
Abstract
The wax gourd (Benincasa hispida), the only species in the genus Benincasa, is an important crop native to Asia that has been widely planted for multi-purpose uses. The first wax gourd draft genome was published three years ago, but it was incomplete and highly-fragmented due to data and technical limitations. Herein, we report a new chromosome-level genome assembly and annotation of B. hispida. We generated 974.87 Mb of unitigs with N50 size of 2.43 Mb via a hybrid assembly strategy by using PacBio long reads and Illumina short reads. We then joined them into scaffolds with Hi-C data, resulting 1862 scaffolds with a total length of 975.62 Mb, and 94.92% of the length (926.05 Mb) is contained in the 12 largest scaffolds corresponding to the 12 chromosomes of B. hispida. We predicted 37,092 protein-coding genes, and 85.05% of them were functionally annotated. This chromosome-level reference genome provides significant improvement to the earlier version of draft genome and would be valuable resource for research and molecular breeding of the wax gourd.
Similar content being viewed by others


Background & Summary
The wax gourd (Benincasa hispida), also known as ash gourd, white gourd, Chinese watermelon or winter gourd, is the only species in the genus Benincasa. It is an annual cucurbits native to Asia, and has been used as a vegetable and herbal medicine for thousands of years in China and India1,2. In the recent decades, the wax gourd is grown in more and more areas throughout the world for multi-purpose uses. It contains many important nutrients, and some metabolites can be used in treating fever and various disorders3,4. Commonly, it is used as an important vegetable and its young leaves, flower buds, immature and mature fruit is cooked and eaten. For its medicinal properties, it has been recognized in the traditional Chinese medicine and Ayurvedic medicine system over thousands of years, and now there are an increasing number of studies reported its medicinal values5,6. Moreover, the wax gourd is widely used in the food industry for making candied fruit, moon cakes and many kinds of pies as a base filling material. It is of great importance to broaden our knowledge of the wax gourd for promoting to fully exploit its benefits.
Development of a high-quality reference genome would be very useful for molecular genetics, molecular breeding and evolutionary studies of the wax gourd. Previously, we have reported a draft genome sequence of the wax gourd variety B227, and we revealed that the 12 chromosomes of wax gourd represent the most ancestral karyotype of the investigated cucurbits1. The B227 assembly is the only published de novo assembly of the wax gourd genome so far. Its contigs and scaffolds were constructed based on Illumina paired-end reads (approx. 28-fold), mate-pair reads (approx. 12-fold) and PacBio long-reads (approx. 15-fold), and the final pseudo-chromosomes were generated by anchoring scaffolds onto a published genetic map7. Though developed by combing data from multi platforms, it remains incomplete and highly-fragmented (contig N50 of 68.5 Kb, scaffold N50 of 3.4 Mb), and may contain mis-ordered or mis-oriented scaffolds due to technical limitations of the genetic map based pseudo-chromosome construction as reported in some plant species8,9,10. There is still much left to be improved about the wax gourd reference genome, and the availability of genomes of different varieties would provide more resources that can help to understand the genetic variations and evolutionary history of the crop.
Through our continuous efforts, a high-quality and near-complete reference genome assembly has been achieved. Herein, we report a chromosome-scale high quality genome assembly of the wax gourd variety pf3 by combined use of high-coverage PacBio long reads (approx. 86-fold), Illumina short reads (approx. 50-fold) and the Hi-C data. The de novo genome assembly and annotation workflow is as showed in the Fig. 1.

Overview of genome sequencing, assembly and annotation. Data information is shown in rectangles, software and tools are indicated in italic.
Methods
Sample selection, library preparation and sequencing
Previously we divided wax gourd germplasm into four groups according to their genomic variation data1, which were the wild group (W), the landrace (L), the two cultivated groups (C1 and C2). The wax gourd variety pf3 used for sequencing in this study is an inbreed line developed by us at the Vegetable Research Institute, Guangdong Academy of Agricultural Sciences in Guangzhou, China. It derived from a cross between a small fruit landrace (belongs to L group) collected from Yunnan and a giant fruit elite line (belongs to C2 group). It shows moderate fruit size with wax, high-yielding potential and good quality of taste (detailed morphology see Fig. 2). Fresh seedlings of the pf3 were used for high-quality DNA extraction followed by construction of PacBio SMRT Bell library, Illumina short-read library and Hi-C library. The Bell library was sequenced on the PacBio Sequel II platform (CLR mode), and then the output raw subreads bam file was converted to fastq format, generating 86.53 Gb data (Table 1). The Illumina short-read and Hi-C library was sequenced on the Illumina NovaSeq-6000 platform(PE150), generating 50.92 Gb and 99.55 Gb clean data respectively. All the DNA extraction, library construction and sequencing procedures were performed by the Novogene Company (Tianjin, China) according to the manufacturer’s protocols.

Morphology of the sequenced wax gourd cv. pf3. (a) The whole plant in the field. (b) Female flower. (c) Male flower. (d) Mature fruit. (e) Transection of mature fruit. (f) Seeds.
RNA sequencing
Root, stem, leaf and flower tissue of the pf3 plants were collected for RNA extraction. Total RNA was extracted from each tissue respectively, and then equal amount of them were pooled together. Thereafter, direct-cDNA sequencing and TruSeq RNA-seq library were constructed using the pooled RNA, and the transcriptomes were sequenced on the Nanopore PromethION and Illumina Hiseq4000 platform by Novogene Company (Tianjin, China), respectively. In total, 50.05 Gb full-length RNA-seq data and 8.33 Gb short-read RNA-seq data were obtained (Table 1). These RNA-seq data were used for whole-genome protein-coding gene prediction.
De novo genome assembly
We first converted raw subreads bam file generated by PacBio sequencer into fastq format using the software BAM2fastx. Statistical analysis showed that the average length of the long reads was 17.68 kb, and the N50 length was 21.01 kb. Then we constructed a primary assembly by using MaSuRCA assembler v4.0.911 with default parameters. The assembler built unitigs (high-confidence contigs) through hybrid assembly strategy with high-coverage PacBio long-read and Illumina short-read data, and generated 1897 unitigs with a total size of 974.87 Mb and unitig N50 of 2.43 Mb (Table 2). We then joined unitigs into scaffolds using Hi-C data via Juicer v1.6 and 3D-DNA v180922 pipeline12 with default parameters. We further visualized the raw scaffolds and conducted manual curation using the Juicebox tool package v1.22.0113. After curation, we obtained 1862 high-accuracy scaffolds with a total length of 975.62 Mb and N50 scaffold size of 70.97 Mb. We designated the assembly pf3 v1.1, and 94.92% of the total length (926.05 Mb) is contained in the 12 largest scaffolds that corresponding to the expected 12 chromosomes of the wax gourd (Fig. 3). By comparison with the B227 assembly we reported previously, great improvement on continuity (contig N50 size of 2.43 Mb vs 68.5 Kb), completeness (975.62 Mb vs 912.95 Mb) and chromosome-anchored size (926.05 Mb vs 859.0 Mb) was achieved in the pf3 v1.1 assembly (Table 2).

Genome features of the wax gourd cv. pf3. (a) GC content (30–50%) across 12 chromosomes. (b) Repeat percentage (60–100%). (c) Gene density (0–1452). (d) SNP density (0–24764). (e) Syntenic blocks of paralogous genes. a-d are drawn in non-overlapping 1 Mb sliding window.
Repeat annotation
We masked and annotated repetitive sequences and transposable elements (TEs) in the pf3 v1.1 assembly through incorporating de novo and homology-based predictions. We built a de novo repeat sequences library by using RepeatModeler v2.0.114, and performed homology-based predictions by using RepeatMasker v4.1.2-p115 with the Arabidopsis repeat sequences database. The output cat.gz file (contains list and alignment of repeat regions found in the assembly) of de novo and homology-based prediction was merged together and subjected to post-process with RepeatMasker package to produce the final repeat annotation. In total, we identified 770.68 Mb repetitive sequences in the pf3 v1.1 assembly, accounting for 78.99% of its total length (Fig. 3b and Table 3).
Gene prediction and functional annotation of the genome
We conducted protein-coding gene prediction with integration of evidence data from ab initio training, transcript and homologous proteins alignment through BRAKER pipeline v2.1.616. We prepared transcript evidence by mapping the full-length RNA-seq and short-read RNA-seq data to pf3 v1.1 assembly by using minimap2 v2.23-r111117 and STAR v2.7.9a18 respectively, followed by sorting alignment bam files with samtools v1.719. In running the pipeline, the soft-masked pf3 v1.1 assembly was used as input genome (–genome option), the sorted RNA-seq alignment files were used as RNA-seq evidence (–bam option), and peptides of the wax gourd B227 assembly and other three cucurbit species (Cucumis sativus ChineseLong v3, Lagenaria siceraria v1 and Cucurbita moschata v1) were used as homologous protein data (–prot_seq option) and aligned to the genome using GenomeThreader v1.7.020 (–prg gth). Briefly, the pipeline started with generating seed genes by GeneMark-ES v4.69_lic with supported by RNA-Seq evidence. Subsequently it performed ab initio training of AUGUSTUS v3.4.021 with the seed genes, RNA-Seq and protein alignment information. Finally, it conducted gene prediction with AUGUSTUS through integrating of the training output, RNA-Seq and homologous protein alignment information. After filtered out genes encoding protein sequence shorter than 50 aa (amino acids), and genes containing internal stop codon, illegal start or stop codon, a total of 37,092 genes were annotated in the pf3 v1.1 assembly (Fig. 3c and Table 4).
Furthermore, we performed functional annotation of the predicted genes through searching proteins against InterPro database (v88.0). We carried out this by submitted protein sequences to InterProScan 5 webservice22 via a perl script (iprscan5_lwp-nodie.pl) with default parameters. We further used PANNZER223 and eggNOG-mapper v2.1.724 to annotate proteins by Gene Ontology (GO) terms. In total, 31,562 genes were functionally annotated, and 22,707 genes were assigned to specific GO term (Table 4).
Comparison of the assembly of B227 and pf3
To infer the synteny and colinearity the pf3 v1.1 assembly and B227 assembly, we ran the Quick Genome Dot Plot plugin (parameters: Blast e-value 1e-3, Num of BlastHits 5) embedded in TBtools v1.09872625. We found that the two assemblies were quite syntenic (Fig. 4a), but there were large-scale inversions at the ends of some chromosomes. We further analysed the genomic rearrangements and local sequence differences between the two assemblies by using the SyRI v1.626 software. We identified a lot of Mb-sized structural rearrangements including inversions, translocations and duplications, and these rearrangements were mainly located at the end of chromosomes (Fig. 4b).

Whole-genome comparison of the pf3 v1.1 with B227 assembly. (a) Dot plot for the syntenic blocks. (b) Chromosome-level local sequence differences.
Considering the close genetic background between the two cultivars, and that the pseudo-chromosome of B227 assembly was developed based on a genetic map, it could be deduced that most of the rearrangements may be not real existing but rather mis-oriented or mis-placed in the B227 assembly. To check this, we further split the B227 assembly and re-constructed scaffolds (pseudo-chromosomes) by using Hi-C data of a genetically very close cultivar B418 that also belongs to C2 group. We detected much higher collinearity between the Hi-C-based B227 assembly and the pf3 v1.1, and most of large rearrangements discovered previously disappeared (Supplementary Fig. S1). We also examined the differences between the Hi-C based and genetic map based assemblies of the B227, and detected several large inversions at end of chromosomes (Supplementary Fig. S2). These evidences suggest that most of rearrangements showed in the Fig. 4d are indeed errors in the genetic map based B227 assembly.
Discovery of genomic variations
To explore genetic variation pattern of the wax gourd germplasm referenced to the newly developed pf3 genome, 31 representative wax gourd accessions (Supplementary Table S1) that we sequenced previously were selected and subjected to mapping and variation discovery procedure. Sequencing data of each accession was mapped onto the pf3 v1.1 assembly by using bwa v0.7.17-r118827 with default parameters, and the alignment files were sorted and indexed with samtools. We called variations of all the 31 accessions together by using bcftools v1.828 with mpileup and call commands. We applied pre-call filtering with -q 30 and -Q 20 to skip poor mapped reads and low-quality bases when run mpileup command, resulting in an initial total of 36,401,973 variant sites. We evaluated summary metrics of the raw variants, and then filtered them by using VCFtools v0.1.1629 and bcftools based on quality score, depth, average mapping quality and other criteria as described in the Supplementary Note 2. We finally got more than 12 million high-quality variations, including 12,366,466 single-nucleotide polymorphisms (SNPs) and 286,201 small insertions and deletions (InDels). The distribution of SNPs across the pseudo-chromosomes was as shown in the Fig. 3d. Furthermore, we investigated numbers of SNPs in subset of samples, we found that the C2 group contained the minimum SNPs among the four groups (Table 5), and there was only 593,107 SNPs between B227 and pf3.
Data Records
The sequencing data, genome assembly and annotation data reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center (NGDC), Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation30,31, under the BioProject accession number PRJCA010475 that is publicly accessible at https://ngdc.cncb.ac.cn/gwh. All the clean genome sequencing data including PacBio long-read, Illumina short-read and Hi-C data, as well as RNA sequencing data including Nanopore full-length RNA-seq and Illumina short-read RNA-seq data, were deposited in the Genome Sequence Archive (GSA) of NGDC under the accession number CRA007486. The pf3 v1.1 assembly and annotation data have been deposited in the Genome Assembly Sequences and Annotations (GWH) of NGDC under accession number GWHBJVO00000000. The DNA and RNA sequencing data were also submitted to the National Center for Biotechnology Information (NCBI) SRA database with accession number SRR23081782, SRR23081783, SRR23081784, SRR23081781 and SRR23096591 under BioProject PRJNA89881932,33,34,35,36. The genome assembly has also been deposited at DDBJ/ENA/GenBank under the accession GCA_027475165.137. Sequencing data of the 30 wax gourd cultivars used for genetic variation discovery are available in the GSA under project accession number PRJCA001140, and sequencing information for these cultivars is summarized in Supplementary Table S1.
Technical Validation
Assessment of the genome assembly
To evaluate the completeness of the wax gourd pf3 v1.1 assembly, we first mapped Illumina short-read and PacBio long-reads data back to the assembly, and analysed the alignment file with Qualimap v.2.2.238. The mapping rate of both libraries was above 98% (Table 2), and more than 96.5% of the assembly have at least 20× coverage of Illumina short-read and PacBio long-reads respectively. We then performed the Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.2.239 with eudicots dataset (n = 2,326) to assess the completeness of the assembly. We identified 2,280 complete BUSCOs (98.02%) out of the 2,326 BUSCO groups, including 2,183 complete and single-copy BUSCOs and 97 complete and duplicated BUSCOs (Table 6). The number of fragmented BUSCOs and missing BUSCOs was 9 (0.4%) and 37 (1.5%), respectively.
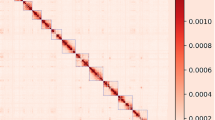
Moreover, we evaluated the result of Hi-C based pseudo-chromosomes construction. We mapped the Hi-C data to the 12 pseudo-chromosomes, and then analysed and visualized with Hicexplorer v3.740. As the heat-map of Hi-C contact displays in Fig. 5, the signal intensities of interaction between the two bins were clearly divided into 12 distinct groups, indicating the high-quality of the pseudo-chromosomes assembly.

Hi-C contact map of the chromosome-level assembly of pf3. The intensity of interactions was calculated using a bin size of 10 K.
Code availability
The versions, settings and options of software tools used in this work are described below, and more detailed explanation is described in the Supplementary Notes section.
(1) MaSuRCA: v4.0.9, default parameters;
(2) Juicer: v1.6, default parameters;
(3) 3D-DNA: v180922, default parameters;
(4) Juicebox tool: v1.22.01, default parameters;
(5) RepeatModeler: v2.0.1, default parameters+;
(6) RepeatMasker: v4.1.2-p1, parameters: -xsmall -gff;
(7) BRAKER: v2.1.6, parameters:–species = Benincasa_hispida–softmasking–prg gth–gth2traingenes–AUGUSTUS_ab_initio–gff3;
(8) minimap2: v2.23-r1111, parameters: whole-genome alignment: -ax asm5 –eqx; mapping PacBio SMRT reads: -ax map-pb;
(9) STAR: v2.7.9a, default parameters;
(10) samtools: v1.7, parameters: view command: -bS, sort command: -O BAM;
(11) GenomeThreader: v1.7.0, default parameters;
(12) GeneMark-ES: v4.69_lic, default parameters;
(13) AUGUSTUS: v3.4.0, default parameters;
(14) InterProScan: v5.56–88.0, parameters: -dp -f tsv;
(15) PANNZER2: web server version, default parameters;
(16) eggNOG-mapper: v2.1.7, default parameters;
(17) TBtools: v1.098726, creating genome dot plot: Quick Genome Dot Plot plugin: evalue 1e-3 Num of BlastHits 5, creating genome circus plot: Advanced Circos (input data prepared via Fasta Stats, One Step MCScanX, Text Merge for MCScanX and Transformat for Micro-Synteny View) with default paremeters;
(18) SyRI: v1.6, default parameters;
(19) bwa: v0.7.17-r1188, parameters: mapping reads: mem -M;
(20) bcftools: v1.8, parameters: mpileup -Ou -q 30 -Q 20 –p; call -m -Ov;
(21) VCFtools: v0.1.16, parameters:–remove-filtered-all–remove-filtered-geno-all–max-missing 1.0–min-alleles 2–max-alleles 2;
(22) Qualimap: v.2.2.2, parameters: bamqc;
(23) BUSCO: v5.2.2, parameters: -m genome -c 40;
(24) Hicexplorer: v3.7, parameters: hicBuildMatrix:–binSize 10000, hicPlotMatrix:–dpi 600.
References
Xie, D. et al. The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nat. Commun. 10, 5158 (2019).
Pandey, A. K., Bhardwaj, D. R., Dubey, R. K., Singh, V. & Pandey, S. Botany, diversity, utilization and improvement of ash gourd (Benincasa hispida Thunb. Ex Murray Cogn)-A review. Ann. Hortic. 8, 1–15 (2015).
Zaini, N. A. M., Anwar, F., Hamid, A. A. & Saari, N. Kundur [Benincasa hispida (Thunb.) Cogn.]: A potential source for valuable nutrients and functional foods. Food Res. Int. 44, 2368–2376 (2011).
Talaei, A., Forouzanfar, F. & Akhondzadeh, S. Medicinal Plants in the Treatment of Obsessive-Compulsive Disorder: A Review. Curr. Drug Discov. Technol. 18, 8–16 (2021).
Doharey, V., Kumar, M., Upadhyay, S. K., Singh, R. & Kumari, B. Pharmacognostical, physicochemical and pharmaceutical paradigm of ash gourd, Benincasa hispida (Thunb.) fruit. Plant Archives. 21, 249–252 (2021).
Islam, M. T. et al. A literature-based update on Benincasa hispida (thunb.) Cogn.: traditional uses, nutraceutical, and phytopharmacological profiles. Oxid. Med. Cell. Longev. 2021, 6349041 (2021).
Jiang, B. et al. High-density genetic map construction and gene mapping of pericarp color in wax gourd using specific-locus amplified fragment (SLAF) sequencing. BMC Genomics. 16, 1035 (2015).
Jibran, R. et al. Chromosome-scale scaffolding of the black raspberry (Rubus occidentalis L.) genome based on chromatin interaction data. Hortic. Res. 5, 8 (2018).
Xie, T. et al. De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana. Mol. Plant. 8, 489–492 (2015).
Jiao, Y. et al. Improved maize reference genome with single-molecule technologies. Nature. 546, 524–527 (2017).
Zimin, A. V. et al. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27, 787–792 (2017).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for hi-c contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. Chapter 4, 4–10 (2009).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, a108 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inform. Software Tech. 47, 965–978 (2005).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 24, 637–644 (2008).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 30, 1236–1240 (2014).
Toronen, P., Medlar, A. & Holm, L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 46, W84–W88 (2018).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 13, 1194–1202 (2020).
Goel, M., Sun, H., Jiao, W. B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Danecek, P. & McCarthy, S. A. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics. 33, 2037–2039 (2017).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics. 27, 2156–2158 (2011).
Chen, M. et al. Genome Warehouse: A Public Repository Housing Genome-scale Data. Genomics Proteomics Bioinformatics. 19, 584–589 (2021).
Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27-D38 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23081781 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23081782 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23081783 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23081784 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23096591 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_027475165.1 (2023).
Okonechnikov, K., Conesa, A. & Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32, 292–294 (2016).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Wolff, J. et al. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 48, W177–W184 (2020).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31972403, 32002044), the Discipline Team Construction Project of GDAAS (202114TD) and the Training Plan for Young and Middle-aged Discipline Leaders of GDAAS (R2020PY-JG003). We thank Dr. Kunshen Wu for his comments and suggestions of improvement to the manuscript.
Author information
Authors and Affiliations
Contributions
Wenlong Luo and Jinqiang Yan contributed equally to this work. Biao Jiang and Dasen Xie conceived and designed the experiments. Jinqiang Yan, Shanwei Luo, Wenrui Liu prepared plant samples, conducted experiments and performed laboratory work. Wenlong Luo performed data analysis and drafted the manuscript. All authors provided feedback on the draft manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, W., Yan, J., Luo, S. et al. A chromosome-level reference genome of the wax gourd (Benincasa hispida). Sci Data 10, 78 (2023). https://doi.org/10.1038/s41597-023-01986-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-01986-7
