
Abstract
The increased incidence of antibiotic resistant ‘superbugs’ has amplified the use of broad spectrum antibiotics worldwide. An unintended consequence of antimicrobial treatment is disruption of the gastrointestinal microbiota, resulting in susceptibility to opportunistic pathogens, such as Clostridium difficile. Paradoxically, treatment of C. difficile infections (CDI) also involves antibiotic use, leaving patients susceptible to re-infection. This serious health threat has led to an urgent call for the development of new therapeutics to reduce or replace the use of antibiotics to treat bacterial infections. To address this need, we have developed colostrum-derived antibodies for the prevention and treatment of CDI. Pregnant cows were immunised to generate hyperimmune bovine colostrum (HBC) containing antibodies that target essential C. difficile virulence components, specifically, spores, vegetative cells and toxin B (TcdB). Mouse infection and relapse models were used to compare the capacity of HBC to prevent or treat primary CDI as well as prevent recurrence. Administration of TcdB-specific colostrum alone, or in combination with spore or vegetative cell-targeted colostrum, prevents and treats C. difficile disease in mice and reduces disease recurrence by 67%. C. difficile-specific colostrum should be re-considered as an immunotherapeutic for the prevention or treatment of primary or recurrent CDI.
Similar content being viewed by others


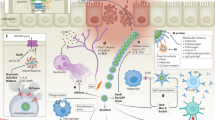
Introduction
There is a desperate worldwide need to minimise the use of antibiotics. Although the treatment of most bacterial infections relies on antibiotic use, resistance has emerged and is now one of our most serious global health threats. Also of concern is the rapid increase in nosocomial antibiotic-associated infections caused by opportunistic pathogens such as Clostridium difficile. C. difficile infection (CDI) is most often associated with antibiotic use as the alteration to the endogenous gastrointestinal microbiota results in increased susceptibility to CDI1. The over-use of antibiotics has been a driver for the astonishing increase in the rate and prevalence of C. difficile, as has the change in the virulence of the causative strains, with BI/NAP1/027 isolates causing increased mortality rates in the UK, USA, Canada and Europe in the last decade1.
C. difficile is a Gram-positive, spore-forming, anaerobic bacterium that infects the gastrointestinal tract and causes an array of clinical symptoms ranging from mild diarrhoea to more severe, often fatal, gastrointestinal disease such as pseudomembranous colitis and toxic megacolon2. The infection cycle of C. difficile is complex because this bacterium produces spores that are highly resistant to environmental assaults, enabling persistence in unfavourable environments3. Spores are the infectious particles ingested by the host, where they germinate into vegetative cells, colonise the large intestine and establish infection1. Disease symptoms occur in response to toxin-mediated damage with up to three secreted toxins, TcdA, TcdB and CDT, variably produced by strains1. TcdA and TcdB are monoglucosyltransferases that modify Rho GTPases leading to disorganisation of the actin cytoskeleton, cell-rounding, death of the intoxicated cell and extensive colonic inflammation4. The relative contribution of these two major toxins to disease pathogenesis has long been contentious, however, many studies have now clearly demonstrated the importance of TcdB in disease1, 5,6,7,8,9 and many strains that produce TcdB but not the other toxins continue to emerge10, 11. Targeting TcdB for disease treatment has resulted in the production of a human monoclonal antibody, bezlotoxumab, which reduced rates of recurrent infection in human clinical trials and has recently obtained FDA approval12. In support of the approach of targeting TcdB, antibodies against TcdB, but not TcdA, protected piglets from gastrointestinal and systemic signs of CDI when administered intraperitoneally13. Moreover, delivery of both anti-TcdA and anti-TcdB neutralising antibodies to either piglets or humans via systemic routes was not beneficial compared to anti-TcdB antibodies alone12, 13 and administration of anti-TcdA antibodies alone may return adverse clinical outcomes13. For these reasons, a universal toxin-based CDI therapeutic must include TcdB as a target and consideration given to the inclusion of a TcdA target.
Rather incongruously, the management of CDI often requires antibiotic administration, usually metronidazole or vancomycin. Although these antibiotics are effective at inhibiting C. difficile, they also prevent the re-establishment of the normal, protective microbiota2. Consequently, 20–30% of patients experience relapses in C. difficile infection after treatment ceases, with many patients suffering multiple relapses2. Without doubt, alternative and rationally designed preventive therapies and treatments that do not require the use of antibiotics are required to manage recurrent C. difficile disease.
Bovine colostrum is the first milk produced after parturition and is perfectly suited to oral administration; it is therefore ideal for treating gastrointestinal infections14. Colostrum provides passive immunity to newborn calves from opportunistic infections and immunisation of dairy cows during gestation with specific antigens results in colostrum containing high concentrations of antigen-specific antibodies. Known as hyperimmune bovine colostrum (HBC), variations of this targeted product, including whole-HBC, immune whey or purified antibodies, have been tested in animals and humans and are effective against many enteric pathogens, including C. difficile 15, 16. Most recently, a whole-HBC product containing antibodies against TcdA and TcdB showed efficacy as a treatment for primary CDI in a gnotobiotic piglet diarrhoea model17. C. difficile-specific HBC products have also previously been shown to be effective at preventing recurrent CDI in human patients18, 19. Importantly, colostrum is resistant to gastrointestinal degradation20 and, unlike antibiotics, does not adversely disrupt the resident microbiota17. This concept is particularly relevant for managing the increasing incidence, severity and recurrence of CDI. As colostrum has the potential to be used as a prevention or treatment of primary disease or recurrence, we examined the use of spore-, vegetative cell- or TcdB-specific colostrum for the prevention and treatment of CDI in a mouse model of infection.
Results
C. difficile-specific antibodies in colostrum are cross-reactive and able to neutralise TcdB cytotoxicity in vitro
Using platform technologies developed by Immuron Ltd, we produced HBC targeting spores by immunisation of pregnant cows with either inactivated whole spores (Spore-HBC) or an extract of the outermost spore layer, the exosporium (Exo-HBC). We also produced HBC targeting vegetative cells by immunisation with either inactivated vegetative cells (Veg-HBC) or a surface layer protein (SLP) preparation (SLP-HBC). Vegetative cell- or spore-specific vaccines were generated from strain DLL3109, a ribotype 027 isolate. TcdB-specific colostrum (TcdB-HBC) was generated by immunisation with the C-terminal binding domain of TcdB, which was prepared as described elsewhere21. C. difficile-specific antibody titres in colostrum were determined by ELISA and in all cases were elevated compared to non-immune bovine colostrum (NI-BC) from unvaccinated cows (Fig. 1). Western blotting showed that Veg-HBC and SLP-HBC antibodies cross-reacted with whole cell lysates or SLP from diverse human and animal strains (Fig. 2a,b). TcdB-HBC antibodies cross-reacted with TcdB isolated from culture supernatants of various strains (Fig. 2c). Binding was specific to TcdB as bands were not detected in supernatant from a non-toxigenic strain (Lane 10, Fig. 2c) and were detected in a lane containing TcdB purified from a C. difficile 027 strain (tgcBIOMICS; Lane 11, Fig. 2c). Spore-HBC and Exo-HBC antibodies cross-reacted with the exosporium extracted from a panel of strains (Fig. 2d,e). Universal cross-reactivity was seen with each antigen across all strains, suggesting that HBC antibodies may have a broad capacity to combat CDI caused by diverse isolates. Importantly, the TcdB-HBC IgG abolished the cytotoxic activity of purified TcdB, with a significant, dose-dependent, reduction in cell death when 25 pg (P < 0.0001) or 5 pg (P < 0.0001) of TcdB was incubated with TcdB-HBC IgG compared to TcdB that had been incubated with NI-BC IgG or PBS (Fig. 2f).

ELISA analysis of C. difficile-specific antibodies in colostrum from cows immunised with C. difficile antigens. ELISA plates were coated with C. difficile spore, exosporium, vegetative cell, SLP or recombinant TcdB antigens to determine the specific colostrum antibody titres of Spore-HBC (a), Exo-HBC (b), Veg-HBC (c), SLP-HBC (d) or TcdB-HBC (e), respectively, compared with colostrum from non-immune cows (NI-BC).

C. difficile-specific antibodies in colostrum are cross-reactive and able to neutralise the cytotoxicity of TcdB. C. difficile whole cell lysates from a variety of strains were used to detect vegetative cell-specific IgG purified from Veg-HBC (a) or SLP-HBC (b), respectively. Crude toxin preparations from the panel of strains indicated were used to detect TcdB-specific IgG purified from TcdB-HBC; control lanes contained purified TcdB from an 027 strain (tgcBIOMICS; Lane 11) while a non-toxigenic strain (CD37) was used to show that binding was toxin-specific (Lane 10) (c). Exosporium proteins isolated from a variety of clinical and animal isolates of C. difficile were used to detect spore-specific IgG purified from Spore-HBC (d) or Exo-HBC (e), respectively. C. difficile strains used are indicated alongside each panel (a–e). To determine if TcdB-HBC contained IgG capable of neutralising TcdB, purified TcdB from strain VPI10463 (Abcam) was incubated for 60 minutes with either PBS, NI-BC IgG or TcdB-HBC IgG before being added to Vero cell monolayers. After a 24 hour incubation, the percentage of cell death was determined by directly visualising cells for cell rounding (f). The experiment was performed in triplicate. Data represent the mean ± SEM and statistical significance was assessed using a two-way ANOVA with a post hoc Bonferroni multiple comparison test. ****Indicates P < 0.0001.
TcdB-specific HBC treats CDI in mice when administered post-infection
The treatment efficacy of the TcdB-HBC was assessed using an infection model. Six hours post-infection, mice were administered NI-BC, TcdB-HBC or vancomycin, the main CDI standard-of-care antibiotic. Untreated and NI-BC-treated mice showed no significant difference in survival, with 0% or 6.7% survival rates (P = 0.3178), respectively, while uninfected and vancomycin-treated mice all survived infection (Fig. 3a). By comparison, 78.6% survival was seen in TcdB-HBC-treated mice (Fig. 3a), with TcdB-HBC- (P < 0.0001) and vancomycin- (P < 0.0001) treated mice showing significantly increased survival rates compared to untreated mice. TcdB-HBC-treated mice showed no statistical difference (P = 0.0628) in survival rate compared to vancomycin-treated mice. Uninfected animals did not lose weight (Fig. 3b) and shed no spores, as expected (Fig. 3c), whereas infected but untreated and NI-BC-treated animals lost up to 15-20% of their body weight (Fig. 3b) and shed high spore numbers (~1.5 × 107 CFU/g, Fig. 3c). Infected and vancomycin-treated mice did not lose weight (Fig. 3b) and shed few spores (Fig. 3c), the latter resulting from vancomycin-mediated vegetative cell death and attenuated spore production. By contrast, TcdB-HBC prevented rapid weight loss (Fig. 3b), yet mice shed spores at numbers comparable to both no-treatment and NI-BC groups (Fig. 3c) and developed mild diarrhoea, suggesting that TcdB activity was not completely neutralised or that TcdA was contributing to disease. Mice from all groups that did not survive infection had severe and comparable damage in the colon (Fig. 3d) and caecum (data not shown). Surviving TcdB-HBC-treated mice had less damage in the colon and caecum than NI-BC and no-treatment control mice, however, increased epithelial damage and inflammation was seen compared to uninfected or vancomycin-treated mice (Fig. 3d and data not shown). Collectively, these data provide strong evidence supporting the use of TcdB-HBC to treat CDI.

HBC-mediated treatment of C. difficile-infected mice. Mice were uninfected or infected with C. difficile spores and treated six hours post-infection with NI-BC, TcdB-HBC, vancomycin or were untreated. Mice were monitored daily for survival (a) and weight loss (b). Weight loss is presented as the % weight relative to the day of infection (day 0 or D0). Numbers in brackets indicate the number of surviving mice at the completion of the experiment compared to (/) the total number of mice at the beginning of the experiment. Faecal spore load was determined 24 hours post-infection and is presented as CFU/gram faeces (log10), with each point representing a single mouse (c). The limit of detection is represented as a dotted line. Error bars represent the mean ± SEM of n = 10–15 mice. ****Indicates P < 0.0001. Representative images of PAS/Alcian blue stained colonic (d) tissue from mice. Square brackets ([) indicate crypt hyperplasia, arrow heads (▲) represent epithelial damage and asterisks (*) represent oedema and inflammation. Scale bars represent 200 µm.
Prophylactic administration of TcdB-specific HBC protects mice from CDI
The capacity of TcdB-HBC, Spore-HBC, Exo-HBC, Veg-HBC or SLP-HBC to act prophylactically was also tested. Animals were given access to either water or colostrum ad libitum two days prior to infection and throughout the trial. NI-BC-treated and untreated mice succumbed rapidly to infection, with no significant difference in survival observed (P = 0.3511; Fig. 4a). Mice that received Spore-HBC lost weight at day 1.5 (Fig. 4b), however, 40% recovered from infection (Fig. 4a) and regained weight (Fig. 4b). Exo-HBC-treated mice did not survive infection, however, death was delayed compared to mice that received no colostrum or NI-BC (Fig. 4a) and weight loss was less rapid (Fig. 4b). Veg-HBC or SLP-HBC-treated mice all succumbed to disease by 1.5–2 days post-infection (Fig. 4a), with rapid weight loss observed during the first 1.5 days (Fig. 4b), similar to untreated mice. Strikingly, 70.8% of TcdB-HBC-treated mice survived infection (Fig. 4a), with less rapid weight loss detected initially and subsequent weight gain reaching levels similar to uninfected and vancomycin-treated groups (Fig. 4b). Spore-HBC- (P < 0.0001), Exo-HBC- (P = 0.0075), TcdB-HBC- (P < 0.0001) and vancomycin- (P < 0.0001) treated mice showed significantly increased survival rates compared to untreated mice whereas mice treated with Veg-HBC- (P = 0.3511) or SLP-HBC- (P = 0.2709) showed no statistical difference in survival compared to untreated mice. When compared to vancomycin-treated mice, Spore-HBC- (P = 0.0042), Exo-HBC- (P < 0.0001), Veg-HBC- (P < 0.0001), or SLP-HBC- (P < 0.0001) showed significantly less survival, however, TcdB-HBC-treated mice showed no significant difference in survival (P = 0.630). All mice (except vancomycin-treated) shed similar, but variable, numbers of spores 24 hours post-infection (Fig. 4c). Non-surviving mice from all groups had severe damage in the colon (Fig. 4d) and caecum (data not shown). Gut histopathology of surviving TcdB-HBC and Spore-HBC-fed mice looked similar to that of uninfected or vancomycin-treated mice, suggesting that these colostrum preparations prevented severe damage (Fig. 4d). The high survival rate of mice administered TcdB-HBC suggests that targeting TcdB is an effective CDI prophylactic target.

HBC-mediated prevention of C. difficile disease in mice. Mice were untreated or were pre-treated for two days with NI-BC, Spore-HBC, Exo-HBC, Veg-HBC, SLP-HBC, TcdB-HBC or vancomycin prior to infection with C. difficile spores. Colostrum or vancomycin was administered daily for the duration of the experiment. Uninfected, untreated mice served as controls. Mice were monitored daily for survival (a) and weight loss (b). Weight loss is presented as the % weight relative to the day of infection (day 0 or D0). Numbers in brackets indicate the number of surviving mice at the completion of the experiment compared to (/) the total number of mice at the beginning of the experiment. Faecal spore load was determined 24 hours post-infection and is presented as CFU/gram faeces (log10), with each point representing a single mouse. The limit of detection is represented as a dotted line (c). Error bars represent the mean ± SEM of n = 10–24 mice. **Indicates P < 0.01, ****Indicates P < 0.0001. Representative images of PAS/Alcian blue stained colonic (d) tissue from mice. Square brackets ([) indicate crypt hyperplasia, arrow heads (▲) represent epithelial damage and asterisks (*) represent oedema and inflammation. Scale bars represent 200 µm.
Prophylactic administration of a mixture of HBC protects mice from CDI
The efficacy of combining colostrum preparations to simultaneously target all stages of the infectious cycle was assessed with either Spore-HBC, Veg-HBC and TcdB-HBC (Mix1-HBC), or Exo-HBC, SLP-HBC and TcdB-HBC (Mix2-HBC) combined in equal ratios (1:1:1). As a control, mice were administered TcdB-specific colostrum that had been diluted 1:3 in non-immune colostrum (Mix3-HBC), which had equivalent amounts of TcdB-specific antibody to Mix2- and Mix3-HBC. Mice were given no treatment or were prophylactically administered either NI-BC, TcdB-HBC, Mix1-HBC, Mix2-HBC or Mix3-HBC. No-treatment and NI-BC-treated mice succumbed to disease by day 2, with no difference in survival (P = 0.796; Fig. 5a) and lost up to 15% of their body weight (Fig. 5b), as expected. By comparison, in this experiment, 60% of TcdB-HBC-treated mice survived infection (Fig. 5a), with surviving mice initially losing, then regaining, weight (Fig. 5b). Spore enumeration 24 hours post-infection was similar between all mice (~106 CFU/gram), indicating no colonisation effects (Fig. 5c). Notably, prophylactic administration of Mix1-HBC and Mix2-HBC protected 70% and 80% of mice, respectively, whereas mice receiving Mix3-HBC all succumbed to disease by day 2 (Fig. 5a). Similar to surviving TcdB-HBC-treated mice, Mix1-HBC- and Mix2-HBC-treated mice initially lost but then regained weight (Fig. 5b). Vancomycin- (P = 0.0012), TcdB-HBC- (P = 0.004), Mix1-HBC- (P = 0.0033) and Mix2-HBC- (P = 0.0052) treated mice showed significantly increased survival rates compared to untreated mice, whereas TcdB-HBC- (P = 0.1174), Mix1-HBC- (P = 0.6998) and Mix2-HBC- (P = 0.4292) treated mice showed no statistical difference in survival compared to vancomycin-treated mice, suggesting that colostrum treatment is comparable to the standard-of-care antibiotic for CDI. Collectively, this data suggests that there is added benefit in combining TcdB-specific colostrum with colostrum that targets spores and vegetative cells. Although not significant, the mixed-colostrum products yielded a higher level of protection in mice compared to TcdB-HBC alone and thus subsequent experiments utilised a colostrum mixture.

Prevention of primary CDI by HBC mixtures. Mice were untreated (no colostrum) or were pre-treated with NI-BC, TcdB-HBC, Mix1-HBC, Mix2-HBC, Mix3-HBC or vancomycin prior to infection. Uninfected, untreated mice were controls. Mice were monitored daily for survival (a) and weight loss (b). Weight loss is presented as the % weight relative to the day of infection (day 0 or D0). Numbers in brackets indicate the number of surviving mice at the completion of the experiment compared to (/) the total number of mice at the beginning of the experiment. Faecal spore load is presented as CFU/gram faeces (log10), with each point representing a single mouse. The limit of detection is represented as a dotted line (c). Error bars represent the mean ± SEM of n = 5–10 mice.
A mixture of HBC protects mice from disease recurrence
Disease recurrence occurs in 20-30% of patients and many current strategies are targeting this important disease aspect1. We therefore tested a combined Exo-HBC, SLP-HBC and TcdB-HBC product in a disease relapse model. Most mice receiving no-colostrum treatment succumbed to disease 8–14 days after vancomycin cessation, with a survival rate of 11.1% (Fig. 6a). Severe diarrhoea and weight loss was seen (Fig. 6b) and high spore numbers were detected on the day animals succumbed (Fig. 6c). By contrast, colostrum-treated mice had a significantly higher survival rate of 77.8% (P = 0.0027; Fig. 6a) and lost little weight (Fig. 6b). Although the surviving mice from this group began shedding spores (Fig. 6c) and had mild diarrhoea, they did not succumb to disease, suggesting that colostrum antibodies did not prevent bacterial growth and colonisation, but effectively neutralised TcdB activity. Overall, this work suggests that an Exo-HBC, SLP-HBC and TcdB-HBC combination product effectively prevents C. difficile disease relapse.

Prevention of recurrent CDI by HBC mixtures. Infected mice were orally treated with vancomycin prior to receiving either vancomycin or vancomycin and a mixture of Exo-HBC, SLP-HBC and TcdB-HBC. After vancomycin treatment ceased, mice either received no treatment or the HBC mixture. Mice were monitored daily for survival (a) and weight loss (b). Weight loss is presented as the % weight relative to the day before, with each point representing a single mouse. Faecal spore load was determined daily after cessation of vancomycin treatment and is presented as CFU/gram faeces (log10) as described above (c). Error bars represent the mean ± SEM of n = 9 mice. **Indicates P < 0.01.
Discussion
We have developed the first oral whole-HBC immunotherapeutic product specifically engineered to neutralise the activity of only one of the C. difficile toxins, TcdB. Until now most vaccines and immunotherapeutics have focussed on TcdA alone or have combined TcdA and TcdB, however, many pathogenic strains do not produce TcdA whereas almost all produce TcdB11. The importance of TcdB, particularly in severe intestinal damage22 and severe8 and systemic disease13, 22, has been established, For these reasons, TcdB was selected as a key target for the development of C. difficile colostrum-based therapeutics, together with vegetative cell and spore targets that represent all important infectious cycle components.
The use of C. difficile-specific colostrum as a prevention or treatment for CDI has yielded promising results when tested in various animal models and in human clinical trials. Colostrum directed against C. difficile culture supernatant or purified TcdA was shown to prevent the enterotoxic effect of toxins in a rat ileal loop model23. Most recently, whole colostrum that was generated by vaccinating cows with both recombinant TcdA and TcdB was shown to successfully treat CDI in a gnotobiotic piglet model when administered after the onset of mild diarrhoea17. Prophylactic administration of colostrum immunoglobulin concentrate containing antibodies against C. difficile culture filtrate (with both TcdA and TcdB present) protected hamsters from CDI24. Moreover, treatment of hamsters with a bovine milk product (MucoMilk) that contained high titres of sIgA, raised against whole C. difficile cells and toxoid prepared from bacterial culture filtrate, prevented disease18. This same product was shown to prevent disease relapse in human patients18, 19, while bovine colostrum containing antibodies raised against C. difficile vegetative cells was shown to be as effective as metronidazole in treating recurrent CDI in humans25. It has also been shown that C. difficile-specific colostrum antibodies survive transit through the human gastrointestinal tract and retain their ability to neutralise toxin activity20, 26. As colostrum has the potential to be used for the prevention or treatment of primary disease or recurrence, we examined the use of spore-, vegetative cell- or TcdB-specific colostrum for the prevention and treatment of CDI in a mouse model of infection.
The TcdB-HBC product developed here showed excellent efficacy, supporting our hypothesis that targeting TcdB is beneficial in preventing and treating primary CDI. Dilution of TcdB-specific colostrum with non-immune colostrum at a ratio of 1:3 reduced the efficacy of the TcdB colostrum, but a similar dilution, using a combination of spore and vegetative cell colostrum, in which the ratio of TcdB antibody remained 1:3, resulted in enhanced survival in mice, suggesting that these antibodies acted synergistically to provide superior protection against CDI. Importantly, our product combining TcdB-, SLP- and Exo-HBC reduced disease relapse in mice by 66.7%. Supporting our approach of targeting TcdB, intravenous infusion of the monoclonal TcdB antibody product bezlotoxumab (Merck) during a phase III clinical trial reduced disease relapse from 28% to 17% in MODIFY I and 26% to 16% in MODIFY II compared to placebo12. The use of bovine milk or modified colostrum to reduce CDI relapse rates in human clinical trials18, 19, 25 also supports the efficacy of a colostrum product. However, the antibodies used in these studies were generated against vegetative cells alone or in combination with TcdA- and TcdB-containing culture supernatants, and whole HBC was not investigated.
Although the development of colostrum based immunotherapeutics for CDI treatment is not a new concept, our study is the first to clearly show a reduction in recurrence with a defined colostrum product. We are the first to test a colostrum-based therapy in a mouse model of CDI as well as being the first to show that colostrum antibodies targeting TcdB alone are sufficient for preventing and treating disease. We have also shown for the first time that combining different batches of colostrum, containing antibodies against individual C. difficile components, generates a product that is superior to targeting a single antigen alone. Finally, our results have proven the efficacy of this type of therapy in three different aspects of disease: prevention and treatment of primary disease and prevention of recurrent CDI, the latter of which represents an important disease aspect currently being targeted in the development of many diverse therapeutic products. For our product to progress from pre-clinical studies to the clinic, the delivery and dose of colostrum required to prevent disease in humans needs to be determined. Although colostrum inherently protects antibodies from gastrointestinal tract degradation, allowing transit to the colon26, encapsulation or sustained-released formulations may improve product efficacy by increasing the amount and longevity of the antibodies in the colon.
The period of recovery of the microbiota following antibiotic treatment for CDI is hypothesised to correspond to the window of susceptibility to recurrence27. The systemic administration of monoclonal TcdA and TcdB antibodies to mice facilitates the normalisation of the gut microbiota during CDI while reducing toxin-mediated damage28, and may prevent recurrent CDI by reducing clinical disease manifestations until the microbiota is replenished and colonisation resistance to C. difficile is restored27. We hypothesise that HBC could be orally administered to patients in parallel with standard-of-care antibiotics, and continued after the cessation of antibiotic treatment to prevent recurrent infection. Colostrum allows transit of the antibodies to the colon26, where they can neutralise toxin activity directly at the site of infection, allowing a healthy microbiota to be restored and repair of the damaged gut to proceed. By comparison, it is unknown how systemically-delivered monoclonal antibodies protect against toxins located in the large intestine, although it has been suggested that toxin-mediated damage to the gut mucosa allows these antibodies to leak into the gut lumen and then neutralise the toxins, protecting the mucosa from further toxin-induced damage29. Extensive toxin-mediated mucosal damage leading to a leaky gut permits extra-intestinal dissemination of toxins and other lumen components, resulting in systemic disease in animal models30. Orally delivered toxin-neutralising antibodies could reduce gut leakage and subsequent systemic effects by lessening the severity of gut damage and decreasing the extra-intestinal dissemination of toxins or other damaging factors.
Our work provides strong proof-of-principle evidence that HBC for the prevention and treatment of C. difficile infections is effective. Oral delivery of antibodies allows easy administration to patients as a treatment for primary or relapsing disease, while the low cost of production (USD$1/gram of colostrum polyclonal antibodies (Immuron Ltd, personnel communication, December, 2016) compared to USD$100/gram for the equivalent production of monoclonal antibodies31) may permit their use prophylactically. Colostrum IgG antibodies can be readily generated in large amounts, with an average of 200 grams of pure IgG extracted from 5 kilograms of colostrum produced from each dairy cow (Immuron Ltd, personnel communication, April, 2017). These features make HBC an ideal product for the treatment of C. difficile and other enteric pathogens, particularly those that are antibiotic resistant, and the successful use of HBC for Escherichia coli, Cryptosporidium, rotavirus and Shigella flexneri infections16 supports this approach.
Methods
Bacterial culture conditions and strains
C. difficile strains were cultured on HIS agar [heart infusion (HI) (Oxoid) containing 1.5% glucose, 0.1% (w/v) L-cysteine, 1.5% (w/v) agar] in an anaerobic chamber (Don Whitley Scientific) at 37 °C. To prepare vegetative cells, spores were germinated on HIS-T agar [HIS agar containing 0.1% (w/v) sodium taurocholate (New Zealand Pharmaceuticals)], subcultured and inoculated into HIS-T broth. A mid-exponential culture (OD600 0.40–0.70) was used to inoculate another HIS-T broth. A 1:100 dilution of this culture (OD600 of 0.50) was used to inoculate a final HIS-T broth and grown to an OD600 of 0.50–0.70. The cells were washed 3 times with PBS (3200 × g for 20 minutes at 4 °C) and sonicated to prepare whole cell lysates for use in Western blotting. For immunisations, cells were fixed using 10% (v/v) formalin in PBS overnight (4 °C), then washed 3 times with PBS. To prepare spores, strains were subcultured on HIS-T agar and inoculated into 500 mL Tryptone Yeast (TY) broth [3.0% tryptone, 2.0% yeast extract and 0.1% sodium thioglycolate] and grown anaerobically (37 °C) for 10 days. Spores were harvested by centrifugation at 10,000 × g for 20 minutes at 4 °C and washed five times with chilled dH2O. To inactivate spores for vaccinations, spores were exposed to gamma irradiation for 200 hours (12, 840 Gray) followed by overnight treatment with 10% (v/v) formalin in PBS at 4 °C and three washes with PBS. For mouse infections, spores were resuspended in PBS containing 0.05% tween-80 and heat shocked at 65 °C for 20 min. Strains used in this study were: DLL3109 (TcdA+TcdB+)32, M7404 (TcdA+TcdB+)33, R20291 (TcdA+TcdB+)34, VPI10463 (TcdA+TcdB+)35, 630∆ERM (TcdA+TcdB+)36, DLL3111 (TcdA+TcdB+), DLL7518 (TcdA−TcdB+), JGS6133 (TcdA+TcdB+)37, AI35 (TcdA−TcdB+)38, 1470 (ATCC 43598; TcdA−TcdB+)39, and CD37 (TcdA−TcdB−)40.
Preparation of exosporium
Exosporium from the spore surface was prepared as previously described41. Briefly, spores were resuspended in extraction buffer (50 mM Tris-HCl, pH 10; 8 M Urea; 2% (v/v) 2-mercaptoethanol) and heated (15 minutes, 90 °C) prior to centrifugation at 13, 000 × g for 10 minutes at room temperature (RT). The supernatant was dialysed against PBS overnight using 6000–8000 MWCO dialysis tubing. For immunisations, the exosporium preparation was fixed using 10% formalin (v/v) in PBS (8 hours, 4 °C) prior to further dialysis and concentrated to 1 mg/mL in PBS using 10,000 MWCO centrifugal filters.
Preparation of surface layer proteins (SLP)
SLP from the vegetative cell surface was prepared as described previously42. Briefly, overnight cultures of vegetative cells were harvested by centrifugation at 3200 × g for 20 minutes (4 °C). Cells were washed once with PBS, then resuspended in 0.04 volumes of 0.2 M glycine pH 2.2 and incubated, mixing end over end, for 30 minutes at RT. The sample was centrifuged at 16, 200 × g for 10 minutes and the supernatant containing S-layer proteins was collected. The supernatant was neutralised using 2 M Tris pH 9.0 and dialysed overnight in PBS. For immunisations, the SLP preparation was fixed, dialysed and concentrated as before.
Partial purification of C. difficile toxins from culture media
A three day old 500 mL TY broth culture was centrifuged (10,000 × g, 15 min, 4 °C) and the toxin-rich supernatant was filter sterilised (0.2 μm filter) and concentrated ten-fold using a 100,000 MWCO concentrating cassette. The supernatants were dialysed using PBS, filter sterilised (0.2 μm filter) and adjusted to 1 mg/mL in PBS.
Construction of a C-terminal TcdB expression plasmid
Using C. difficile strain 630 genomic DNA as a template, a PCR product corresponding to the C-terminal end of TcdB was amplified as previously described21 and cloned into pGEM-T Easy (Promega); the resulting vector was used as the template for further PCR. The TcdB fragment was amplified from this vector using DLP865 (5′ ATGCCATATGGAAGAAAATAAGGTGTCACAAG 3′) and DLP866 (5′ ATGCCTCGAGTTGAGCTGTATCAGGATCA 3′), which are the same as OL169 and OL17021, except that the BamHI and EcoRI restriction sites were changed to NdeI and XhoI to facilitate cloning into pET30b. This final vector, pDLL217, was introduced into E. coli BL21 (DE3) cells for protein expression.
Recombinant protein expression and purification
E. coli cells were cultured overnight in Terrific broth supplemented with 50 µg/mL of kanamycin at 37 °C with shaking on a rotating platform at 200 rpm. Bacteria from these cultures were inoculated into 200 mL of Terrific broth (1:20 dilution), which was grown until the OD600nm reached 1.2. Expression was induced by adding 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG; Promega) for five hours (37 °C), with shaking at 250 rpm. Cells were pelleted by centrifugation at 2,500 × g for 15 minutes at 4 °C, resuspended in lysis buffer [100 mM sodium phosphate pH 7.4, 0.15 M NaCl] and frozen at −80 °C. For protein purification, cells were washed twice in wash buffer I [100 mM sodium phosphate pH 7.4, 0.15 M NaCl, 1% (v/v) Triton X-100, 1 mM 2-mercaptoethanol] followed by one wash with wash buffer II [100 mM sodium phosphate pH 7.4, 0.15 M NaCl, 1 mM 2-mercaptoethanol]. Cell pellets were resuspended in resuspension buffer [100 mM sodium phosphate pH 7.4, 0.15 M NaCl, 8 M urea, 10 mM 2-mercaptoethanol] and incubated at 25 °C for 1 hour with shaking at 100 rpm. Cell pellets were compacted by centrifugation at 30, 000 × g for 20 minutes at 10 °C and the filtered insoluble fraction was loaded onto a His Trap column (GE Healthcare Biosciences) at 1 mL/minute. The column was washed with His Trap buffer [100 mM sodium phosphate pH 7.4, 0.15 M NaCl, 20 mM imidazole, 8 M urea, 1 mM 2-mercaptoethanol] and the protein eluted from the column in 5 column volumes using His Trap elution buffer [100 mM sodium phosphate pH 7.4, 0.15 M NaCl, 0.5 M imidazole, 8 M urea, 1 mM 2-mercaptoethanol]. Eluted protein was loaded onto a Hi Prep 26/10 desalting column (GE Healthcare Biosciences) and 2 mL fractions collected in desalting buffer [100 mM sodium phosphate pH 7.4, 0.15 M NaCl, 8 M urea]. Fractions containing the protein of interest were pooled, diluted to 30 mL with desalting buffer and dialysed (4 °C) against a series of buffers (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 10% (v/v) glycerol) containing successively decreasing amounts of urea (4 M, 2 M, 1 M, 0 M) using 10 kDa cut-off dialysis tubing. The protein was concentrated to 1 mg/mL and frozen at −80 °C.
Immunisations
Animal handling and experimentation was performed in accordance with Victorian Government guidelines (Department of Economic Development, Jobs, Transport & Resources. All experimental protocols were approved by the Immuron Ltd animal ethics committee (ethics numbers A17 and A18). Pregnant Holstein dairy cows received three intramuscular vaccinations beginning eight weeks prior to the expected calving date. Immunisations were administered every two weeks, with the final dose administered four weeks prior to calving. Vaccines were prepared by emulsifying 1 mL of each antigen with the same volume of Montanide ISA206VG adjuvant (Tall Bennett). Cows received 1 × 109 spores per dose (3 cows), 2 × 1010 vegetative cells per dose (3 cows), 0.5 mg (6 cows) or 1 mg of TcdB (3 cows) per dose, 0.5 mg of exosporium per dose (3 cows), or 0.5 mg of SLP per dose (3 cows).
Colostrum preparation and purification of IgG antibody
Colostrum was collected up to 12 hours post calving. Fat was removed by centrifugation at 10,000 × g for 30 minutes (4 °C) and the colostrum pasteurised at 63.5 °C for 30 minutes. Following rapid chilling on ice, the colostrum was centrifuged at 10,000 × g for 30 minutes at 4 °C and concentrated to approximately 60% of the original volume using a 30, 000 MWCO cassette. The original volume of colostrum was restored by the addition of dH2O and diafiltration performed by concentrating the colostrum again to approximately 60% of the original volume. Concentrated colostrum was then freeze dried at −80 °C and reconstituted to 10% (w/v) in dH2O. IgG antibody was purified from the colostrum using a protein G column (GE Healthcare) according to the manufacturer’s instructions.
Measurement of C. difficile-specific IgG levels in colostrum
C. difficile-specific IgG binding titres in whole HBC were measured by enzyme-linked immunosorbent assay (ELISA). Wells of a 96-well Maxisorp plate were coated with 100 µl of the immunisation antigens [recombinant TcdB (1 μg/mL), vegetative cells (106/mL), SLP (1 µg/mL), spores (105/mL) or exosporium (1 µg/mL)] in 50 mM carbonate-bicarbonate buffer [pH 9.6] and incubated overnight (4 °C). Plates were washed five times with PBS-0.1% (v/v) tween-20 (PBST), blocked for two hours (RT) with 5% (w/v) casein in PBS and washed again. Reconstituted colostrum was diluted 1:250 in 0.5% (w/v) casein in PBST. Five four-fold serial dilutions were performed and 100 μl of colostrum was added to triplicate wells and incubated at RT for two hours. As a background control, wells received 0.5% casein in PBST. Plates were washed and 100 μl of goat anti-bovine IgG-peroxidase conjugate, diluted 1:2000 in PBST, was added to each well and incubated for one hour (RT). Following washing, colour was developed by the addition of 100 μl/well of 3,3′,5,5;-tetramethylbenzidine (TMB). The reaction was stopped by 50 μl/well of 2 M H2SO4. Absorbance was read at 450 nm. The endpoint titre was determined as the reciprocal of the highest analyte dilution that gave a value two times higher than the mean background control reading.
SDS-PAGE and Western blotting
Proteins (10 µg) were separated by 12.5% (v/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. Proteins were detected using purified IgG colostrum antibodies (1:500 dilution of 30 mg/mL stock), followed by a goat anti-bovine IgG-HRP antibody (1:2000 dilution). Antigen-antibody complexes were detected using a Western Lightning Chemiluminescence reagent kit and visualised using a Chemidoc system (Biorad).
TcdB neutralisation assays
Vero cells were cultured as described8. For neutralisation assays, cells were seeded in 96-well plates at 1 × 104 cells/well and incubated for 24 hours at 37 °C in 5% CO2. Purified TcdB from strain VPI10463 (Abcam) was diluted in MEM-α containing 1% (v/v) FCS to 6.4 ng/mL, 1.25 ng/mL, 0.25 ng/mL and 0.05 ng/mL. Toxin preparations were incubated (60 minutes) with either PBS or a 1:20 dilution of a 20 mg/mL stock of purified non-immune IgG or TcdB-specific IgG. The toxin or toxin-antibody complexes were then added to the Vero cells (100 μl/well), resulting in final toxin concentrations of 640 pg, 125 pg, 25 pg and 5 pg. Cells were incubated (24 hours) before being observed by microscopy (Olympus 1X71 inverted microscope) and scored for cytopathic effect (CPE). Percentage cell death was determined as the percentage of cells undergoing cell rounding.
Murine models for disease prevention and treatment
Animal handling and experimentation was performed in accordance with Victorian State Government regulations and approved by the Monash University Animal Ethics Committee (Monash University AEC no. SOBSB/M/2010/25 and MARP/2014/134). Male, 6–7 week old, C57BL/6 J mice (Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia) were pre-treated with an antibiotic cocktail in the drinking water for seven days as described22, followed by two days of cefaclor alone. Antibiotic treatment ceased one day prior to infection with C. difficile strain DLL3109 (103 spores/mouse) by oral gavage. For colostrum prophylaxis studies, mice were administered colostrum in the drinking water two days prior to infection, continuing for the trial duration. For treatment studies, mice received 200 µl of colostrum (10% w/v) or 100 µl of vancomycin (6 mg/mL) by oral gavage six hours post-infection followed by immediate access to colostrum (10% w/v) or vancomycin (0.4 mg/mL) in the drinking water. Mice were monitored twice daily for disease signs (weight loss, behavioural and physical changes, and diarrhoea). Faecal pellets were collected 24 hours post-infection and resuspended in PBS (100 mg/mL), heat shocked (30 minutes, 65 °C) and plated for spore enumeration as described43. Animals were humanely killed by CO2 overdose or cervical dislocation when defined endpoints were met as previously defined22. The colon and caecum from each mouse were swiss-rolled44 and fixed in 4% (w/v) phosphate buffered formaldehyde solution, sectioned transversely and stained with Periodic Acid Schiffs (PAS) and Alcian Blue. Tissues were scanned using Aperio ScanScope and imaged using Aperio ImageScope and assessed for histopathological damage as described22.
Murine model for prevention of disease recurrence
Mice were pre-treated with antibiotics and infected as above. Ten hours following infection, all mice were administered 100 µl of vancomycin (6 mg/mL) by oral gavage and then given access ad libitum to either vancomycin (0.4 mg/mL) alone in the drinking water (group A) or colostrum (15% (w/v)) containing 0.4 mg/mL of vancomycin (group B). These solutions were replenished daily. Mice were monitored daily for signs of infection and faecal samples were enumerated for the presence of spores. Once levels of C. difficile in the faeces reached undetectable levels, vancomycin treatment ceased (day 8) and mice were housed individually to assess disease relapse. At this point, mice were given either plain water (group A) or colostrum (group B). Mice were then weighed and faecal samples collected daily to detect spore shedding and disease relapse. Mice were humanely euthanised according to animal ethics guidelines if they lost 10% body weight in 24 hours or met other disease criteria as previously defined22.
Statistical Analysis
Statistical analysis was performed using Prism 6 software (GraphPad Software). The in vitro toxin neutralisation assay was analysed by two-way ANOVA with a post hoc Bonferroni multiple comparison test. The Kaplan-Meier survival curves were assessed using a log-rank (Mantel-Cox) test. Differences in data values were considered significant at a P value of <0.05.
Data Availability
All data generated or analysed during this study are included in this published article.
References
Smits, W. K., Lyras, D., Lacy, D. B., Wilcox, M. H. & Kuijper, E. J. Clostridium difficile infection. Nature reviews. Disease primers 2, 16020, doi:10.1038/nrdp.2016.20 (2016).
Seekatz, A. M. & Young, V. B. Clostridium difficile and the microbiota. J Clin Invest 124, 4182–4189, doi:10.1172/JCI72336 (2014).
Awad, M. M., Johanesen, P. A., Carter, G. P., Rose, E. & Lyras, D. Clostridium difficile virulence factors: Insights into an anaerobic spore-forming pathogen. Gut Microbes 5, 579–593, doi:10.4161/19490976.2014.969632 (2014).
Chen, S., Sun, C., Wang, H. & Wang, J. The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins. Toxins 7, 5254–5267, doi:10.3390/toxins7124874 (2015).
Kuehne, S. A., Cartman, S. T. & Minton, N. P. Both, toxin A and toxin B, are important in Clostridium difficile infection. Gut Microbes 2, 252–255, doi:10.4161/gmic.2.4.16109 (2011).
Kuehne, S. A. et al. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713, doi:10.1038/nature09397 (2010).
Kuehne, S. A. et al. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis 209, 83–86, doi:10.1093/infdis/jit426 (2014).
Lyras, D. et al. Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–1179, doi:10.1038/nature07822 (2009).
Carter, G. P., Awad, M. M., Kelly, M. L., Rood, J. I. & Lyras, D. TcdB or not TcdB: a tale of two Clostridium difficile toxins. Future Microbiol 6, 121–123, doi:10.2217/fmb.10.169 (2011).
Drudy, D., Fanning, S. & Kyne, L. Toxin A-negative, toxin B-positive Clostridium difficile. Int J Infect Dis 11, 5–10, doi:10.1016/j.ijid.2006.04.003 (2007).
King, A. M., Mackin, K. E. & Lyras, D. Emergence of toxin A-negative, toxin B-positive Clostridium difficile strains: epidemiological and clinical considerations. Future Microbiol 10, 1–4, doi:10.2217/fmb.14.115 (2015).
Wilcox, M. H. et al. Bezlotoxumab for Prevention of Recurrent Clostridium difficile Infection. N Engl J Med 376, 305–317, doi:10.1056/NEJMoa1602615 (2017).
Steele, J., Mukherjee, J., Parry, N. & Tzipori, S. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J Infect Dis 207, 323–330, doi:10.1093/infdis/jis669 (2013).
Pakkanen, R. & Aalto, J. Growth factors and antimicrobial factors of bovine colostrum. Int Dairy J 7, 285–297, doi:10.1016/S0958-6946(97)00022-8 (1997).
Kelly, G. S. Bovine colostrums: a review of clinical uses. Altern Med Rev 8, 378–394 (2003).
Steele, J., Sponseller, J., Schmidt, D., Cohen, O. & Tzipori, S. Hyperimmune bovine colostrum for treatment of GI infections: a review and update on Clostridium difficile. Hum Vaccin Immunother 9, 1565–1568, doi:10.4161/hv.24078 (2013).
Sponseller, J. K. et al. Hyperimmune bovine colostrum as a novel therapy to combat Clostridium difficile infection. J Infect Dis 211, 1334–1341, doi:10.1093/infdis/jiu605 (2015).
van Dissel, J. T. et al. Bovine antibody-enriched whey to aid in the prevention of a relapse of Clostridium difficile-associated diarrhoea: preclinical and preliminary clinical data. J Med Microbiol 54, 197–205, doi:10.1099/jmm.0.45773-0 (2005).
Numan, S. C., Veldkamp, P., Kuijper, E. J., van den Berg, R. J. & van Dissel, J. T. Clostridium difficile-associated diarrhoea: bovine anti-Clostridium difficile whey protein to help aid the prevention of relapses. Gut 56, 888–889, doi:10.1136/gut.2006.119016 (2007).
Warny, M. et al. Bovine immunoglobulin concentrate-Clostridium difficile retains C. difficile toxin neutralising activity after passage through the human stomach and small intestine. Gut 44, 212–217 (1999).
Letourneur, O., Ottone, S., Delauzun, V., Bastide, M. C. & Foussadier, A. Molecular cloning, overexpression in Escherichia coli, and purification of 6x his-tagged C-terminal domain of Clostridium difficile toxins A and B. Protein Expr Purif 31, 276–285 (2003).
Carter, G. P. et al. Defining the Roles of TcdA and TcdB in Localized Gastrointestinal Disease, Systemic Organ Damage, and the Host Response during Clostridium difficile Infections. MBio 6, e00551, doi:10.1128/mBio.00551-15 (2015).
Kelly, C. P. et al. Anti-Clostridium difficile bovine immunoglobulin concentrate inhibits cytotoxicity and enterotoxicity of C. difficile toxins. Antimicrobial agents and chemotherapy 40, 373–379 (1996).
Lyerly, D. M., Bostwick, E. F., Binion, S. B. & Wilkins, T. D. Passive immunization of hamsters against disease caused by Clostridium difficile by use of bovine immunoglobulin G concentrate. Infect Immun 59, 2215–2218 (1991).
Mattila, E. et al. A randomized, double-blind study comparing Clostridium difficile immune whey and metronidazole for recurrent Clostridium difficile-associated diarrhoea: efficacy and safety data of a prematurely interrupted trial. Scand J Infect Dis 40, 702–708, doi:10.1080/00365540801964960 (2008).
Kelly, C. P. et al. Survival of anti-Clostridium difficile bovine immunoglobulin concentrate in the human gastrointestinal tract. Antimicrobial agents and chemotherapy 41, 236–241 (1997).
Warn, P. et al. Disease progression and resolution in rodent models of Clostridium difficile infection and impact of antitoxin antibodies and vancomycin. Antimicrobial agents and chemotherapy 60, 6471–6482, doi:10.1128/AAC.00974-16 (2016).
Dzunkova, M. et al. The Monoclonal Antitoxin Antibodies (Actoxumab-Bezlotoxumab) Treatment Facilitates Normalization of the Gut Microbiota of Mice with Clostridium difficile Infection. Frontiers in cellular and infection microbiology 6, 119, doi:10.3389/fcimb.2016.00119 (2016).
Cohen, O. R. et al. Systemically administered IgG anti-toxin antibodies protect the colonic mucosa during infection with Clostridium difficile in the piglet model. PLoS One 9, e111075, doi:10.1371/journal.pone.0111075 (2014).
Steele, J. et al. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis 205, 384–391, doi:10.1093/infdis/jir748 (2012).
IAVIReport. Making it to Manufacturing. 18, No. 2 (2014).
Richards, M. et al. Severe infection with Clostridium difficile PCR ribotype 027 acquired in Melbourne, Australia. Med J Aust 194, 369–371 (2011).
Carter, G. P. et al. Binary toxin production in Clostridium difficile is regulated by CdtR, a LytTR family response regulator. J Bacteriol 189, 7290–7301, doi:10.1128/JB.00731-07 (2007).
Stabler, R. A. et al. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J Bacteriol 188, 7297–7305, doi:10.1128/JB.00664-06 (2006).
Dove, C. H. et al. Molecular characterization of the Clostridium difficile toxin A gene. Infect Immun 58, 480–488 (1990).
Hussain, H. A., Roberts, A. P. & Mullany, P. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J Med Microbiol 54, 137–141, doi:10.1099/jmm.0.45790-0 (2005).
Carter, G. P. et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 7, e1002317, doi:10.1371/journal.ppat.1002317 (2011).
Squire, M. M. et al. Novel molecular type of Clostridium difficile in neonatal pigs, Western Australia. Emerg Infect Dis 19, 790–792, doi:10.3201/eid1905.121062 (2013).
Delmee, M. & Avesani, V. Virulence of ten serogroups of Clostridium difficile in hamsters. J Med Microbiol 33, 85–90, doi:10.1099/00222615-33-2-85 (1990).
Smith, C. J., Markowitz, S. M. & Macrina, F. L. Transferable tetracycline resistance in Clostridium difficile. Antimicrobial agents and chemotherapy 19, 997–1003 (1981).
Sylvestre, P., Couture-Tosi, E. & Mock, M. A collagen-like surface glycoprotein is a structural component of the Bacillus anthracis exosporium. Mol Microbiol 45, 169–178 (2002).
Wright, A. et al. Proteomic analysis of cell surface proteins from Clostridium difficile. Proteomics 5, 2443–2452, doi:10.1002/pmic.200401179 (2005).
Lyon, S. A., Hutton, M. L., Rood, J. I., Cheung, J. K. & Lyras, D. CdtR Regulates TcdA and TcdB Production in Clostridium difficile. PLoS Pathog 12, e1005758, doi:10.1371/journal.ppat.1005758 (2016).
Moolenbeek, C. & Ruitenberg, E. J. The “Swiss roll”: a simple technique for histological studies of the rodent intestine. Laboratory animals 15, 57–59 (1981).
Acknowledgements
The authors acknowledge the facilities, scientific and technical assistance of the Monash Histology Platform, Department of Anatomy and Developmental Biology, Clayton, Monash University and the Monash Protein Production Unit, Clayton, Monash University for the expression and purification of recombinant Toxin B protein. This work was supported by the Australian Research Council (Linkage project LP110200752) and Research Connections (Grants CF14/3721 and RC47582).
Author information
Authors and Affiliations
Contributions
M.L.H., B.A.C. and D.L. were involved in concept and design, data analysis and interpretation, manuscript drafting and critical manuscript revision for important intellectual content. M.L.H., B.A.C., K.E.M., S.A.L. and M.L.J. were involved in data acquisition and statistical analysis. J.I.R. and D.L. obtained funding and helped with study design. D.L. was responsible for study supervision. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing Interests
This study was partly funded by our industry partner, Immuron Limited, who provided input into study design, but had no role in data collection and analysis, interpretation or decision to publish or preparation of the manuscript. Patent 20160083457 resulted from this work.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hutton, M.L., Cunningham, B.A., Mackin, K.E. et al. Bovine antibodies targeting primary and recurrent Clostridium difficile disease are a potent antibiotic alternative. Sci Rep 7, 3665 (2017). https://doi.org/10.1038/s41598-017-03982-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03982-5
This article is cited by
-
Cephamycins inhibit pathogen sporulation and effectively treat recurrent Clostridioides difficile infection
Nature Microbiology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
