
Abstract
A new method has been developed for the isolation of 223,224,225Ra, in high yield and purity, from a proton irradiated 232Th matrix. Herein we report an all-aqueous process using multiple solid-supported adsorption steps including a citrate chelation method developed to remove >99.9% of the barium contaminants by activity from the final radium product. A procedure involving the use of three columns in succession was developed, and the separation of 223,224,225Ra from the thorium matrix was obtained with an overall recovery yield of 91 ± 3%, average radiochemical purity of 99.9%, and production yields that correspond to physical yields based on previously measured excitation functions.
Similar content being viewed by others
Introduction
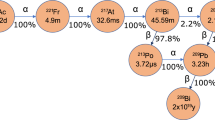
Radium-223 chloride (t1/2 11.4d) is available as a U.S. FDA approved pharmaceutical for the treatment of bone metastases under the trademark Xofigo© 1. Ionic radium is a bone seeking agent, due to its chemical similarity to calcium. It preferentially accumulates in the rapidly forming cells in bone metastases1, 2. Radium-223 is the first alpha emitting isotope that obtained FDA approval for the treatment of cancer. Other radium isotopes of interest for preclinical research are 224Ra (t1/2 3.6d) and 225Ra (t1/2 14.9d). Radium-224 has alpha-therapeutic properties for use similar to that of 223Ra, however, it has a shorter half-life. It thus could be more suitable for molecular targets with a shorter biological half-life than that of 223Ra3 or as a source for the 212Pb/212Bi generator4, 5. Radium-225 is of interest due to its decay to 225Ac (t1/2 9.92d), another alpha emitting isotope relevant to the treatment of cancer6, 7. There are also non-medical interests in radium isotopes. For instance, 225Ra is of interest in the search for an atomic electric-dipole moment8, 9.
These radium isotopes are challenging to produce directly, and so are commonly obtained from their radioactive parents’ decay chains. Radium-223 is derived from 227Ac (t1/2 21.77y) via 227Th (t1/2 18.7d). Actinium-227 comes only from the decay of 235U, whose supply is tightly-controlled10, or via neutron irradiation of 226Ra11. Radium-224 forms via the alpha decay of 228Th (t1/2 1.91 a). Radium-225 is a decay product of 229Th (t1/2 7400 a) and may theoretically be produced by neutron, proton or photon bombardment of radioactive 226Ra targets.
Actinium-225 production via spallation of thorium targets is currently being explored at Los Alamos National Laboratory (LANL), Brookhaven National Laboratory (BNL) and Oak Ridge National Laboratory (ORNL)12. The amount of 225Ac produced by this method represents a small fraction of the total activity produced. Many of the additional radionuclides generated have the potential for use in a variety of medical applications13,14,15. Included in these side products are 223,224,225Ra, and the predicted yields of 223,224,225Ra along with 227Th (parent radionuclide of 223Ra) are in the GBq/mAh range13, 16. These yields are large enough to justify the investigation of pathways isolating these radium isotopes from the proton irradiated thorium matrix. The separation of radium from thorium has been reported in the literature17,18,19,20,21, and in a recent study the recovery of 223Ra from proton irradiated thorium target has been described in more detail22 In this recent study by Vasiliev et al. radium sorption on extraction chromatography “SR resin” has been investigated, and measured HClO4 concentration dependent capacity factors for several fission products including radium were reported. The study also presented exemplary SR resin/HNO3 and cation exchanger resin/HClO4 elution profiles in order to demonstrate radium separation from barium and other fission products. A potential draw-back in this approach is the necessity of an HDEHP/toluene liquid-liquid extraction step for initial bulk thorium removal, which was implemented prior to chromatography-based activation/fission product recovery. Liquid-liquid extraction procedures introduce larger amounts of contaminated solvent waste and complicate remote or automated handling for the purpose of scale-up to routine large-scale radium production. Furthermore, the use of organic solvents like toluene is a complicating factor in the establishment of radium isotopes for human drug applications.
In the present work, we introduce a novel, aqueous recovery process that solely relies on multiple steps of solid-support sorption. For a seamless integration of an additional isotope recovery “module”, we modified our previously developed method for the isolation of 225Ac to co-extract radium isotopes (namely 223,224,225Ra)12. As the production of 225Ac remains our main priority, it is important to obtain these radium isotopes in high purity and yield with minimal impact on 225Ac recovery. Described within is a method to obtain these radium isotopes in high yield and radiochemical purity (>99.9%) as an ancillary process to the recovery of 225Ac.
Results
Thorium metal proton irradiation
The thorium target irradiation resulted in the formation of activation products 223,224,225Ra along with several fission products. The production yield of 223,224,225Ra (10 days post irradiation) in the dissolved 10 g target pre-separation are shown in Table 1. The theoretical cumulative yields for 223,224,225Ra (10 days post irradiation), predicted by published cross section measurements13, 16, are also shown in Table 1. The discrepancies of the measured values from the predicted values likely stem from the difficulty in integrating gamma peaks with high backgrounds and multiple contributions from various sources, uncertainty in cross-section measurements, and large dilution factors for HPGe measurements14, 23, 24.
Chemical Separation
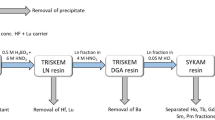
Cation exchange column contact resulted in the separation of the majority of the fission products, fractions 1–4 (Fig. 1). The addition of 6 M nitric acid removed 97 ± 6% of radium along with barium, actinium, cesium, and lanthanides (fraction 5). Loading this fraction onto a TEHDGA) column resulted in the elution of ≥99% of the radium, barium, and cesium isotopes while retaining the remaining isotopes (Fig. 2). Three column bed volumes were chosen to assess Ra/Ba separation behavior in a 0.32 M citrate pH 5.5 mobile phase (or eluent). As shown in Fig. 3, the 1 and 2 mL sized columns resulted only in partial separation of Ra from Ba accompanied by substantial losses. The 3 mL column, however, resulted in an effective separation of radium from barium and a total radium recovery yield of 91 ± 3% with a radiochemical purity of ≥99.9%. Cesium isotopes eluted in the initial loading effluent and also in the 0.1 M HNO3 washes. Due to the broad elution peak of radium when using 0.32 M citrate, radium was eluted with 6 M HNO3 once barium was no longer detected in the 0.32 M citrate fractions collected from the 3 mL column. The fully integrated separation scheme, as adapted from Radchenko et al.12 now including the newly developed Ra/Ba separation module, is shown in Fig. 4.

Cation Exchange Chromatography.

TEHDGA Extraction Chromatography.

Cation exchange separation of radium/barium separation using 0.32 M Citrate pH 5.5 with (A) 1 mL, (B) 2 mL, and (C) 3 mL of AG 50WX8 resin. In C, radium was eluted with 6 M HNO3. Plotted lines are eye guides only.

Final 225Ac separation schematic with recovery of radium isotopes.
Discussion
This method uses a three column approach to the separation of radium isotopes from a proton irradiated thorium matrix adding only one additional column to the 225Ac separation scheme. The benefit to this process over the previously published process is the elimination of large volumes of radioactive organic wastes22. Mixed waste (radioactive organic solutions) are a limitation to many facilities due to the high cost and difficulty of disposal.
One concern for separating radium isotopes from fission products is potential contamination with the strontium isotopes; 89Sr and 90Sr. These isotopes are of concern due to their long half-lives (50.6 days and 28.8 years respectively) and lack of a strong gamma line for detection. Strontium and barium both demonstrate stronger complexation with citrate than radium25. Separation of strontium, barium, and radium with citrate media has been performed by Thompson et al. and it is shown that strontium elutes before barium, which in turn elutes before radium26. Therefore using this method would effectively remove contaminating strontium isotopes before radium elution.
The importance of supplying radionuclides with high purity and specific activity, especially therapeutic radioisotopes, has been increasingly relevant in the field of nuclear medicine. The results described within show that radium isotopes can be obtained as a by-product of 225Ac production with an average radiochemical purity ≥99.9% as determined by activity. Yield estimates establish that quantities up to 0.3 Ci of 223Ra and 0.1 Ci of 225Ra13 and 0.2 Ci of 224Ra16 can be made and recovered from thorium targets in tandem with anticipated Ci-scale 225Ac production at LANL or BNL isotope facilities. Further, the citric acid flow through from the cation exchange column can be reprocessed 21 days after initial purification to obtain 223Ra from the decay of 227Th. Quantities as high as 4 Ci of 223Ra would be available during this second processing13. As the radium recovery is performed downstream of the 225Ac recovery process, there is no effect on the processing time or yield of 225Ac.
Isotopic impurities of radium cannot be separated chemically, however, there are many uses for radium isotopes that do not require isotopic purity. For instance pure 225Ac can be obtained from this source after 18 days have passed. Once the 225Ac has grown in, separation from the radium isotopes can be obtained employing the TEHDGA column as shown in Fig. 2. Additionally, since 223Ra is FDA approved for radiotherapy, there has been interest in developing radium chelates for use in targeted alpha therapy27. For the purposes of designing and testing chelates, an isotopically pure form of 223Ra would not be necessary. Furthermore, the non-medical interest of 225Ra for the measurement of an atomic electric-dipole moment also does not require isotopically pure 225Ra. The radium recovered from 225Ac processing could provide a consistent supply of radium for these purposes.
Materials and Methods
Chemicals
Thorium metal targets were manufactured at the Los Alamos National Laboratory (LANL). Small pieces of thorium metal (purity >99% as determined via X-ray fluorescence spectroscopy) were obtained from LANL’s internal inventory. The raw material was arc melted and rolled into sheets with mean thickness of 0.50 ± 0.02 mm for the use as proton beam targets.
All experiments were performed in triplicate unless stated otherwise. All chemicals were used without further purification. Nitric acid and hydrochloric acid, both Optima grade, were purchased from Fisher Scientific (Pittsburgh, PA, USA). Citric acid (99.9%) was obtained from Sigma–Aldrich (St Louis, MO, USA), and deionized water (≥18 MΩcm) was prepared on site with a Millipore water purification system. Cation exchange resin AG 50WX8, 200–400 mesh, was obtained from Biorad (Hercules, CA, USA). Branched DGA resin (N,N,N′,N′-tetrakis-2-ethylhexyldiglycolamide; abbreviated TEHDGA) 100–150 µm, was purchased from Eichrom Inc. (Lisle, IL, USA).
Gamma-ray spectrometry
The radioactivity measurements made at ORNL to determine radium yields in proton irradiated targets were conducted using a well-shielded, Canberra Model GC2020 High-Purity Germanium detector with a relative efficiency of 20%. A PC-based multichannel analyzer utilizing Canberra Genie 2000 software was coupled to the detector. The measured resolution of the detector was 2.0 keV at 1.33 MeV. Energy and efficiency calibrations were completed using a γ ray source traceable to the National Institute of Standards and Technology (NIST). Spectra collection times varied from one-hour counts for initial sample dilutions to 36-hour counts for severely decayed samples. Sample to detector geometry was varied to reduce the detector dead time below 5%. Each peak in the γ ray spectra was fitted using the non-linear least squares fit method (Canberra, 2009). When possible, multiple γ ray peaks were used to quantify the activity at end of bombardment (EOB) for each radioisotope through a weighted average method.
For separation experiments gamma-ray spectrometry was conducted using an EG&G Ortec Model GMX-35200-S HPGe detector system in combination with a Canberra Model 35-Plus multichannel analyzer. Detector diameter was 50.0 mm; detector length 53.5 mm; Be window thickness 0.5 mm; outer dead-layer thickness 0.3 µm. Detector response function determination and evaluation were performed using standards of radionuclide mixtures containing 241Am, 109Cd, 57Co, 139Ce, 203Hg, 113Sn, 137Cs, 88Y, 60Co, traceable to the National Institute of Standards and Technology (NIST) and supplied by Eckert & Ziegler, Atlanta, GA, USA. The detector was a p-type Al-windowed HPGe detector with a measured FWHM at 1333 keV of approximately 2.2 keV and a relative efficiency of about 10%. Relative total source activity uncertainties ranged from 2.6% to 3.3%. Counting dead time was kept below 10%.
Irradiation Conditions
A thorium metal target, 10 g mass, 640 mg/cm2 mass thickness, was irradiated at the Isotope Production Facility (IPF), Los Alamos National Laboratory (LANL, NM, USA). The target was encapsulated in Inconel cladding and placed into the high energy “A” slot (90 MeV incident energy) of the IPF target assembly. IPF targetry and 4π water cooling have been described previously28, 29. The target was irradiated in a proton beam with average intensity of 230 µA for 22.5 hours. The target was then transported to the IPF Hot Cell Facility where it was removed from the Inconel shell and shipped to Oak Ridge National Laboratory (ORNL) for chemical processing.
Chemical Separation
Cation exchange column
The chemical separation was adapted as follows from previous work in order to extract the radium isotopes with little impact on 225Ac recovery12, 24, 30. The irradiated 10 g target material was dissolved in 200 mL 10 M HCl and 250 μL of 2 M HF with heating (~70 °C) for approximately 2 hours. Three aliquots of the dissolved target were used to spike three separate solutions of ~1 g of 232Th metal dissolved in 40 mL 10 M HCl spiked with 40 μL 2 M HF. Each aliquot represented a mixture of all radionuclides previously identified in the target12 and each solution was processed independently according to the following steps. The solution was brought to near dryness and reconstituted in 205 mL of 1 M citrate, pH 2, and passed through a cation exchange column containing 5 mL AG50 × 8 resin, 200–400 mesh, which was preconditioned with a 1 M citrate, pH 2, solution. The eluate from the column was collected, fraction 1, then an additional 50 mL 1 M citrate, pH 2, was used to wash the column, fraction 2. The column was then washed with 10 mL 1 M nitric acid to remove residual citrate, fractions 3 (5 mL) and 4 (5 mL). Twenty-five milliliters of 6 M nitric acid was then added to the column and collected in fraction 5.
TEHDGA resin
Fraction 5 from each of the replications was then loaded onto separate TEHDGA resin columns, 1 mL 50–100 µm, preconditioned with 6 M nitric acid. The eluate was collected, fraction 6 (25 mL). The column was then washed with an additional 10 mL 6 M nitric acid, fractions 7 (5 mL) and 8 (5 mL). Next, 20 mL of 10 M nitric acid was added to the column and collected, fractions 9–12 (5 mL volume each). Finally, the column was washed with 5 mL 0.1 M nitric acid, fraction 13. All collected fractions were measured using HPGe spectrometry to identify the isotopic and elemental species present.
Radium/Barium Separation
For the radium/barium separation, a method by Power et al. was adapted as described below31. Fraction 6–8 from each of the reiterations were brought to dryness, reconstituted in 5 mL 0.1 M HNO3 and then passed through cation columns containing 1 mL (0.8 cm inner diameter), 2 mL (0.8 cm inner diameter), or 3 mL (0.8 cm inner diameter) of AG 50WX8 resin, 200–400 mesh, and the effluent was collected, fraction 14 (5 mL). For the 1 mL column, an additional 5 mL of 0.1 M HNO3 was added to wash the column, fraction 15, followed by five fractions of 0.32 M citrate pH 5.5, fractions 16–20 (5 mL each). For the 2 mL column, two additional 5 mL washes of 0.1 M HNO3 were added to the column and collected to result in fractions 15 (5 mL) and 16 (5 mL), followed by eight 5 mL fractions of 0.32 M citrate pH 5.5 (fractions 17–24, 5 mL each). For the 3 mL volume sized column, three additional 5 mL washes of 0.1 M HNO3 were added to the column and collected: fractions 15–17, followed by seven 5 mL fractions of 0.32 M citrate pH 5.5, fractions 18–24. Radium was eluted from the 3 mL column using two 5 mL fractions of 6 M HNO3, fractions 25 and 26. All collected fractions were measured using HPGe spectrometry in order to identify the radionuclidic species present.
Conclusion
A novel, all aqueous methodology for the recovery of Ra isotopes from proton irradiated thorium matrices has been presented in our study. The results presented within show that radium isotopes can be recovered in high yield and radiochemical purity in a process that is ancillary to 225Ac production. With the scale-up to 100 g thorium targets, hundreds of millicuries of radium isotopes would be available for recovery. The radium product obtained by this method is composed of 223Ra, 224Ra, and 225Ra and is suitable for chemistry applications (such as chelate development) and the nuclear physics community focused on measurement of the atomic electric-dipole moment. Additionally, the 225Ra produced via this method would be suitable as a generator of pure 225Ac for clinical applications as the other radium impurities do no decay to actinium isotopes.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Kluetz, P. G. et al. Radium Ra 223 Dichloride Injection: US Food and Drug Administration Drug Approval Summary. Clin Cancer Res 20, 9–14, doi:10.1158/1078-0432.Ccr-13-2665 (2014).
Bruland, O. S., Jonasdottir, T. J., Fisher, D. R. & Larsen, R. H. Radium-223: From radiochemical development to clinical applications in targeted cancer therapy. Current Radiopharmaceuticals 1, 203–208, doi:10.2174/1874471010801030203 (2008).
Piotrowska, A., Leszczuk, E., Bruchertseifer, F., Morgenstern, A. & Bilewicz, A. Functionalized NaA nanozeolites labeled with 224,225Ra for targeted alpha therapy. Journal of Nanoparticle Research 15, 2082, doi:10.1007/s11051-013-2082-7 (2013).
Mirzadeh, S. & Knapp, F. F. Biomedical radioisotope generator systems. J Radioan Nucl Ch Ar 203, 471–488, doi:10.1007/Bf02041524 (1996).
Su, F. M., Beaumier, P., Axworthy, D., Atcher, R. & Fritzberg, A. Pretargeted radioimmunotherapy in tumored mice using an in vivo Pb-212/Bi-212 generator. Nucl Med Biol 32, 741–747, doi:10.1016/j.nucmedbio.2005.06.009 (2005).
Boll, R. A., Malkemus, D. & Mirzadeh, S. Production of actinium-225 for alpha particle mediated radioimmunotherapy. Appl Radiat Isotopes 62, 667–679, doi:10.1016/j.apradiso.2004.12.003 (2005).
Maslov, O. D., Sabel’nikov, A. V. & Dmitriev, S. N. Preparation of 225Ac by 226Ra photonuclear reaction on an electron accelerator, MT-25 microtron. Radiochemistry 48, 195–197 (2006).
Spevak, V., Auerbach, N. & Flambaum, V. V. Enhanced T-odd, P-odd electromagnetic moments in reflection asymmetric nuclei. Phys Rev C 56, 1357–1369, doi:10.1103/PhysRevC.56.1357 (1997).
Parker, R. H. et al. First Measurement of the Atomic Electric Dipole Moment of Ra-225. Phys Rev Lett 114, doi:ARTN 233002 10.1103/PhysRevLett.114.233002 (2015).
Shishkin, D. N., Krupitskii, S. V. & Kuznetsov, S. A. Extraction generator of 223Ra for nuclear medicine. Radiochemistry 53, 404–406, doi:10.1134/S1066362211040126 (2011).
Kukleva, E., Kozempel, J., Vlk, M., Micolova, P. & Vopalka, D. Preparation of Ac-227/Ra-223 by neutron irradiation of Ra-226. J Radioanal Nucl Ch 304, 263–266, doi:10.1007/s10967-014-3432-3 (2015).
Radchenko, V. et al. Application of ion exchange and extraction chromatography to the separation of actinium from proton-irradiated thorium metal for analytical purposes. J Chromatogr A 1380, 55–63, doi:10.1016/j.chroma.2014.12.045 (2015).
Weidner, J. W. et al. Proton-induced cross sections relevant to production of Ac-225 and Ra-223 in natural thorium targets below 200 MeV. Appl Radiat Isotopes 70, 2602–2607, doi:10.1016/j.apradiso.2012.07.006 (2012).
Engle, J. W. et al. Cross sections from proton irradiation of thorium at 800 MeV. Phys Rev C 88, doi:ARTN 014604 10.1103/PhysRevC.88.014604 (2013).
Radchenko, V. et al. Formation cross-sections and chromatographic separation of protactinium isotopes formed in proton-irradiated thorium metal. Radiochim Acta 104, 291–304, doi:10.1515/ract-2015-2486 (2016).
Hogan, J. J., Gadioli, E., Gadiolierba, E. & Chung, C. Fissionability of Nuclides in the Thorium Region at Excitation-Energies to 100 Mev. Phys Rev C 20, 1831–1843, doi:10.1103/PhysRevC.20.1831 (1979).
Alhassanieh, O., Abdul-Hadi, A., Ghafar, M. & Aba, A. Separation of Th, U, Pa, Ra and Ac from natural uranium and thorium series. Appl Radiat Isotopes 51, 493–498, doi:10.1016/S0969-8043(99)00068-8 (1999).
Oliveira, J. M. & Carvalho, F. P. Sequential Extraction Procedure for Determination of Uranium, Thorium, Radium, Lead and Polonium Radionuclides by Alpha Spectrometry in Environmental Samples. Czech J Phys 56, D545–D555 (2006).
Burcik, I. Separation of Radium, Thorium, Uranium and Plutonium on Ostion Anion-Exchanger in Atmospheric Precipitation Samples. J Radioan Nucl Ch Ar 121, 285–293, doi:10.1007/Bf02041415 (1988).
Henriksen, G., Hoff, P., Alstad, J. & Larsen, R. H. Ra-223 for endoradiotherapeutic applications prepared from an immobilized Ac-227/Th-227 source. Radiochim Acta 89, 661–666, doi:10.1524/ract.2001.89.10.661 (2001).
Abou, D. S., Pickett, J., Mattson, J. E. & Thorek, D. L. J. A Radium-223 microgenerator from cyclotron-produced trace Actinium-227. Appl Radiat Isotopes 119, 36–42, doi:10.1016/j.apradiso.2016.10.015 (2017).
Vasiliev, A. N. et al. Recovery of Ra-223 from natural thorium irradiated by protons. Radiochim Acta 104, 539–547, doi:10.1515/ract-2015-2549 (2016).
Engle, J. W. et al. Ac, La, and Ce radioimpurities in Ac-225 produced in 40-200 MeV proton irradiations of thorium. Radiochim Acta 102, 569–581, doi:10.1515/ract-2013-2179 (2014).
Griswold, J. R. et al. Large scale accelerator production of (225)AC: Effective cross sections for 78–192 MeV protons incident on Th-232 targets. Appl Radiat Isotopes 118, 366–374, doi:10.1016/j.apradiso.2016.09.026 (2016).
Schubert, J. Complexes of Alkaline Earth Cations Including Radium with Amino Acids and Related Compounds. J Am Chem Soc 76, 3442–3444, doi:10.1021/ja01642a021 (1954).
Tompkins, E. R. Separation of Radium from Barium by the Use of an Ion-Exchange Column Procedure. J Am Chem Soc 70, 3520–3522, doi:10.1021/ja01190a515 (1948).
Henriksen, G., Hoff, P. & Larsen, R. H. Evaluation of potential chelating agents for radium. Appl Radiat Isotopes 56, 667–671, doi:10.1016/S0969-8043(01)00282-2 (2002).
Nortier, F. M. & Hong, B. The isotope production facility at TA-53. 6–10 (Los Alamos National Laboratory, 2010).
Ballard, B. et al. Selenium-72 formation via Br-nat(p,x) induced by 100 MeV Protons: Steps towards a novel Se-72/As-72 generator system. Appl Radiat Isotopes 70, 595–601, doi:10.1016/j.apradiso.2012.01.018 (2012).
Mirzadeh, S. Accelerator Produced 225Ac via Proton Spallation of 232Th: a Joint Research Program among ORNL, LANL, and BNL (2014).
Power, W. H., Kirby, H. W., Mccluggage, W. C., Nelson, G. D. & Payne, J. H. Separation of Radium and Barium by Ion Exchange Elution. Anal Chem 31, 1077–1079, doi:10.1021/ac60150a002 (1959).
Acknowledgements
We gratefully recognize the United States Department of Energy, Office of Science, Isotope Development and Production for Research and Application subprogram within Office of Nuclear Physics and the LANL LDRD program (LDRD 20160439ER) for financial support.
Author information
Authors and Affiliations
Contributions
V.R., M.F., and K.J. contributed the overall idea; V.R., T.M. and M.F. designed the chemical separation process. T.M. wrote the main manuscript text and prepared the figures. R.C. and T.M. prepared Table 1. A.O., R.C., R.B., J.G., L.W. and S.M. prepared and characterized sample aliquots at ORNL J.E. and F.N. calculated nuclear reaction cross sections. M.B. and E.B. reviewed the manuscript and helped with editorial input. M.F. reviewed, edited the final manuscript version and submitted it to the journal.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mastren, T., Radchenko, V., Owens, A. et al. Simultaneous Separation of Actinium and Radium Isotopes from a Proton Irradiated Thorium Matrix. Sci Rep 7, 8216 (2017). https://doi.org/10.1038/s41598-017-08506-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08506-9
This article is cited by
-
Eco-friendly and cost-effective adsorbent derived from blast furnace slag with black liquor waste for hazardous remediation
Environmental Science and Pollution Research (2023)
-
Feasibility of a novel photoproduction of 225Ac and 227Th with natural thorium target
Scientific Reports (2022)
-
Evaluations of molecular modeling and machine learning for predictive capabilities in binding of lanthanum and actinium with carboxylic acids
Journal of Radioanalytical and Nuclear Chemistry (2022)
-
Guest Edited Collection: Radioisotopes and radiochemistry in health science
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
