
Abstract
The nucleosome, composed of DNA and a histone core, is the basic structural unit of chromatin. The fission yeast Schizosaccharomyces pombe has two genes of histone H2A, hta1+ and hta2+; these genes encode two protein species of histone H2A (H2Aα and H2Aβ, respectively), which differ in three amino acid residues, and only hta2+ is upregulated during meiosis. However, it is unknown whether S. pombe H2Aα and H2Aβ have functional differences. Therefore, in this study, we examined the possible functional differences between H2Aα and H2Aβ during meiosis in S. pombe. We found that deletion of hta2+, but not hta1+, causes defects in chromosome segregation and spore formation during meiosis. Meiotic defects in hta2+ deletion cells were rescued by expressing additional copies of hta1+ or by expressing hta1+ from the hta2 promoter. This indicated that the defects were caused by insufficient amounts of histone H2A, and not by the amino acid residue differences between H2Aα and H2Aβ. Microscopic observation attributed the chromosome segregation defects to anaphase bridge formation in a chromosomal region at the repeats of ribosomal RNA genes (rDNA repeats). These results suggest that histone H2A insufficiency affects the chromatin structures of rDNA repeats, leading to chromosome missegregation in S. pombe.
Similar content being viewed by others


Introduction
In eukaryotes, genomic DNA is organized as chromatin, which comprises arrays of nucleosomes. A nucleosome contains a ~150 base pair (bp) DNA wrapped around a histone octamer, which is composed of two molecules of histones H2A, H2B, H3, and H41. In addition to these canonical histones, studies have identified a variety of histone variants in metazoans, which are used differentially on the basis of the diverse functions of chromatin2,3. On the other hand, the chromatin of the fission yeast Schizosaccharomyces pombe comprises a small number of histone species: two protein species of histone H2A (H2Aα and H2Aβ), one protein species each of histones H2B, H3, and H44,5, and two histone variants, Pht1 and Cnp1. Pht1 is the ortholog of histone H2A.Z6, and Cnp1 is the ortholog of CENP-A, the centromere-specific histone H3 variant7. These findings indicate that in S. pombe, canonical histones (H2Aα/H2Aβ, H2B, H3, and H4) and two histone variants are sufficient to achieve the broad functions of chromatin.
The S. pombe genome comprises multiple genes for the simple protein constituents of canonical histones: two genes (hta1+ and hta2+) for histone H2A, one gene (htb1+) for histone H2B, three genes (hht1+, hht2+, and hht3+) for histone H3, and three genes (hhf1+, hhf2+, and hhf3+) for histone H44,5. hta1+ and hta2+ encode two protein species of histone H2A, H2Aα and H2Aβ, respectively, which differ in three amino acid residues. hht1+, hht2+, and hht3+ encode histone H3 with an identical amino acid residue. hhf1+, hhf2+, and hhf3+ encode histone H4 with an identical amino acid residue. Therefore, in S. pombe, histone H2A uniquely has a variant set of proteins among the canonical histone proteins.
It is unknown whether S. pombe H2Aα and H2Aβ have functional differences. As mentioned before, S. pombe H2Aα and H2Aβ differ in three amino acid residues. However, both bear the C-terminal stretch characteristic of metazoan H2A.X containing the serine residue S128/S127, which is phosphorylated by ataxia telangiectasia mutated/ataxia telangiectasia and Rad3-related (ATM/ATR) kinases and is required as a DNA damage checkpoint and for DNA damage repair8. H2Aα and H2Aβ also share another C-terminal serine residue, S121, which is phosphorylated by Bub1 kinase and is required for recruiting shugoshin proteins9 and the N-terminal region required for condensin binding10. Therefore, no obvious functional differences between H2Aα and H2Aβ have been reported in vegetative cell cycles, although studies have reported that hta2 expression levels are up-regulated but hta1 expression levels remain low during meiosis11. Therefore, in this study, we examined the possible functional differences between H2Aα and H2Aβ during meiosis in S. pombe.
Results
hta2 + deletion causes meiotic defects
To examine the functional differences between H2Aα and H2Aβ, we constructed two strains with hta1+ and hta2+ deletions each (∆hta1 and ∆hta2). Cells of both ∆hta1 and ∆hta2 strains were viable and formed colonies comparable to those of wild type (WT) cells at 20 °C–36 °C (Fig. 1A). However, ∆hta2, but not ∆hta1, cells formed asci containing abnormal spores (<4 spores and/or premature spores with a thin spore wall) (Fig. 1B,C). Add-back of the hta2+ gene in ∆hta2 cells restored normal spore formation (Fig. 1D), confirming that the sporulation defect resulted from hta2+ deletion. These results indicated that hta2+ is specifically required for meiosis and sporulation.

∆hta2 cells form abnormal spores. (A) Spot assay comparing the growth of WT (TGO350), ∆hta1 (TGO351), and ∆hta2 (TGO352) cells. Dilution series (1/5 dilution) of cell suspensions were spotted on complete YES medium and grown for 11 days at 20 °C, 4 days at 26 °C, 3 days at 30 °C, or 3 days at 36 °C. (B) Abnormal spore formation in ∆hta2 cells. Meiosis was induced in WT (TGO350) and ∆hta2 (TGO352) cells, and the cells were fixed 2 days later. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI). Bright-field images of the same cells are also shown (“Phase”). Scale bar, 5 µm. (C) Frequency of asci containing abnormal spores in WT (TGO350), ∆hta1 (TGO351), and ∆hta2 (TGO352) cells. Meiosis was induced in those cells, and the frequency of asci containing abnormal spores (<4 spores and/or premature spores with a thin spore wall) was measured 2 days later. At least 200 asci were examined for each strain; the mean values from three independent experiments are shown. Error bars represent the standard deviation. (D) Frequency of asci containing abnormal spores in ∆hta2 (TGO485) and ∆hta2 add-backed of hta2+ (TGO487; “∆hta2 + hta2+”) cells. Meiosis was induced in those cells, and the frequency of asci containing abnormal spores was measured as in (C).
To examine the meiotic defects in ∆hta2 cells, we observed their nuclear behavior during meiosis progression in living S. pombe cells expressing green fluorescent protein fused to the nuclear localization signal (GFP-NLS) as a marker for the nucleus (Fig. 2). In S. pombe meiosis, fusion of two haploid nuclei was followed by the so-called “horsetail” nuclear movements (the elongated diploid nucleus moves back and forth within the cell), which in turn was followed by two consecutive nuclear divisions to form four spores, as shown for the WT example (Fig. 2A,C).

Prolonged meiosis in ∆hta2 cells. (A) Extended horsetail stage in ∆hta2 cells. Time-lapse images of meiosis progression from nuclear fusion to meiosis I in WT (TGO350) and ∆hta2 (TGO352) cells. The nucleus was labeled with GFP-NLS. Numbers indicate the time elapsed since nuclear fusion. MI indicates meiosis I timing. Scale bar, 5 µm. (B) Duration of the horsetail stage in WT (TGO350), ∆hta2 (TGO352), ∆cds1 (TGO542), and ∆cds1 ∆hta2 (TGO543) cells. At least 26 cells were examined for each strain; the mean values are shown. Error bars represent the standard deviation. (C) Extended period from anaphase I to prometaphase II in ∆hta2 cells. Time-lapse images of meiosis progression from the end of the horsetail stage to meiosis II in WT (TGO647) and ∆hta2 (TGO648) cells. Microtubules were labeled with GFP-tubulin. Numbers indicate the time elapsed since prometaphase I. “Prometa I”, “Ana I”, “Prometa II”, and “Ana II” indicate the timing of prometaphase I, anaphase I, prometaphase II, and anaphase II, respectively. Scale bar, 5 µm. (D) Durations from the end of the horsetail stage to prometaphase I (HT end–prometa I), prometaphase to anaphase I (prometa I–ana I), anaphase I to prometaphase II (ana I–prometa II), and prometaphase II to anaphase II (prometa II–ana II) in WT (TGO647) and ∆hta2 (TGO648) cells. At least 32 cells were examined for each strain; the mean values are shown. Error bars represent the standard deviation.
We first found extension of the horsetail stage in ∆hta2 cells (Fig. 2A). It was extended to 1.5 times in ∆hta2 cells than in WT cells (155 min in WT vs. 241 min in ∆hta2 cells) (Fig. 2B). Because the horsetail stage is reported to be extended upon DNA replication checkpoint activation12, we examined whether this extension in ∆hta2 cells was rescued by deleting the cds1+ gene for the DNA replication checkpoint. However, we still observed extension of the horsetail stage in ∆cds1 ∆hta2 cells (Fig. 2B), indicating that the DNA replication checkpoint is not involved in the extension of the horsetail stage observed in ∆hta2 cells.
We also found extension of the meiosis I–II duration in ∆hta2 cells. Therefore, we measured the meiosis I–II duration in cells expressing GFP-tubulin as a marker for the spindle to determine which meiotic period was extended. The prometaphase I-anaphase I duration was not extended in ∆hta2 cells (Fig. 2C,D), suggesting that the spindle assembly checkpoint is not activated. However, the anaphase I–prometaphase II duration was extended to 1.7 times in ∆hta2 cells than in WT cells (26 min in WT vs. 43 min in ∆hta2 cells) (Fig. 2C,D).
We observed the characteristic appearance of the spindles crossing over each other in the second division in ∆hta2 cells (Fig. 2C; 75 min in ∆hta2 cells). This characteristic spindle appearance was caused by abnormal nuclear division in meiosis I, in which the nucleus apparently divided into two daughter nuclei but reunited into a single nucleus in ∆hta2 cells (50 min in Fig. 3A). This reunion of divided nuclei in meiosis I was observed in most ∆hta2 cells but not in ∆hta1 cells (Fig. 3B). Add-back of the hta2+ gene in ∆hta2 cells resulted in the recovery of normal nuclear division in meiosis I (Fig. 3C), confirming that this nuclear division defect was due to the deletion of hta2+. These results indicated that hta2+ is required for normal meiosis progression and nuclear division in meiosis I.

Abnormal nuclear division in meiosis I in ∆hta2 cells. (A) Time-lapse images of meiosis I progression in WT (TGO350) and ∆hta2 (TGO352) cells. The nucleus was labeled with GFP-NLS. Numbers indicate the time elapsed after anaphase I onset. Scale bar, 5 µm. (B) Frequency of cells showing reunion of divided nuclei in meiosis I in WT (TGO350), ∆hta1 (TGO351), and ∆hta2 (TGO352) cells. At least 20 cells were examined for each strain; the mean values from three independent experiments are shown. Error bars represent the standard deviation. (C) Frequency of cells showing reunion of divided nuclei in meiosis I in ∆hta2 (TGO485) and ∆hta2 add-backed of hta2+ (TGO487; “∆hta2 + hta2”) cells. At least 30 cells were examined for each strain; the mean values from three independent experiments are shown. Error bars represent the standard deviation. (D) Time-lapse images of meiosis I progression in WT (TGO728) and ∆hta2 (TGO729) cells. The chromatin was labeled with H2B-GFP. Numbers indicate the time elapsed after anaphase I onset. Scale bar, 5 µm.
Among the observed meiotic defects in ∆hta2 cells, we analyzed a failure in nuclear division (reunion of divided nuclei) that may be the major cause of the sporulation defect.
hta2 + deletion causes chromosome bridge formation during nuclear division
To characterize the reunion of divided nuclei in ∆hta2 cells, we observed the behavior of chromosomes during meiotic nuclear divisions using GFP-tagged histone H2B (H2B-GFP), as shown in Fig. 3D. ∆hta2 cells exhibited an anaphase chromosome bridge between two segregated masses of chromosomes (20–40 min in Fig. 3D, right). These chromosomes were reunited at the end of meiosis I (40–60 min in Fig. 3D, right) and segregated in meiosis II (80 min in Fig. 3D, right). This result suggested that entangled chromosomes in ∆hta2 cells resulted in the reunion of divided nuclei.
Anaphase chromosome bridges form at rDNA repeats in ∆hta2 cells
As an anaphase chromosome bridge was observed, we assumed that chromosomes were entangled near the telomere. Therefore, we observed the behaviors of the nhe1 (formerly called sod2) and B1 (see the Methods section) loci near the telomeres of chromosomes I and II, respectively, that were visualized using the LacI-GFP/lacO system (Fig. 4A). Both loci segregated to the poles in ∆hta2 cells and were never observed within the anaphase chromosome bridge between the poles in 32 cells for nhe1 and 27 cells for B1 that we examined (Fig. 4B,C, right), indicating that the chromosomes were not entangled at the end of chromosome I or II. Nevertheless, the two divided nuclei reunited, suggesting that the chromosomes were entangled elsewhere.

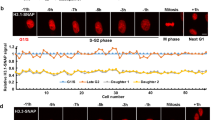
rDNA does not separate in meiosis I in ∆hta2 cells. (A) Schematic representing the positions of nhe1, B1, and rDNA loci on three chromosomes. The telomere–nhe1 distance is ~51 kb, and the telomere–B1 distance is ~110 kb. (B) Time-lapse images of meiosis I progression in WT (CT2121-4) and ∆hta2 (TGO462) cells. The nhe1 locus was labeled with the LacI-GFP/lacO system. The numbers on the left of the images indicate the time elapsed after anaphase I onset, and the numbers under the images indicate the frequency of reunion of divided nuclei in meiosis I and total number of cells observed. Scale bar, 5 µm. (C) Time-lapse images of meiosis I progression in WT (YW537) and ∆hta2 (TGO463) cells. The B1 locus was labeled with the LacI-GFP/lacO system. The numbers under the images indicate the frequency of reunion of divided nuclei in meiosis I and total number of cells observed. Scale bar, 5 µm. (D) Time-lapse images of meiosis I progression in WT (TGO566) and ∆hta2 (TGO578) cells. The rDNA and nucleolus were labeled with the LacI-GFP/lacO system (green, “rDNA”) and Nuc1-mCherry (red, “Nuc1-mCh”), respectively. The numbers on the left of the images indicate the time elapsed after anaphase I onset, and the numbers under the images indicate the frequency of reunion of divided nuclei in meiosis I and total number of cells observed. Scale bar, 5 µm.
Therefore, we next observed the behaviors of rDNA repeats located at both ends of chromosome III (Fig. 4A), visualized using the LacI-GFP/lacO system (see the Methods section). The rDNA repeats are surrounded by the nucleolus, and so we also visualized the nucleolus using mCherry tagged to a nucleolar protein Nuc1 (Nuc1-mCherry). The nucleus was also stained faintly by Nuc1-mCherry, allowing us to observe both the nucleus and nucleolus simultaneously. In WT cells, rDNA repeats and the nucleolus segregated normally during the two nuclear divisions (Fig. 4D, left). Conversely, an example of ∆hta2 cells (Fig. 4D, right) demonstrated that rDNA repeats and the nucleolus remained unsegregated in the middle of the cell, and a part of the nucleolus segregated into two nuclei (10–50 min in Fig. 4D, right); the segregated parts of the nucleolus moved towards one another and reunited at the center (60–80 min in Fig. 4D, right). These results indicated that anaphase chromosome bridges were formed at the rDNA repeats in ∆hta2 cells.
Although we observed the phenotype of the reunion of divided nuclei in the majority of ∆hta2 cells, we also observed the formation of two daughter nuclei in a small part of the cells. Therefore, we counted the populations of these classes of phenotypes in Nuc1-GFP-expressing cells. We expected four patterns of the phenotypes shown in Fig. 5A and categorized the images into the following four patterns: normal nuclear division with divided nucleoli (pattern 1), nuclear division with the nucleolus remaining in one of the divided nuclei (pattern 2), reunion of divided nuclei with the connected nucleolus (pattern 3), and reunion of divided nuclei with the nucleolus remaining in one of the divided nuclei (pattern 4). In WT cells, we observed normal segregation of Nuc1-GFP (pattern 1) in 104 out of 105 cells examined (Fig. 5B). In contrast, the majority of ∆hta2 cells (91%) showed the reunion of divided nuclei with the connected nucleolus (pattern 3); in some cells (9%), the nucleus divided into two, and the nucleolus was observed only in one of the divided nuclei (pattern 2) (Fig. 5B). In pattern 2 cells, nondisjunction of chromosome III must occur, suppressing the pattern 3 phenotype. We did not observe any examples of the reunion of divided nuclei with the nucleolus in one of the nuclei (pattern 4). These findings indicated that all ∆hta2 cells observed showed defects in meiosis I (pattern 2 or 3).

Characterization of the Nuc1 segregation pattern in ∆hta2 cells. (A) Time-lapse images of meiosis I progression in WT (TGO443) and ∆hta2 (TGO444) cells. The nucleolus was labeled with Nuc1-GFP. Patterns of segregation were classified into four categories based on nuclear division and Nuc1 segregation: normal nuclear division with divided nucleoli (pattern 1), nuclear division with the nucleolus remaining in one of the divided nuclei (pattern 2), reunion of divided nuclei with the connected nucleolus (pattern 3), and reunion of divided nuclei with the nucleolus remaining in one of the divided nuclei (pattern 4). Numbers indicate the time elapsed after anaphase I onset. Scale bar, 5 µm. (B) Frequency of cells showing the patterns 1–4 in meiosis I in WT (TGO443) and ∆hta2 (TGO444) cells. At least 28 cells were examined for each strain; the mean values from three independent experiments are shown. Error bars represent the standard deviation. (C) Time-lapse images of meiosis I and II progression in ∆hta2 pattern 2 cells shown in (A). The nucleolus was labeled with Nuc1-GFP. Numbers indicate the time elapsed after anaphase I onset. Scale bar, 5 µm.
We observed that in the second meiotic division of pattern 2 ∆hta2 cells (Fig. 5C), the nucleus containing the nucleolus failed to divide (the right nucleus), while the nucleus without the nucleolus divided into two nuclei (the left nucleus). This behavior of nuclear division in meiosis II was observed in all pattern 2 cells examined (8 in eight cases). These findings supported our idea that the anaphase chromosome bridge formed at rDNA repeats led to the reunion of divided nuclei.
Defects in ∆hta2 cells are rescued by increased hta1 expression
As the mRNA levels of hta2+, but not of hta1+, are upregulated in meiosis11, we estimated the H2Aα and H2Aβ levels by measuring the fluorescence intensity of the GFP-fused proteins (H2Aα-GFP and H2Aβ-GFP, respectively) during meiosis. We found that the fluorescence intensity of H2Aβ-GFP in the nucleus increases more strikingly compared to that of H2Aα-GFP during meiosis progression (Fig. 6A,B). We also compared the fluorescence intensities of H2Aα-GFP and H2Aβ-GFP in the nucleus at anaphase I onset. We found that the fluorescence intensity of H2Aβ-GFP was 2.6-fold higher than that of H2Aα-GFP (Fig. 6C). These results were consistent with those of a previous transcriptional study11. Similarly, we measured the fluorescence intensity of H2B-GFP and found that the fluorescence intensity of H2B-GFP in the nucleus also increased during meiosis but remained low in ∆hta2 cells (Fig. 6D–F). These results suggested that the histone H2A and H2B levels in the nucleus increase during meiosis and reduce in the absence of hta2+. Therefore, it is possible that histone H2A levels are insufficient in ∆hta2 cells during meiosis and that reduced histone H2A levels result in the reduction of histone H2B, possibly leading to insufficiency of the H2A–H2B dimer.

Amounts of histone H2Aβ increase in meiosis. (A) Time-lapse images of meiosis progression from nuclear fusion to meiosis I in WT cells expressing H2Aα-GFP (TGO804) or H2Aβ-GFP (TGO808). Numbers indicate the time elapsed since nuclear fusion. MI indicates meiosis I timing. Scale bar, 5 µm. (B) Time course of the nuclear intensity of H2Aα-GFP (TGO804) and H2Aβ-GFP (TGO808). (C) Nuclear intensity of H2Aα-GFP (TGO804) or H2Aβ-GFP (TGO808) before anaphase I onset in WT cells. For (B) and (C), at least nine cells were examined for each strain; the mean values are shown. Error bars represent the standard deviation. (D) Time-lapse images of meiosis progression from nuclear fusion to meiosis I in WT (TGO728) and ∆hta2 (TGO729) cells expressing H2B-GFP. Numbers indicate the time elapsed since nuclear fusion. MI indicates meiosis I timing. Scale bar, 5 µm. (E) Time course of the nuclear intensity of H2B-GFP in WT (TGO728) and ∆hta2 (TGO729) cells. (F) Nuclear intensity of H2B-GFP before anaphase I onset in WT (TGO728) and ∆hta2 (TGO729) cells. For (E and F), at least 15 cells were examined for each strain; the mean values are shown. Error bars represent the standard deviation.
To investigate whether histone H2A insufficiency is a cause of meiotic defects in ∆hta2 cells, we constructed ∆hta2 strains bearing additional copies of hta1+. The strain bearing one additional copy of hta1+ showed slightly decreased abnormal spore formation and nuclear division (“hta1+ ×1” in Fig. 7A). Strikingly, increasing the copies of hta1+ more remarkably eliminated these defects (“hta1+ ×2” and “hta1+ ×3” in Fig. 7B). We also examined this by expressing hta1+ under the hta2 promoter by replacing the hta2 coding region with the hta1 coding region (see the Methods section; “hta2::hta1” in Fig. 7B). We observed that abnormal spore formation and nuclear division in ∆hta2 cells was eliminated in hta2::hta1 cells (Fig. 7B). These results indicated that the phenotypes uniquely observed in ∆hta2 cells were not due to differences in amino acid residues between H2Aα and H2Aβ but due to histone H2A insufficiency.

Additional expression of hta1+ rescues defects in ∆hta2 cells. (A) Frequency of asci containing abnormal spores and reunion of divided nuclei in meiosis I in ∆hta2 cells expressing no extra copy (TGO629; “∆hta2”), one extra copy (TGO630; “∆hta2 + hta1+ ×1”), two extra copies (TGO631; “∆hta2 + hta1+ ×2”), or three extra copies (TGO632; “∆hta2 + hta1+ ×3”) of hta1+. Abnormal spore formation and nuclear division in ∆hta2 cells were suppressed by expression of additional copies of hta1+. At least 200 asci or 31 cells were examined for spore formation or nuclear division, respectively, for each strain; the mean values from three independent experiments are shown. Error bars represent the standard deviation. (B) Frequency of asci containing abnormal spores and reunion of divided nuclei in meiosis I in WT (TGO350), ∆hta2 (TGO352), and hta2::hta1 (TGO399) cells. Abnormal spore formation and nuclear division were not observed in cells that replaced the hta2 ORF with the hta1 ORF (“hta2::hta1”). At least 200 asci or 29 cells were examined for spore formation or nuclear division, respectively, for each strain; the mean values from three independent experiments are shown. Error bars represent the standard deviation. (C) Frequency of cells showing patterns 1–3 in meiosis I in WT (TGO477), ∆hta2 (TGO521), ∆1 ∆3 (TGO522), ∆1 ∆3 ∆hta2 (TGO523), ∆1 ∆hta2 (TGO525), or ∆3 ∆hta2 (TGO526) cells. rDNA was labeled with Nuc1-GFP. Patterns of segregation were classified into four categories based on nuclear division and Nuc1 segregation, as in Fig. 5A. The number of cells examined is shown at the bottom of the graph.
Reduction of histone H3 and H4 partially rescues the chromosome segregation errors in ∆hta2 cells
To examine involvement of histone H3 and H4 in ∆hta2 cells, we deleted 1 or 2 of the 3 genes for histone H3 and H4. Among the three pairs of histone H3 and H4 genes, we deleted a pair of the hhf1+ and hht1+ genes (∆1) and/or a pair of the hhf3+ and hht3+ genes (∆3). Cells of ∆1 ∆hta2 and ∆3 ∆hta2 showed a high frequency of abnormal nuclear division (pattern 2 and 3) at a level similar to that in ∆hta2 cells; however, this defect was partially rescued in ∆1 ∆3 ∆hta2 cells (Fig. 7C), suggesting that depletion of histone H3 and H4 rescues histone H2A insufficiency to a limited extent. Thus, excess amounts of histone H3 and H4 as a consequence of H2A and H2B insufficiency may be a cause of chromosome segregation defects.
Histone H2A insufficiency also causes chromosome bridge formation at rDNA repeats during mitosis
Finally, we examined whether histone H2A insufficiency causes chromosome bridge formation at rDNA repeats during mitosis. We constructed a strain in which hta2+ expression could be conditionally shut down using the nmt1 promoter under the ∆hta1 background (i.e., hta2 expression could be induced by the absence of thiamin and be repressed by the addition of thiamin). As expected, this strain did not form a colony on the plate containing thiamin (Fig. 8A). Because ~80% of the cells were in the G2 phase in the S. pombe asynchronous culture, most cells had sufficient histone H2A to perform one nuclear division. Therefore, we focused on the second nuclear division after the addition of thiamin to examine the cells depleted of H2A. Although most cells proceeded with a normal second nuclear division in the absence of thiamin (Fig. 8B,C), they did not enter the second nuclear division in the presence of thiamin (Fig. 8C). In some of the cells that did enter the second nuclear division, the nuclear division failed as observed by Nuc1-GFP (Fig. 8B, “Abnormal”). In these cells, the nucleus apparently divided into two daughter nuclei (10–20 min in Fig. 8B, “Abnormal”) but returned to the center of the cells (25 min in Fig. 8B, “Abnormal”), as observed in ∆hta2 cells, although the reunion of divided nuclei was disturbed by septation (30 min in Fig. 8B, “Abnormal”). These results indicate that histone H2A insufficiency causes chromosome segregation errors at rDNA repeats during mitosis as well, suggesting that this phenotype is not meiosis-specific.

Depletion of histone H2A in mitosis. (A) Spot assay comparing the growth of WT (TGO443), ∆hta1 (TGO575), Pnmt1:hta2 (TGO572), and ∆hta1 Pnmt1:hta2 (TGO579) cells. Dilution series (1/5 dilution) of cell suspensions were spotted on EMM2 with (“+Thiamin”) or without (“−Thiamin”) thiamin and grown for 3 days at 30 °C. (B) Time-lapse images of progression of normal and abnormal mitosis in ∆hta1 Pnmt1:hta2 (TGO579) cells. The nucleolus was labeled with Nuc1-GFP. Numbers indicate the time elapsed after anaphase onset. Scale bar, 5 µm. (C) Frequency of normal and abnormal nuclear divisions at the second mitosis during observation in ∆hta1 Pnmt1:hta2 (TGO579); frequency of arrested cells (no second mitosis during a 12 h observation) is also shown. At least 72 cells were examined for each condition; the mean values from three independent experiments are shown. Error bars represent the standard deviation.
Discussion
In this study, we demonstrated that deletion of hta2+ causes meiotic defects but no remarkable defects in vegetative growth. This does not indicate that histone H2Aβ produced from hta2+ has meiosis-specific functions; instead, the defects occur due to histone H2A insufficiency. This finding is supported by the fact that when increased amounts of H2Aα is produced by hta1+, it can replace histone H2Aβ. Therefore, to date, no functional differences between H2Aα and H2Aβ have been found.
We first considered the involvement of condensin in chromosome segregation defects in ∆hta2 cells. Studies have reported that histone H2A binds to condensin10; therefore, histone H2A insufficiency might cause insufficient loading of condensin on to chromatin, leading to chromosome segregation defects. However, we concluded that condensin is not involved because a mutant of histone H2Aβ (K13A R18A K21A) that does not bind to condensin10 rescued the defects in ∆hta2 cells (Supplementary Fig. S1). Similarly, a mutant of histone H2Aβ (S121A) that does not recruit shugoshin9 rescued the defects in ∆hta2 cells (Supplementary Fig. S1). In addition, a mutant of H2Aβ (S127A) that lacks the phosphorylation site required for DNA damage repair8 also rescued the defects in ∆hta2 cells (Supplementary Fig. S1), indicating that condensin loading was not involved in chromosome segregation defects in ∆hta2 cells.
We then considered the involvement of cohesin in chromosome segregation defects in ∆hta2 cells. In meiosis, mitotic cohesin Rad21 is replaced to a large extent with meiotic cohesin Rec813; however, Rad21 remains at the chromosomal regions of rDNA repeats during the horsetail stage14. The frequency of Rad21 localization to rDNA repeat regions was increased in ∆hta2 cells (Supplementary Fig. S2). This increased localization of Rad21 could be a cause for the observed defects. However, the frequency of abnormal nuclear division in ∆rad21 ∆hta2 cells was high and at a level similar to that in ∆hta2 (Supplementary Fig. S2), indicating that increased localization of Rad21 at the rDNA repeat regions was not a cause of chromosome segregation defects in ∆hta2.
The characteristic phenotype of the reunion of divided nuclei during meiosis I as observed in ∆hta2 cells has also been reported in the ∆dbl2 mutant15. However, unlike that in the ∆hta2 mutant, rDNA repeats were not involved in the ∆dbl2 mutant as observed by Nuc1-GFP (Supplementary Fig. S3), indicating that the role of histone H2A is unrelated to the Dbl2 pathway, which regulates the resolution of recombination intermediates during meiosis15.
Alternatively, transcription of rRNA genes might cause chromosome segregation defects at rDNA repeats. Chromosome regions at rDNA repeats have sparse nucleosomes, which occupy only the intervening sequences between 18S and 28S rRNA coding sequences16. Upon entry into meiosis, rRNA transcription might be repressed by the occupation of nucleosomes on rRNA genes. Anaphase chromosome bridge formation at rDNA repeats was also observed in a cdc14 mutant in Saccharomyces cerevisiae17,18. Cdc14 is a mitotic phosphatase19 and inhibits rRNA transcription by polymerase I to separate the rDNA repeats20. Inhibition of rRNA transcription by thiolutin rescues chromosome bridge formation at rDNA repeats in a cdc14 mutant in S. cerevisiae21. Therefore, histone H2A insufficiency might lead to failure of transcriptional repression for rRNA genes and consequently cause chromosome segregation failure at rDNA repeats in S. pombe. However, thiolutin treatment did not suppress chromosome bridge formation at rDNA repeats in ∆hta2 cells (Supplementary Fig. S4), suggesting that transcription of rDNA repeats is not the cause of chromosome bridge formation in S. pombe.
Histone stoichiometry is considered important for the fidelity of chromosome segregation in S. cerevisiae22. In S. cerevisiae, canonical histone proteins are encoded by multiple genes. The increased copy number of histone genes results in chromosome loss22, while a decrease in the copy number of the genes causes slow growth and G2/M arrest23,24,25. The dosage of histone proteins affects the cell sensitivity to DNA damaging agents: an excess amount of histones enhances sensitivity, whereas a reduced amount increases resistance26 and histone H4 depletion induces hyper-recombination, collapse of replication forks, and activation of the spindle assembly checkpoint, leading to genome instability27,28,29. Thus, it is likely that histone imbalance is responsible for the defects associated with chromosome instability in S. pombe as well.
Considering that chromosome regions at rDNA repeats have sparse nucleosomes16, these regions may be sensitive to histone imbalance. Measurements of nucleosome occupancy at rDNA repeats in strains bearing histone imbalance may reveal the cause of chromosome segregation errors associated with histone imbalance.
Methods
Strains and culture media
The S. pombe strains used in this study are listed in Supplementary Table S1. The growth media and basic genetic techniques for S. pombe have been described in previous studies30. The complete yeast extract with supplements (YES) medium (i.e., YE medium supplemented with 225 mg/L of adenine, leucine, histidine, uracil, and lysine) was used for spot assay (Fig. 1A). Edinburgh Minimal Media (EMM2) containing nutritional supplements (150 mg/L of adenine, 200 mg/L of leucine, and 75 mg/L of lysine), when necessary, was used for routine culture. EMM2 lacking nitrogen sources (EMM2-N) containing nutritional supplements (150 mg/L of adenine, 200 mg/L of leucine, and 75 mg/L of lysine), when necessary, was used to induce meiosis. The frequency of abnormal nuclear division at the first meiosis in ∆hta2 cells is high (~90%) if meiosis is induced on an EMM2-N plate but moderate (40–60%) on a molt extract plate. EMM2 was supplemented with 2 µM thiamin to repress the nmt1 promoter and depleted of thiamin to induce the nmt1 promoter31.
Strain constructions
S. pombe strain expressing GFP-NLS (NLS from the SV40 T-antigen) was constructed as follows: First, the nmt1 promoter and the GFP coding region of pCST832 were replaced with the nda3 promoter (−620 to −1 nucleotides [nt]) and the GFP-NLS coding region33, respectively. Next, the resulting plasmid, pTG3, was integrated into the chromosome at the lys1 locus. Integration was confirmed by polymerase chain reaction (PCR).
S. pombe strains expressing hta1 from the lys1, leu1, and aur1 loci were constructed as follows: First, the hta1 gene fragment containing its promoter (from −626 nt), coding region, and terminator (601 nt after the stop codon) was ligated into the integration vectors pYC3634, pYC2833, and pYC33; pYC33 is a derivative of pYC36 and has the aur1R gene fragment (TaKaRa Bio Inc., Shiga Prefecture, Japan) containing its partial coding region (72 nt to the stop codon) and terminator (639 nt after the stop codon) instead of the lys1-N fragment. Next, the resulting plasmids, namely, pYC36-hta1, pYC28-hta1, and pYC33-hta1, were integrated into the chromosome at the lys1, leu1, and aur1 loci, respectively. Integration was confirmed by PCR.
S. pombe strain expressing hta2 from the lys1 locus was constructed as follows: First, the hta2 gene fragment containing its promoter (from −561 nt), coding region, and terminator (603 nt after the stop codon) was ligated into the integration vector pYC36. Next, the resulting plasmid, pYC36-hta2, was integrated into the chromosome at the lys1 locus. Integration was confirmed by PCR.
S. pombe strain expressing htb1 from the lys1 locus was constructed as follows: First, the htb1 gene fragment containing its promoter (from −471 nt), coding region, and terminator (693 nt after the stop codon) was ligated into the integration vector pYC36. Next, the resulting plasmid, pYC36-htb1, was integrated into the chromosome at the lys1 locus. Integration was confirmed by PCR.
S. pombe strains deleted of hta1 and hta2 gene (∆hta1 and ∆hta2, respectively) were constructed using PCR-based gene targeting35,36. Gene deletion was confirmed by PCR and sequencing.
S. pombe strain expressing hta1 under the hta2 promoter (hta2::hta1) was constructed as follows: First, the hta2 coding region of pYC36-hta2 was replaced with the hta1 coding region. Next, the hta2::hta1 fragment from the resulting plasmid, pYC36-P2-hta1-T2, was amplified by PCR and transformed to an hta2 gene deletion strain (∆hta2::ura4). Finally, transformants were selected by 5-fluoroorotic acid (5-FOA) and confirmed by sequencing.
S. pombe strain expressing hta2 under the nmt1 promoter (Pnmt1:hta2) was constructed as follows: First, the nmt1 promoter of pCST8 was inserted in the SacI site of pAG3237. Next, the resulting plasmid, pAG32-Pnmt1, was used for PCR-based gene targeting to integrate the nmt1 promoter fragment at the hta2 locus. Integration was confirmed by PCR.
S. pombe strain expressing H2Aα-GFP was constructed as follows: First, the hta1-GFP fragment containing the hta1 promoter (from −626 nt), hta1 and GFP coding regions, and the hta1 terminator (601 nt after the stop codon) was cloned into plasmid pHSG299 using the In-Fusion HD Cloning Kit. Next, the hta1-GFP fragment from the resulting plasmid, pHSG-hta1-GFP, was amplified by PCR and transformed to an hta1 gene deletion strain (∆hta1::ura4). Finally, transformants were selected by 5-FOA and confirmed by sequencing.
S. pombe strain expressing H2Aβ-GFP was constructed as follows: First, the hta2-GFP fragment containing the hta2 promoter (from −561 nt), hta2 and GFP coding regions, and the hta2 terminator (603 nt after the stop codon) was cloned into plasmid pHSG299 using the In-Fusion HD Cloning Kit. Next, the hta2-GFP fragment from the resulting plasmid, pHSG-hta2-GFP, was amplified by PCR and transformed to an hta2 gene deletion strain (∆hta2::ura4). Finally, transformants were selected by 5-FOA and confirmed by sequencing.
S. pombe strain expressing H2B-GFP was constructed as follows: First, the htb1-GFP fragment from the htb1-GFP plasmid38 was amplified by PCR and then transformed to an htb1 gene deletion strain (∆htb1::ura4) integrated pYC36-htb1. Then, transformants were selected by 5-FOA and confirmed by sequencing.
In this study, chromosomal loci were visualized using the lac repressor (LacI)/lac operator (lacO) recognition system39,40,41. To track the dynamics of the telomere-proximal locus of chromosome II, tandem repeats of lacO arrays were integrated in the position of chromosome II between 110,029 and 110,061 using two-step integration42. This locus was named B1, and integration was confirmed by PCR.
To track the dynamics of rDNA in live cells, 5x lacO arrays were integrated into the repetitive 28s ribosomal RNA (rRNA) gene cluster using the I-PpoI cut-and-refill method43,44 and visualized with LacI-GFP. I-PpoI is a homing endonuclease encoded by the group I intron45. In S. pombe, in addition to other eukaryotes, the 28s rRNA gene contains a unique 15 bp target sequence of I-PpoI. Induction of I-PpoI makes double-strand breaks at rDNA repeats and kills the cell unless the target site is repaired and mutated. Next, a 28s rRNA gene and TsLSU-5x lacO containing a DNA fragment, in which the I-PpoI recognition site was interrupted by TsLSU-5x lacO insertion (cloned from plasmid p5xlacOTtLSU43, a gift from Dr. Yu), were cloned into plasmid pREP4146 using the In-Fusion HD Cloning Kit, and the resulting plasmid was named pY12. pY12 was transformed to LacI-GFP-containing cells and selected on a leucine dropout plate. The resulting cells were then transformed with pSS1244, a tetracycline-inducible I-PpoI expression plasmid (a gift from Dr. Sanders). The I-PpoI-resistant cell, in which the I-PpoI recognition site was disrupted and in which 5x lacO was inserted, was selected on an ahTET-containing plate44 and confirmed with colony PCR and sequencing. GFP fluorescence at rDNA was confirmed using a microscope. The cells were cultured in a rich medium for at least 20 generations to drop out plasmids pY12 and pSS12, which were subsequently selected on a 5-FOA plate.
Live-cell fluorescence microscopy
Microscopy images were obtained using a DeltaVision microscope system (GE Healthcare, Chicago, IL, USA) equipped with a Plan Apo 60x oil-immersion objective lens (numerical aperture [NA] = 1.4; Olympus Corporation, Tokyo, Japan) and a CoolSNAP HQ2 CCD (Photometrics, Tucson, AZ, USA). For time-lapse observation, we mounted living cells on 35 mm glass-bottomed culture dishes (MatTek Corporation, Ashland, MA, USA) coated with 0.2 mg/mL of soybean lectin (Sigma-Aldrich Corporation, St. Louis, MO, USA) and observed them at 26 °C. Briefly, we took a set of images of 11 focal planes at 0.3 µm intervals every 5 min. To measure the meiosis duration, we took a set of images of seven focal planes at 0.5 µm intervals every 5 min. To quantify H2A-GFP and H2B-GFP, we took a set of images of 11 focal planes at 0.4 µm intervals every 5 min and measured nuclear fluorescence intensities, as described previously47, with the following threshold values to draw two-dimensional (2D) polygons: 1800 for H2Aα-GFP, 3800 for H2Aβ-GFP, and 3500 for H2B-GFP. We used a semiconductor light source instead of a mercury arc; therefore, we detected almost no progressive decline of light output during our observation for quantification. All images, except those in Figs 1B, 3D and 6A,D, were processed using the denoising algorithm48 and then projected with a maximum intensity method. The images in Fig. 1B were projected with a maximum intensity method without denoising, and those in Figs 3D and 6A,D were projected with a summation method without denoising. Projection and quantification were performed using softWoRx software (GE Healthcare).
Statistical analyses
All statistical analyses were performed using R (www.r-project.org). For between-group comparison, we used two-sided, unpaired Student’s t-test. For multiple-group comparison, we used two-sided Tukey’s (Tukey–Kramer). Supplementary Table S2 summarizes the types of test methods and P-values in each analysis. A significance level (α) was set at 0.05 in all analyses, and significance was indicated by asterisks in all graphs: *P < 0.05, **P < 0.01, and ***P < 0.001. NS stands for “not significant” (P ≥ 0.05).
References
Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997).
Talbert, P. B. & Henikoff, S. Histone variants — ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell. Biol. 11, 264–275 (2010).
Cheema, M. S. & Ausió, J. The structural determinants behind the epigenetic role of histone variants. Genes 6, 685–713 (2015).
Choe, J., Schuster, T. & Grunstein, M. Organization, primary structure, and evolution of histone H2A and H2B genes of the fission yeast Schizosaccharomyces pombe. Mol. Cell. Biol. 5, 3261–3269 (1985).
Matsumoto, S. & Yanagida, M. Histone gene organization of fission yeast: a common upstream sequence. EMBO J. 4, 3531–3538 (1985).
Carr, A. M. et al. Analysis of a histone H2A variant from fission yeast: evidence for a role in chromosome stability. Mol. Gen. Genet. 245, 628–635 (1994).
Takahashi, K., Chen, E. S. & Yanagida, M. Requirement of Mis6 centromere connector for localizing a CENP-A-like protein in fission yeast. Science 288, 2215–2219 (2000).
Nakamura, T. M., Du, L.-L., Redon, C. & Russell, P. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol. Cell. Biol. 24, 6215–6230 (2004).
Kawashima, S. A., Yamagishi, Y., Honda, T., Ishiguro, K. & Watanabe, Y. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 327, 172–177 (2010).
Tada, K., Susumu, H., Sakuno, T. & Watanabe, Y. Condensin association with histone H2A shapes mitotic chromosomes. Nature 474, 477–483 (2011).
Mata, J., Lyne, R., Burns, G. & Bähler, J. The transcriptional program of meiosis and sporulation in fission yeast. Nature Genet. 32, 143–147 (2002).
Ruan, K. et al. Meiotic nuclear movements in fission yeast are regulated by the transcription factor Mei4 downstream of a Cds1-dependent replication checkpoint pathway. Genes Cells 20, 160–172 (2015).
Watanabe, Y. & Nurse, P. Cohesin Rec8 is required for reductional chromosome segregation at meiosis. Nature 400, 461–464 (1999).
Yokobayashi, S., Yamamoto, M. & Watanabe, Y. Cohesins determine the attachment manner of kinetochores to spindle microtubules at meiosis I in fission yeast. Mol. Cell. Biol. 23, 3965–3973 (2003).
Polakova, S. et al. Dbl2 regulates Rad51 and DNA joint molecule metabolism to ensure proper meiotic chromosome segregation. PLoS Genet. 12, e1006102 (2016).
Gal, C., Moore, K. M., Paszkiewicz, K., Kent, N. A. & Whitehall, S. K. The impact of the HIRA histone chaperone upon global nucleosome architecture. Cell Cycle 14, 123–134 (2015).
D’Amours, D., Stegmeier, F. & Amon, A. Cdc14 and condensin control the dissolution of cohesin-independent chromosome linkages at repeated DNA. Cell 117, 455–469 (2004).
Sullivan, M., Higuchi, T., Katis, V. L. & Uhlmann, F. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell 117, 471–482 (2004).
Stegmeier, F. & Amon, A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu. Rev. Genet. 38, 203–232 (2004).
Clemente-Blanco, A. et al. Cdc14 inhibits transcription by RNA polymerase I during anaphase. Nature 458, 219–222 (2009).
Tomson, B. N., D’Amours, D., Adamson, B. S., Aragon, L. & Amon, A. Ribosomal DNA transcription-dependent processes interfere with chromosome segregation. Mol. Cell. Biol. 26, 6239–6247 (2006).
Meeks-Wagner, D. & Hartwell, L. H. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell 44, 43–52 (1986).
Rykowski, M. C., Wallis, J. W., Choe, J. & Grunstein, M. Histone H2B subtypes are dispensable during the yeast cell cycle. Cell 25, 477–487 (1981).
Kolodrubetz, D., Rykowski, M. C. & Grunstein, M. Histone H2A subtypes associate interchangeably in vivo with histone H2B subtypes. Proc. Natl Acad. Sci. USA 79, 7814–7818 (1982).
Han, M., Chang, M., Kim, U.-J. & Grunstein, M. Histone H2B repression causes cell-cycle-specific arrest in yeast: effects on chromosomal segregation, replication, and transcription. Cell 48, 589–597 (1987).
Liang, D., Burkhart, S. L., Singh, R. K., Kabbaj, M.-H. M. & Gunjan, A. Histone dosage regulates DNA damage sensitivity in a checkpoint-independent manner by the homologous recombination pathway. Nucleic Acids Res. 40, 9604–9620 (2012).
Prado, F. & Aguilera, A. Partial depletion of histone H4 increases homologous recombination-mediated genetic instability. Mol. Cell. Biol. 25, 1526–1536 (2005).
Clemente-Ruiz, M. & Prado, F. Chromatin assembly controls replication fork stability. EMBO Reports 10, 790–796 (2009).
Murillo-Pineda, M., Cabello-Lobato, M. J., Clemente-Ruiz, M., Monje-Casas, F. & Prado, F. Defective histone supply causes condensin-dependent chromatin alterations, SAC activation and chromosome decatenation impairment. Nucleic Acids Res. 42, 12469–12482 (2014).
Moreno, S., Klar, A. & Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823 (1991).
Maundrell, K. nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J. Biol. Chem. 265, 10857–10864 (1990).
Chikashige, Y. et al. Meiotic proteins Bqt1 and Bqt2 tether telomeres to form the bouquet arrangement of chromosomes. Cell 125, 59–69 (2006).
Chikashige, Y. et al. Fission yeast APC/C activators Slp1 and Fzr1 sequentially trigger two consecutive nuclear divisions during meiosis. FEBS Letters 591, 1029–1040 (2017).
Chikashige, Y., Kurokawa, R., Haraguchi, T. & Hiraoka, Y. Meiosis induced by inactivation of Pat1 kinase proceeds with aberrant nuclear positioning of centromeres in the fission yeast Schizosaccharomyces pombe. Genes Cells 9, 671–684 (2004).
Bähler, J. et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951 (1998).
Sato, M., Dhut, S. & Toda, T. New drug-resistant cassettes for gene disruption and epitope tagging in Schizosaccharomyces pombe. Yeast 22, 583–591 (2005).
Goldstein, A. L. & McCusker, J. H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553 (1999).
Matsuda, A. et al. Highly condensed chromatins are formed adjacent to subtelomeric and decondensed silent chromatin in fission yeast. Nature Comm. 6, 7753 (2015).
Robinett, C. C. et al. S. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 135, 1685–1700 (1996).
Straight, A. F., Belmont, A. S., Robinett, C. C. & Murray, A. W. GFP tagging of budding yeast chromosomes reveals that protein- protein interactions can mediate sister chromatid cohesion. Curr. Biol. 6, 1599–1608 (1996).
Nabeshima, K. et al. Dynamics of centromeres during metaphase-anaphase transition in fission yeast: Dis1 is implicated in force balance in metaphase bipolar spindle. Mol. Biol. Cell 9, 3211–3225 (1998).
Yamamoto, A. & Hiraoka, Y. Monopolar spindle attachment of sister chromatids is ensured by two distinct mechanisms at the first meiotic division in fission yeast. EMBO J. 22, 2284–2296 (2003).
Li, P., Jin, H., Hoang, M. L. & Yu, H. G. Tracking chromosome dynamics in live yeast cells: coordinated movement of rDNA homologs and anaphase disassembly of the nucleolus during meiosis. Chromosome Res. 19, 1013–1026 (2011).
Sunder, S., Greeson-Lott, N. T., Runge, K. W. & Sanders, S. L. A new method to efficiently induce a site-specific double strand break in the fission yeast Schizosaccharomyces pombe. Yeast 29, 275–291 (2012).
Lin, J. & Vogt, V. M. I-PpoI, the endonuclease encoded by the group I intron PpLSU3, is expressed from an RNA polymerase I transcript. Mol. Cell. Biol. 18, 5809–5817 (1998).
Maundrell, K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123, 127–130 (1993).
Ding, D.-Q. et al. Meiotic cohesins modulate chromosome compaction during meiotic prophase in fission yeast. J. Cell Biol. 174, 499–508 (2006).
Boulanger, J., Kervrann, C. & Bouthemy, P. A simulation and estimation framework for intracellular dynamics and trafficking in video-microscopy and fluorescence imagery. Med. Image Anal. 13, 132–142 (2009).
Acknowledgements
We would like to thank Dr. Hong-Guo Yu and Dr. Steven L. Sanders for providing plasmids, Yuko Takayama and the National Bio-Resource Project, Japan for providing strains, and Dr. Atsushi Matsuda for the denoising software. This work was supported by JSPS KAKENHI Grants: JP15K18538 to TGY; JP17KT0116 and JP17K07249 to YC; JP17H03636 and JP18H05528 to TH; JP17H01444 and JP18H05533 to YH.
Author information
Authors and Affiliations
Contributions
T.G.Y., Y.C., T.H. and Y.H. conceived and designed the experiments. T.G.Y., D.Q.D. and Y.N. performed experiments. T.G.Y., Y.N. and Y.H. analyzed the data. T.G.Y., D.Q.D., Y.N., Y.C., T.H. and Y.H. contributed reagents/materials/analysis tools. T.G.Y., D.Q.D., Y.C., T.H. and Y.H. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamamoto, T.G., Ding, DQ., Nagahama, Y. et al. Histone H2A insufficiency causes chromosomal segregation defects due to anaphase chromosome bridge formation at rDNA repeats in fission yeast. Sci Rep 9, 7159 (2019). https://doi.org/10.1038/s41598-019-43633-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43633-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
