleavage site may be explained by insufficient sampling in bats. On the basis of comparison of the sequences around the cleavage site between S1 and S2 (Extended Data Fig. 3), it has been suggested that the furin cleavage site in SARS-CoV-2 could originate from recombination events between SARS-CoV-2-related coronaviruses co-circulating in bats2,47, meaning that BANAL-116, BANAL-247, bat RmYN02 (ref. 2) and bat RacCS203 (ref. 3) coronaviruses may share a common history with SARS-CoV-2. Alternatively, the furin cleavage site could have been acquired through passages of the virus in an alternative host or during an early poorly symptomatic unreported circulation in humans. Finally, the epidemiological link between these bat viruses and the first human cases remains to be established.
As expected from the high affinity for hACE2 of the S ectodomain of BANAL-236, pseudoviruses expressing it were able to efficiently enter human cells expressing endogenous hACE2 using an ACE2-dependent pathway. However, alternative routes of entry may still exist, especially in cells that do not express ACE2 (ref. 48). Entry was blocked by a serum neutralizing SARS-CoV-2. The RaTG13 strain, the closest to SARS-CoV-2 known previously, had never been isolated. By contrast, preliminary studies show that BANAL-236 replicated in primate VeroE6 cells with a small plaque phenotype compared to that of SARS-CoV-2. Further analysis may indicate more clearly which steps shape infectivity.
To conclude, our results pinpoint the presence of new bat sarbecoviruses that seem to have the same potential for infecting humans as early strains of SARS-CoV-2. Guano collectors, certain ascetic religious communities who spend time in or very close to caves and tourists visiting caves are particularly at risk of being exposed. Further investigations are needed to assess whether such exposed populations have been infected, symptomatically or not, by one of these viruses, and whether infection could confer protection against subsequent SARS-CoV-2 infections. In this context, it is noteworthy that SARS-CoV-2 with the furin site deleted replicates in hamsters and in transgenic mice expressing hACE2, but leads to less severe disease and protects from rechallenge with wild-type SARS-CoV-2 (ref. 18).
Methods
Ethical and legal statements
The bat study was approved by the wildlife authorities of the Department of Forest Resource Management (DFRM), and the Ministry of Agriculture and Forestry (MAF), Lao People’s Democratic Republic, no. 2493/DFRM, issued on 21 May 2020 and no. 0755/MAF issued on 2 June 2020. All animals were captured, handled and sampled following previously published protocols and ASM guidelines52,53. Exportation from Laos and importation in France were conducted according to national regulations. Human serum samples used for neutralization assays were already available54 and selected on the basis of their status regarding seroneutralization of SARS-CoV-2. They were collected following a protocol approved by the Lao National Ethics Committee for Health Research (NECHR; reference no. 052/2020).
Biosafety
Both the Institut Pasteur du Laos (IPL) and the Faculty of Environmental Science have extensive experience in safely collecting bats (appropriate biosafety training and personal protective equipment (PPE) for collectors). In this study, training before field work was organized once for field work participants. The aim was to teach participants the transmission risks of infectious agents from bats and how to identify, assess and mitigate these risks, as well to practice the use of PPE.
During field collection, sampling stations were selected to minimize potential exposure to infectious agents and stress on the animals during the handling time by selecting: an area easy to disinfect; an area out of view of the general public; a location that would not cause exposure of the general population (such as a picnic area); and procedures that reduce time and stress on bats caused by handling. For handling bats and bat samples, the following minimum PPE was required: eye protection, an N95 respirator, long clothing/coverall and latex gloves (2 pairs). All waste was disposed of in biohazard bags and was transported to a disposal site of IPL in Vientiane capital. Each sample box containing 81 cryovial tubes was placed inside an individual plastic ziplock and transported from the field to IPL on dry ice in a polystyrene foam box. At IPL, samples were stored in a specific −80 °C freezer until analysis.
For initial sample analysis at IPL, samples were transferred to a BSL-3 room for nucleic acid extraction. Initial coronavirus screening by nested RT-PCR was then performed under internal regulation on biosafety and security of IPL. Sample-extracted products (50 µl) were stored in NucleoSpin 8 sample boxes (8 × 12) and sent to IP-Paris packed in individual plastic ziplock bags on dry ice. Aliquots of anal swab samples were sent individually triple packaged on dry ice in a separate box.
All experiments on potentially infectious samples performed at Institut Pasteur (Paris) were conducted in BSL-3 laboratories according to procedures adapted for respiratory viruses.
Bat sampling areas and sample collection
Trapping sessions were conducted at four sites, in the Fueng and Meth districts, Vientiane province, and in the Namor and Xay districts, Oudomxay province, between July 2020 and January 2021 (Fig. 1a and Supplementary Tables 1 and 2). Bats were captured using four-bank harp traps55 and mist nets set in forest patches between rice fields or orange/banana plantations and karst limestone formations, for 5–8 nights depending on accessibility. Harp traps were set across natural trails in patches of forest understorey. Mist nets were set across natural trails, at the edges of forests, at entrances of caves and in areas near cave entrances, as well as in open areas or those with high forest canopy. Bats were morphologically identified following morphological criteria55,56,57. Other data such as forearm length (FA), sex, developmental stage (adult or juvenile) and reproductive condition (pregnant or lactating) were also recorded. Bats were sampled for saliva, faeces and/or urine, and blood before release at the capture site. Species identification of PCR-positive individuals was confirmed by sequencing the mitochondrial cytochrome oxidase 1 (ref. 20).
Initial coronavirus screening
Total RNA was extracted from faecal samples using the NucleoSpin 8 virus kit (Macherey Nagel). cDNA was synthesized using the Maxima H minus first strand cDNA synthesis kit (Thermo Scientific) and random hexamers following the manufacturer’s instructions. The presence of coronaviruses was tested by a nested RT-PCR approach using PCR master mix (Promega) and by targeting the RNA-dependent RNA Polymerase gene using combinations of degenerate and non-degenerate consensus primers as previously described20. PCR products of the expected size were directly sequenced on both strands by Sanger sequencing using the nested PCR primers. The sequences obtained were confirmed by similarity analysis using the NCBI BLASTn search (http://www.ncbi.nlm.nih.gov/BLAST).
Primer design for Betacoronavirus enrichment before NGS
Betacoronavirus enrichment was performed at the genus level by adapting a previously described protocol58 based on k-mers for targeted-sequence enrichment before NGS. Briefly, 2,000 complete Betacoronavirus genomes were downloaded from the GenBank and GISAID databases and then clustered to a 95% sequence identity using CD-HIT-EST (ref. 59). Overall, 185 representative sequences of all betacoronaviruses were used for further analysis. Owing to the high diversity in the genus Betacoronavirus, the full genomes belonging to the subgenera Sarbecovirus, Nobecovirus, Merbecovirus and Embecovirus were separately aligned using MAFFT multiple sequence alignment software and used to design 13-mer spiked primers for each cluster. The genomic position of the full set of primers was extracted from some representative subgenera sequences and close primers were removed. Finally, 416 spiked primers were synthesized by Eurofins Genomics.
Sample preparation for sequencing
Reverse transcription was performed using the mix of spiked primers and random hexamers at a 10:1 ratio using the SuperScript IV First-Strand Synthesis System (Invitrogen). After a denaturation step at 95 °C for 3 min in the presence of dNTPs (500 µM), DTT (5 mM) and RNaseOUT inhibitor, the reverse transcription reaction was incubated for 10 thermal cycles consisting of 6 steps at 8 °C for 12 s, 15 °C for 45 s, 20 °C for 45 s, 30 °C for 30 s, 35 °C for 2 min and 42 °C for 3 min, followed by a final incubation step at 42 °C for 20 min, as previously described60. Double-stranded cDNA was generated using the Sequenase 2.0 DNA Polymerase kit (Applied Biosystems) in the presence of dNTPs and then purified using the Beckman Coulter Agencourt AMPure XP.
For samples with a low nucleic acid content, a random amplification step was performed using the MALBAC Single Cell WGA kit (Yikon Genomics, Promega). The amplified product was then purified using AMPure XP Beads, eluted in a final volume of 20 μl of low TE (10 mM Tris-HCl (pH 8,0), 0,1 mM EDTA), and quantified with the Qubit DNA HS Assay (Life Technologies, Thermo Fisher Scientific).
Libraries were generated using the NEBNext Ultra II DNA Library Prep kit (New England Biolabs) after a fragmentation step using the Covaris M220 Focused-ultrasonicator using microTUBE-15 (peak incident power = 18 W, duty factor = 20%, cycles per burst = 50, treatment time = 60 s). The PCR-amplified libraries were cleaned up using 0.9× AMPure XP Beads and checked on the 2100 Bioanalyzer system with the High-Sensitivity DNA kit (Agilent Technologies) and quantified with the Qubit DNA HS Assay. Finally, the dual-multiplexed libraries were pooled (six samples per pool) and run on the Illumina NextSeq500 platform with High Output Kit version 2.5 (150 cycles).
Amplicon sequencing
In addition to enrichment-based sequencing, cDNA was amplified using the AmpliSeq for Illumina SARS-CoV-2 Research Panel (catalogue number 20020496), applying 26 amplification cycles in PCR1 and 9 cycles in PCR2. Primers at the end of the amplicons were partially digested during the library preparation, following the manufacturer’s instructions. Libraries were barcoded individually using the Illumina UD dual indices and normalized with the AmpliSeq Library Equalizer for Illumina (catalogue number 20019171), and then pooled and sequenced on the Illumina NextSeq500 instrument using a Mid Output version 2.5 kit (SR 150 cycles). To eliminate residual PCR primer sequences, raw reads were trimmed by 15 bases at each end and special attention was paid to checking that internal sequences corresponding to primer regions in overlapping amplicons did not derive from primer sequences of the multiplex PCR. As an internal control, we verified that sequences of the complete genome of sample BANAL-236 obtained from the enrichment-based sequencing approach and from the AmpliSeq approach were identical.
Genome assembly and finishing
Raw reads from the enrichment-based sequencing were processed with an in-house bioinformatics pipeline (Microseek; Bigot T. et al., unpublished observations) comprising quality check and trimming, read normalization, de novo assembly, and ORF prediction of contigs and singletons, followed by three levels of taxonomic assignation61. Sequences identified as Sarbecovirus were then mapped onto appropriate reference sequences using CLC Genomics Workbench 20.0 (Qiagen). Trimmed reads from the amplicon sequencing were mapped to the SARS-CoV-2 genome first, and then mapped again (refined mapping) to the closest genome relative. When needed, complete genomes were obtained by conventional PCR and Sanger sequencing. Briefly, viral RNA was reverse transcribed using SuperScript IV reverse transcriptase (Invitrogen) and cDNA was subsequently used to fill the gaps in the genomes using Phusion High Fidelity DNA polymerase (New England Biolabs) and specific primers flanking the missing regions. Positive PCR products were further purified and sequenced by Sanger sequencing at Eurofins Genomics.
Recombination and phylogenetic analyses
Identification of recombination events occurring during the evolutionary history of bat sarbecoviruses was performed using the IDPlot package62, a web-based workflow that includes multiple sequence alignment and phylogeny-based breakpoint prediction using the GARD algorithm from the HyPhy genetic analysis suite63. First, a comprehensive analysis comprising 106 sequences and covering all non-human Sarbecovirus and Sarbecovirus-related complete genomes available in GenBank and GISAID databases was performed, including prototype strains of SARS-CoV-2 isolated in 2019. Special attention was paid to including bat-borne and pangolin-borne viral sequences to maximize the ability to capture a large diversity of the sarbecoviruses. Then, a reduced set of 36 sequences was chosen because of their phylogenetic proximity to SARS-CoV-2, and the GARD algorithm was run to identify recombination breakpoints in Laotian and representative human, bat and pangolin sarbecoviruses. Breakpoint coordinates were confirmed by performing phylogenetic analyses on the corresponding fragments using PhyML implemented through the NGPhylogeny portal50. Branch support was evaluated with the aBayes parameter.
Generation of lentiviral pseudoviruses
The synthetic genes encoding spike in BANAL-236 and in SARS-CoV-2 (strain BetaCoV/France/IDF0372/2020, GISAID accession number EPI_ISL_406596) were cloned into the pVAX1 vector with a cytoplasmic tail truncation of 19 amino acids. Pseudotyped lentiviral particles were prepared using HEK-293T cells (ATCC CRL-3216) seeded in 10-cm dishes. HEK-293T cells were co-transfected with 5 µg of spike-encoding plasmid, 10 µg of lentiviral backbone plasmid expressing the firefly luciferase (pHAGE-CMV-Luc2-IRES-ZsGreen-W) and 3.3 µg of each lentiviral helper plasmid expressing HIV Gag-Pol (HDM-Hgpm2), Tat (HDM-tat1b) and Rev (pRC-CMV-Rev1b) using calcium phosphate precipitation64. The medium was replaced 5 h post-transfection by 6 ml of DMEM without fetal calf serum (FCS) and phenol red. Pseudotyped particles were collected 48 h post-transfection, clarified by centrifugation at 2,500g for 5 min and frozen at −80 °C. Mock pseudotyped lentivirus was generated as above but in the absence of an S-expressing plasmid.
Spike-pseudotyped lentivirus entry assays
HEK-293T cells stably expressing hACE2 were transduced in suspension by mixing 50 µl of threefold serial dilutions of S-pseudotyped lentiviruses with 50 µl of cells at 4 × 105 cells per ml in 96-well white culture plates65. At 60–72 h post-transduction, 100 µl of Bright-Glo luciferase substrate (Promega) was added to the wells and luminescence was measured using a Berthold Centro XS luminometer.
Neutralization assays
Sera neutralizing SARS-CoV-2 and non-neutralizing sera were described in ref. 54. They were decomplemented at 56 °C for 30 min and 2.5 µl was incubated with 0.5 µl of S-pseudotyped lentiviruses in a final volume of 50 µl DMEM–10% FCS without phenol red in 96-well white culture plates. After 30 min at room temperature, 50 µl of hACE2-expressing HEK-293T cells in suspension at 4 × 105 cells per ml was mixed into the wells. Luminescence was measured at 60–72 h post-transduction as described above. Neutralization was calculated using the following formula: 1 − (RLU in presence of serum/(mean of RLU in absence of serum determined in 12 wells − 3 × s.d.).
Virus isolation and multiplication
Rectal swabs were inoculated in duplicate in 24-well plates containing VeroE6 cells (ATCC CRL-1586) (1/5 dilution in 100 µl DMEM without FCS supplemented with 1% penicillin–streptomycin, 1% Fungizone and 1 µg ml−1 TPCK-treated trypsin). After 1 h of adsorption at 37 °C, inoculum was removed and 1 ml of the medium described above was added. At 3 and 4 days after infection, CPE was monitored and 100 µl of supernatant was collected for RNA extraction. Quantitative RT-PCR targeting a conserved sequence in the E gene was performed as described previously66. Culture supernatant (C1) was collected at day 4 and titrated by plaque assay on VeroE6 overlaid with 0.5% carboxymethylcellulose containing 1 µg ml−1 TPCK-treated trypsin. A viral stock was prepared by amplification on VeroE6 cells at an MOI of 10−4. Culture supernatant (C2) was collected at day 4 when a massive CPE was observed and titrated by plaque assay on VeroE6 as described above. A C3 viral stock was produced by a subsequent viral amplification at an MOI of 10−4 for 3 days on VeroE6. RNA was extracted from the viral stock and submitted to random NGS analysis using the SMARTer Stranded Total RNA-Seq Kit version 3 - Pico Input Mammalian (Takara Bio). Raw reads were processed with the Microseek pipeline, as described above.
For infection experiments, VeroE6 or the human cell lines Calu-3 (lung cells, ATCC HTB-55) and Caco-2 (intestinal cells, ATCC HTB-37) were infected in triplicate in 24-well plates with viral stocks of BANAL-236 (C3) or SARS-CoV-2 (strain BetaCoV/France/IDF0372/2020) at an MOI of 0.0001 and 0.01, respectively. Human soluble ACE2 was pre-incubated at 25 µg ml−1 (ref. 67) for 30 min with the viral inoculum before VeroE6 infection. Infections were carried out without TPCK in the medium described above for 3 and 4 days. Supernatants were recovered at 0, 3 and 4 days post-infection and genome copy number and viral titres were quantified as described above.
Protein expression and purification
BANAL-52/103, BANAL-236 and SARS-CoV-2 RBDs (residues 233–524, with carboxy-terminal 8×His-Strep and AVI tags) and hACE2 peptidase domain (residues 19–615, with C-terminal 8×His tag) were expressed in Expi293F cells at 37 °C and 8% CO2 (GnTI- Expi293, ThermoFisher Scientific). Cell culture supernatants were collected 5 days post transfection and purified by affinity chromatography followed by size-exclusion chromatography (SEC) using a 200 10/300 GL column pre-equilibrated in 20 mM Tris-HCl pH 8.0, 100 mM NaCl.
For crystallization experiments, the same constructs were expressed in Expi293F GnTI cells. The protein tags were cleaved overnight with thrombin and deglycosylated with EndoH. The RBD was mixed with a 1.3 molar excess of hACE2 and the complex was purified by SEC.
Biolayer interferometry
Purified Avi-tagged RBD was biotinylated using a BirA biotin–protein ligase kit according to the manufacturer’s instructions (Avidity). The biotinylated RBDs at 100 nM were immobilized to SA sensors. A 1:2 dilution series of hACE2 starting at 100 nM in PBS–BSA buffer was used in cycles of 200-s association followed by 200-s dissociation steps to determine protein–protein affinity. The data were baseline-subtracted and the plots were fitted using the Pall FortéBio/Sartorius analysis software (version 12.0). Data were plotted in Prism 9.1.0.
Crystallization and data collection
Crystals of the complex BANAL-236 RBD–hACE2 were obtained at 4 °C in sitting drops by mixing 200 nl of the protein complex at 8 mg ml−1 with 200 nl of reservoir solution containing 0.2 M lithium sulfate, 0.1 M Tris 8.5, 30% w/v PEG 4000. The crystals were soaked in reservoir solution containing 20% glycerol as a cryoprotectant before being flash-frozen in liquid nitrogen. X-ray diffraction data were collected on the beamline PROXIMA 1 at the SOLEIL synchrotron (St Aubin, France) and reduced using the XDS package68. The structure of the complex was determined by molecular replacement with Phaser software69 using the coordinates of the SARS-CoV-2 RBD in complex with hACE2 as a search template (Protein Data Bank (PDB) 6M0J). The model was manually corrected in COOT (ref. 70) and refined with phenix.refine (ref. 71). The final coordinates were deposited in the PDB with the accession code 7PKI.
MD simulations of RBD–hACE2 complexes
Generation of homology models
Homology models of the complexes between the RBDs of BANAL-236 and BANAL-52/103 and hACE2 were constructed using the X-ray structure of the complex between the SARS-CoV-2 RBD and hACE2 (PDB code 6M0J; resolution 2.45 Å) as a template using MODELLER version 10.1 (ref. 72). These models included the fragments S19–D615 and T333–G526 of hACE2 and RBD, respectively, which were the regions resolved in the template. In these regions, BANAL-236 and BANAL-52/103 RBDs have a sequence identity to SARS-CoV-2 RBD equal to 96.9% and 97.4%, respectively. The alignment reported in Extended Data Fig. 3 was used. The zinc and chloride atoms in the template were retained during homology modelling; N-acetylglucosamine (NAG) and water residues were removed. Seven disulfide bonds were detected by MODELLER in the template (three in hACE2 and four in the RBD) and enforced in the generation of the homology models using CHARMM-like distance and dihedral angles restraints. For each construct, 100 homology models were built and ranked on the basis of the normalized DOPE score73. The top three scoring models of each complex were used as starting points of three independent MD simulations, as described in the following section.
Set-up, equilibration and production of the MD simulations
The six homology models described in the previous section along with the X-ray structure of the SARS-CoV-2 RBD–hACE2 complex (PDB code 6M0J) were used as input to the CHARMM-GUI server74. In the case of the X-ray structure, for consistency with the homology models, the zinc and chloride atoms in 6M0J were retained whereas the NAG and water residues were removed. The seven systems were solvated in a triclinic box of initial xyz dimensions of ~13.5 nm × 9.2 nm × 8.3 nm. Potassium and chloride ions were added to ensure charge neutrality at a salt concentration of 0.15 M. The total number of atoms was ~104,000. Further details of the systems are reported in Supplementary Table 4. The CHARMM36m force field75 was used for the protein and ions, and the TIP3P model76 was used for the water molecules. CHARMM36m force field parameters for the seven pairs of cysteines linked by disulfide bonds were used. The CHARMM-GUI models were first energy-minimized using the steepest descent algorithm. After minimization, the systems were equilibrated using a 1-ns-long simulation in the NPT ensemble followed by a 1-ns-long simulation in the NVT ensemble. The temperature T was set at 300 K and the pressure P was set to 1 atm using the Bussi–Donadio–Parrinello thermostat77 and the Berendsen barostat78, respectively. During equilibration, harmonic restraints on the positions of the protein backbone and sidechain heavy atoms were applied. For each system studied, production simulations were performed in the NVT ensemble for 1 µs. A time step of 2 fs was used together with LINCS constraints on hydrogen bonds79. The van der Waals interactions were gradually switched off at 1.0 nm and cut off at 1.2 nm; the particle mesh Ewald method was used to calculate electrostatic interactions with a cutoff at 1.2 nm (ref. 80). Production simulations were performed at room temperature for consistency with the biolayer interferometry experiments.
Details of the analysis
To evaluate the stability of the starting model during the production simulations, we calculated the backbone RMSD with respect to the energy-minimized structure for each frame of the trajectories. The RMSD was calculated separately for the residues in the RBD (Extended Data Fig. 4a), the hACE2 (Extended Data Fig. 4b), the RBD–hACE2 interface (Extended Data Fig. 4c) and the entire complex (Extended Data Fig. 4d). Interfacial residues were defined as the residues in one subunit closer than 0.8 nm to the residues in the other subunit in the X-ray structure of the complex between the SARS-CoV-2 RBD and hACE2. RMSD calculations were performed using the driver utility of PLUMED version 2.7 (ref. 81). To characterize conformational heterogeneity at the RBD–hACE2 interface, we performed a cluster analysis of the MD trajectories using as similarity metrics the backbone RMSD of the interfacial residues and the GROMOS clustering approach82 with a cutoff equal to 0.2 nm. The nine trajectories were first concatenated, clustering was then performed, and finally the population of each cluster was calculated separately for each trajectory (Extended Data Fig. 4e). To estimate the binding energy between RBD and hACE2, we used the InterfaceAnalyzer tool in ROSETTA version 3.11 (ref. 83; Extended Data Fig. 6a) and the AnalyseComplex tool in FoldX version 4 (ref. 84; Extended Data Fig. 6b). To identify relevant interactions at the RBD–hACE2 interface that could contribute to the binding affinity of the complex, we quantified the frequency of formation of inter-subunit salt bridges (Fig. 3b) and hydrogen bonds (Extended Data Fig. 7) during the course of the MD simulations. For each frame of the trajectories, we used PLUMED to calculate the distances between the side-chain charged groups of aspartic acids (OD1/OD2), glutamic acids (OE1/OE2), lysines (NZ) and arginines (NH1/NH2). An inter-subunit salt bridge was then defined as formed if the distance between groups with opposite charge was lower than 0.32 nm. We confirmed this calculation using the Salt Bridges tool available in VMD (ref. 85). To monitor the formation of inter-subunit hydrogen bonds, we used the Hydrogen Bond Analysis module of the MDAnalysis library version 1.0.0 (ref. 86). A donor–acceptor distance and angular cutoff of 0.3 nm and 150° were used to define the formation of a hydrogen bond. We confirmed this calculation using the Hydrogen Bonds tool available in VMD.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Sequence data that support the findings of this study have been deposited in the GenBank and GISAID (https://www.gisaid.org/) databases with the following accession numbers: MZ937000/EPI_ISL_4302644 (BANAL-52), MZ937001/EPI_ISL_4302645 (BANAL-103), MZ937002/EPI_ISL_4302646 (BANAL-116), MZ937003/EPI_ISL_4302647 (BANAL-236) and MZ937004/EPI_ISL_4302648 (BANAL-247). Raw sarbecovirus NGS reads and Sanger sequencing .ab1 files have been deposited in the National Center for Biotechnology Information Sequence Read Archive repository under the BioProject code PRJNA796968 (SAMN24959173–SAMN24959177). The crystal structure presented in this manuscript has been deposited in the PDB with the accession code 7PKI. The GROMACS topology and input files as well as the analysis scripts used are freely available on PLUMED-NEST (www.plumed-nest.org) under accession ID plumID:21.037. Source data are provided with this paper.
Code availability
MODELLER version 10.1 (https://salilab.org/modeller/) and GROMACS version 2020.4 (http://www.gromacs.org) were used for MD simulations. XDS (version 5 February 2021), CCP4 (v 7.0), COOT (version 0.8.6) and PHENIX (version 1.19.2-4158) software programs were used for X-ray diffraction data processing, model building and refinement of the complex between the BANAL-236 RBD and hACE2. PyMOL (version 2.4.2) was used for structural image rendering. PLUMED version 2.7 (https://www.plumed.org), ROSETTA version 3.11 (https://www.rosettacommons.org/), FoldX version 4 (http://foldxsuite.crg.eu/), MDAnalysis version 1.0.0 (https://www.mdanalysis.org/), MDTraj version 1.9.5 (https://www.mdtraj.org/1.9.5/index.html) and in-house scripts available at https://github.com/maxbonomi/bat-MD were used for MD simulations. hyphy 2.5.31, PhyML and MAFFT implemented through NGPphylogeny (https://ngphylogeny.fr/) and Simplot 3.5.1 were used for phylogenetic and recombination analyses. MSSPE 1 was used for Betacoronavirus primer enrichment design and is available at https://github.com/chiulab/MSSPE-design. MICROSEEK is an in-house pipeline that uses ALIENTRIMMER version 2.0 (https://gitlab.pasteur.fr/GIPhy/AlienTrimmer) for read trimming/clipping, BBNORM from BBMAP version 38.86 package (https://sourceforge.net/projects/bbmap/) for coverage normalization, MEGAHIT version 1.2.9 (https://github.com/voutcn/megahit) for assembly, an in-house ORF-finder (https://figshare.com/articles/code/translateReads_py/7588592), and then DIAMOND version 2.0.4 (https://github.com/bbuchfink/diamond/) and NCBI BLAST version 2.12.0+ (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.12.0/) both for sequence searching.
Change history
07 July 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41586-022-05048-7
References
Delaune, D. et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 12, 6563 (2021).
Zhou, H. et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 184, 4380–4391 (2021).
Wacharapluesadee, S. et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 12, 972 (2021).
Murakami, S. et al. Detection and characterization of bat sarbecovirus phylogenetically related to SARS-CoV-2, Japan. Emerg. Infect. Dis. 26, 3025–3029 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Rahalkar, M. C. & Bahulikar, R. A. Lethal pneumonia cases in Mojiang miners (2012) and the mineshaft could provide important clues to the origin of SARS-CoV-2. Front. Public Health https://doi.org/10.3389/fpubh.2020.581569 (2020).
Liu, P. et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 16, e1008421 (2020).
Xiao, K. et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286–289 (2020).
Wahba, L. et al. An extensive meta-metagenomic search identifies SARS-CoV-2-homologous sequences in pangolin lung viromes. mSphere 5, e00160-20 (2020).
Letko, M., Marzi, A. & Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020).
Wang, Q. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904 (2020).
Delaune, D. et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 12, 6563 (2021).
Jackson, B. et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 184, 5179–5188 (2021).
Rochman, N. D. et al. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl Acad. Sci. USA 118, e2104241118 (2021).
Liu, K. et al. Binding and molecular basis of the bat coronavirus RaTG13 virus to ACE2 in humans and other species. Cell 184, 3438–3451 (2021).
Aicher, S.-M. et al. Species-specific molecular barriers to SARS-CoV-2 replication in bat cells. Preprint at bioRxiv https://doi.org/10.1101/2021.05.31.446374 (2021).
Johnson, B. A. et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 591, 293–299 (2021).
Liu, K. et al. Cross-species recognition of SARS-CoV-2 to bat ACE2. Proc. Natl Acad. Sci. USA 118, e2020216118 (2020).
Chu, D. K. W. et al. Avian coronavirus in wild aquatic birds. J. Virol. 85, 12815–12820 (2011).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5, 1403–1407 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Laffeber, C., de Koning, K., Kanaar, R. & Lebbink, J. H. G. Experimental evidence for enhanced receptor binding by rapidly spreading SARS-CoV-2 variants. J. Mol. Biol. 433, 167058 (2021).
Lei, C. et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 11, 2070 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292 (2020).
Niu, S. et al. Molecular basis of cross-species ACE2 interactions with SARS-CoV-2-like viruses of pangolin origin. EMBO J. 40, e107786 (2021).
Zhang, Y. et al. SARS-CoV-2 rapidly adapts in aged BALB/c mice and induces typical pneumonia. J. Virol. 95, e02477–20 (2021).
Huang, K. et al. Q493K and Q498H substitutions in Spike promote adaptation of SARS-CoV-2 in mice. EBioMedicine 67, 103381 (2021).
Zhang, S. et al. Bat and pangolin coronavirus spike glycoprotein structures provide insights into SARS-CoV-2 evolution. Nat. Commun. 12, 1607 (2021).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Hu, B. et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698 (2017).
Ge, X.-Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
Latinne, A. et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 11, 4235 (2020).
Tan, Y., Schneider, T., Leong, M., Aravind, L. & Zhang, D. Novel immunoglobulin domain proteins provide insights into evolution and pathogenesis of SARS-CoV-2-related viruses. mBio https://doi.org/10.1128/mBio.00760-20 (2020).
Su, Y. C. F. et al. Discovery and genomic characterization of a 382-nucleotide deletion in ORF7b and ORF8 during the early evolution of SARS-CoV-2. mBio 11, e01610-20 (2020).
Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303, 1666–1669 (2004).
Conceicao, C. et al. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol. 18, e3001016 (2020).
Damas, J. et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl Acad. Sci. USA 117, 22311–22322 (2020).
Cohen, J. Wuhan coronavirus hunter Shi Zhengli speaks out. Science 369, 487–488 (2020).
Ge, X.-Y. et al. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol. Sin. 31, 31–40 (2016).
Clements, R., Sodhi, N. S., Schilthuizen, M. & Ng, P. K. L. Limestone karsts of Southeast Asia: imperiled arks of biodiversity. BioScience 56, 733–742 (2006).
Hassanin, A., Tu, V. T., Curaudeau, M. & Csorba, G. Inferring the ecological niche of bat viruses closely related to SARS-CoV-2 using phylogeographic analyses of Rhinolophus species. Sci. Rep. 11, 14276 (2021).
Soisook, P. et al. A taxonomic review of Rhinolophus stheno and R. malayanus (Chiroptera: Rhinolophidae) from continental Southeast Asia: an evaluation of echolocation call frequency in discriminating between cryptic species. Acta Chiropt. 10, 221–242 (2008).
Francis, C. Field Guide to the Mammals of South-east Asia 2nd edn (Bloomsbury Wildlife, 2019).
Makarenkov, V., Mazoure, B., Rabusseau, G. & Legendre, P. Horizontal gene transfer and recombination analysis of SARS-CoV-2 genes helps discover its close relatives and shed light on its origin. BMC Ecol. Evol. 21, 5 (2021).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020).
Lytras, S. The Sarbecovirus origin of SARS-CoV-2’s furin cleavage site. Virological https://virological.org/t/the-sarbecovirus-origin-of-sars-cov-2-s-furin-cleavage-site/536/6 (2021).
Puray-Chavez, M. et al. Systematic analysis of SARS-CoV-2 infection of an ACE2-negative human airway cell. Cell Rep. 36, 109364 (2021).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166 (2019).
Lemoine, F. et al. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 47, W260–W265 (2019).
Lole, K. S. et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160 (1999).
PREDICT One Health Consortium. Protocol for Bat and Rodent Sampling Methods (2013).
Sikes, R. S., Gannon, W. L. & the Animal Care and Use Committee of the American Society of Mammalogists. Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J. Mammal. 92, 235–253 (2011).
Virachith, S. et al. Low seroprevalence of COVID-19 in Lao PDR, late 2020. Lancet Reg. Health West Pac. 13, 100197 (2021).
Francis, C. A comparison of mist nets and two designs of harp traps for capturing bats. J. Mammal. 70, 865–870.
Corbet, G. B. & Hill, J. E. Mammals of the Indomalayan Region: a Systematic Review (Oxford Univ. Press & Natural History Museum, 1992).
Csorba, G., Ujhelyi, P. & Thomas, N. Horseshoe Bats of the World (Chiroptera: rhinolophidae) (Alana Books, 2003).
Deng, X. et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 5, 443–454 (2020).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Regnault, B. et al. Deep impact of random amplification and library construction methods on viral metagenomics results. Viruses 13, 253 (2021).
Bratuleanu, B. E. et al. The virome of Rhipicephalus, Dermacentor and Haemaphysalis ticks from Eastern Romania includes novel viruses with potential relevance for public health. Transbound. Emerg. Dis. https://doi.org/10.1111/tbed.14105 (2021).
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H. & Frost, S. D. W. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901 (2006).
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H. & Frost, S. D. W. GARD: a genetic algorithm for recombination detection. Bioinformatics 22, 3096–3098 (2006).
Crawford, K. H. D. et al. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses 12, E513 (2020).
Anna, F. et al. High seroprevalence but short-lived immune response to SARS-CoV-2 infection in Paris. Eur. J. Immunol. 51, 180–190 (2021).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 25, 2000045 (2020).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913 (2020).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Sali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Shen, M.-Y. & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 (2006).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
The PLUMED consortium. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 16, 670–673 (2019).
Daura, X. et al. Peptide folding: when simulation meets experiment. Angew. Chem. Int. Ed. 38, 236–240 (1999).
Stranges, P. B. & Kuhlman, B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 22, 74–82 (2013).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 (2011).
Acknowledgements
We thank S. Mohamed Ali, N. Da Rocha, A. Brisebarre, T. Xaybounsou and S. Chonephetsarath for their help at the bench; A. Haouz and the staff of the protein crystallogenesis facility at Institut Pasteur for help with crystallization trials; P. England from the Molecular Biophysics facility at Institut Pasteur for his support and access to the BLI equipment; P. Legrand from the PX1 beamline at Synchrotron SOLEIL for data collection support; and P. Guardado-Calvo for discussion and support with BLI experiments. NGS was performed with the help of the Biomics Platform, Center for Technological Resources and Research, Institut Pasteur, Paris, France, supported by France Génomique (ANR-10-INBS-09-09), Inclusive Blockchain Insurance using Space Assets and the Illumina COVID-19 Projects’ offer. The work was granted access to the High Performance Computing resources of the Institute for Development and Resources in Intensive Scientific Computing under the allocation 2020-101592 made by Grand Équipement National de Calcul Intensif. We thank the Ministry of Health and the Ministry of Natural Resources and Environments, Lao People’s Democratic Republic, for their authorization of the field work and the Faculty of Environmental Science for its authorization of the field research collaboration. The work was supported by an Institut Pasteur 'Covid Taskforce' and in part by the H2020 project 101003589 (RECOVER) and Labex IBEID (ANR-10-LABX62-IBEID) grants. Field and laboratory work at IPL was also supported by a UK embassy grant (grant no. INT 2021/LOV C19 02) and a Luxembourg Development special grant (grant no. LAO/030·202324).
Author information
Authors and Affiliations
Contributions
S.T., K.V., P.T.B. and M.E. conceived the study design. K.V., B.D., K.L., N.P., D.S., V.X. and P. Paphaphanh carried out sample procurement and bat species identification. S.M. and F.D. performed virus isolation, entry and neutralization assays. B.R., D.C. and P. Pérot prepared the NGS libraries and carried out the NGS. S.T., B.R. and T.B. carried out the genome assembly, recombination and phylogenetic analyses. V.L., S.S., K.L. and N.P. implemented the pan-coronavirus PCR testing. E.B.S. and F.A.R. performed and analysed the structure and binding studies. M.B., Y.K. and M.N. performed and analysed the MD simulations. M.E. and S.T. wrote the manuscript with input from all other authors.
Corresponding author
Ethics declarations
Competing interests
All authors of the manuscript are listed as inventors on US provisional patent number entitled 'New isolated bat SARS-COV able to infect human cells and harboring receptor-binding domains close to SARS-COV-2'. The patent covers medical applications (including diagnostics and vaccination) for the sequences described in the manuscript.
Peer review
Peer review information
Nature thanks Michael Worobey and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Spike identity matrices at the genus level of representative sarbecoviruses.
Amino-acid (lower) and nucleotide (upper) identity matrices of Laotian and representative human, bat, and pangolin sarbecoviruses. Spike N-terminal (NTD), Receptor-binding (RBD) and S2 nucleotide and amino-acid sequences were aligned with MAFFT, and identity matrices were constructed using CLC Main Workbench 21.0.4 (Qiagen). Matrices were colored according to the identity scale, from 25% (red) to 100% (green) of nucleotide or amino-acid identity.
Extended Data Fig. 2 Alignment of the spike RBD domain.
Protein alignment of the Receptor Binding Domain (RBD) of Laotian and representative human, bat and pangolin sarbecoviruses. Sequences were aligned with MAFFT in G-iNS-I mode. Residues interacting with human ACE2 receptor are highlighted in grey. The domain used for interactions modeling, based on the X-ray structure 6M0J (residues T333 to G526), is highlighted by a black line.
Extended Data Fig. 3 Nucleotide and amino-acid alignments of the furin cleavage site region.
Complete nucleotide and amino-acid spike sequences of representative bat SARS-CoV-2-like coronaviruses were downloaded from GenBank and GISAID and aligned with MAFFT (G-INS-I parameter) (A & C). Alignments were manually edited as proposed by Zhou2 and Lytras47 with CLC Main Workbench (Qiagen) (B & D). Alignments of the furin cleavage region are presented at the nucleotide (A & B) and the amino-acid (C & D) level, respectively.
Extended Data Fig. 4 Analysis of the stability and conformational heterogeneity of RBD–hACE2 complexes.
Time series (left column) and violin plots (right column) of backbone Root Mean Square Deviation (RMSD) from the initial, energy-minimized model calculated on the residues in RBD (A), hACE2 (B), at the interface of RBD and hACE2 (C), and on the entire complex (D). In the violin plots, the white circle corresponds to the median value, the black rectangle extends from the first to the third quantiles, and the thin black line represents the 95% confidence intervals. Population of the 3 most significant clusters visited during the course of the MD simulations (E). The analysis is performed for 9 different MD simulations: 3 replicates of the SARS-CoV-2 (shades of green), BANAL-236 (shades of red), and BANAL-52/103 (shades of blue) RBD–hACE2 complexes.
Extended Data Fig. 5 Additional analysis of the MD simulations of the BANAL-52/103 RBD–hACE2 complex.
Comparison of the time series of interface RMSD during the course of two MD simulations of the BANAL-52/103 RBD–hACE2 complex with short (BANAL-52/103-CoV.1) and long (BANAL-52/103-CoV.1*) equilibration phase. The large fluctuations of the interface RMSD are due to the flexibility of the RBD loop between residues S443 and Y449 (insets, in yellow). When these residues were not included in the calculation of the interface RMSD, the time series displayed a more stable behavior (BANAL-52/103-CoV.1-L and BANAL-52/103-CoV.1*-L).
Extended Data Fig. 6 Estimation of RBD–hACE2 binding energy.
Time series (left column) and violin plots (right column) of the RBD–hACE2 binding energy estimated using ROSETTA (A) and FoldX (B). In the violin plots, the white circle corresponds to the median value, the black rectangle extends from the first to the third quantiles, and the thin black line represents the 95% confidence intervals. The analysis is performed for 9 different MD simulations: 3 replicates of the SARS-CoV-2 (shades of green), BANAL-236 (shades of red), and BANAL-52/103 (shades of blue) RBD–hACE2 complexes.
Extended Data Fig. 7 Analysis of the inter-subunits hydrogen bonds at the interface of RBD and hACE2.
Frequency of formation of hydrogen bonds at the interface of RBD and hACE2 in the knob (A), base (B), and tip regions (C). The analysis is performed for 9 different MD simulations: 3 replicates of the SARS-CoV-2 (shades of green), BANAL-236 (shades of red), and BANAL-52/103 (shades of blue) RBD–hACE2 complexes.
Extended Data Fig. 8 Stick representation.
Stick representation of segments D364-S375 of BANAL-236 (left panel) and SARS-CoV-2 (right panel) RBDs. A 2Fo-Fc composite omit map (contoured at 3σ) is shown for this region in BANAL-236 RBD.
Extended Data Fig. 9 Isolation of BANAL-236 on VeroE6 cells.
(A) CPE observed on VeroE6 4 days after inoculation at an MOI of 10-4 from the C1. (B) Uninfected VeroE6 cells layer. (C) Plaque assay performed from the C2 stock on VeroE6 cells. (D) Comparative CPE observed on VeroE6 cells infected with SARS-CoV-2 ('Wuhan', top panel) or BANAL-236 (bottom panel) in absence (left) or in presence (right) of soluble ACE2. Photos were taken with a EVOS XL Core microscope at x10. A single experiment performed in triplicate is shown for each cell line.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Tables 1–8, Figs. 1–5 and References.
Supplementary Data
This file contains the full wwPDB X-ray structure validation report.
Source data
Rights and permissions
About this article

Cite this article
Temmam, S., Vongphayloth, K., Baquero, E. et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 604, 330–336 (2022). https://doi.org/10.1038/s41586-022-04532-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04532-4
This article is cited by
-
Recombination analysis on the receptor switching event of MERS-CoV and its close relatives: implications for the emergence of MERS-CoV
Virology Journal (2024)
-
Broad protection against clade 1 sarbecoviruses after a single immunization with cocktail spike-protein-nanoparticle vaccine
Nature Communications (2024)
-
Inhibitory effects of senkyuchachosan on SARS-CoV-2 papain-like protease activity in vitro
Journal of Natural Medicines (2024)
-
Emergence of SARS and COVID-19 and preparedness for the next emerging disease X
Frontiers of Medicine (2024)
-
Identification of a novel adenovirus in liver tissue sample of the Great Himalayan leaf-nosed bat (Hipposideros armiger)
Brazilian Journal of Microbiology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
