
Abstract
The accumulation of brain amyloid β (Aβ) is one of the main pathological hallmarks of Alzheimer’s disease (AD). However, the role of brain amyloid deposition in the development of AD and the genetic variants associated with this process remain unclear. In this study, we sought to identify associations between Aβ deposition and an a priori evidence based set of 1610 genetic markers, genotyped from 505 unrelated individuals (258 Aβ+ and 247 Aβ−) enrolled in the Australian Imaging, Biomarker & Lifestyle (AIBL) study. We found statistically significant associations for 6 markers located within intronic regions of 6 genes, including AC103796.1-BDNF, PPP3R1, NGFR, KL, ABCA7 & CALHM1. Although functional studies are required to elucidate the role of these genes in the accumulation of Aβ and their potential implication in AD pathophysiology, our findings are consistent with results obtained in previous GWAS efforts.
Similar content being viewed by others

Introduction
Alzheimer’s disease is the most prevalent cause of dementia in elderly populations (age > 65 years). Currently affecting more than 40 million people worldwide, this number is projected to increase at least three-fold by 2050, with the continuing growth and ageing of the population. Hallmarks of disease pathology generally appear several years prior to the onset of clinical symptoms. Although the slow progression provides opportunities for preclinical therapeutic interventions, our ability to accurately detect the disease remains limited.
The accumulation of Aβ occurs at a variable rate early in the development of AD, starting over 20 years before the onset of cognitive decline and structural brain atrophy1,2. The process is a well-recognised histopathological hallmark of AD, and Aβ deposition is necessary for the pathologic diagnosis of the disease. However, the formation of Aβ plaques alone is not sufficient to cause cognitive dysfunction. Individuals with high Aβ accumulation but no or minimal cognitive deficits have been observed in several studies3. In addition, recent studies have shown that the presence of substantial Aβ deposition had low specificity for predicting the development of AD4,5. These observations reflect the intricate contribution of Aβ formation in the development of AD, and the need for more research in the developmental processes of the disease.
This complex pathogenesis of AD involves multiple external risk factors and comorbidities with varying susceptibilities based upon genetic backgrounds. In recent years, genome-wide association studies (GWAS) have identified more than 20 genetic risk loci robustly associated with the disease6,7,8,9,10. Large meta-analyses such as the one conducted by the International Genomics of Alzheimer’s Project (IGAP)11, have played a key role in enhancing our ability to predict the risk of disease onset and expanding our knowledge around the aetiology of the disease. However, most case-control GWAS have focused on identifying variants associated with the pathologic clinical diagnosis of AD compared with cognitively normal participants, even though there is a large discrepancy in the specificity in this diagnosis. Further, there is growing evidence that susceptibility to Aβ-associated decline or rate of progression may be due to either vulnerability or resilience imparted by an individual’s genetic background12,13,14,15.
In this study, a total of 505 patients enrolled in the Australian Imaging, Biomarker & Lifestyle (AIBL) study, were genotyped for an a priori evidence based targeted selection of Single Nucleotide Polymorphisms (SNPs), with rationale to identify possible Aβ specific variants, from a large list of AD-related candidate genes.
Results
We conducted an association analysis using 1610 genetic markers from 505 unrelated participants of the AIBL study (258 cases and 247 controls). Comparing demographic and clinical characteristics between Aβ status, Aβ+ participants were older (72.3 [SD:6.94] vs 69.1 [SD:6.3], p < 0.0001) and were more likely to have an APOE ε4 allele (40% vs 12%, p < 0.0001, Table 1). As expected, there were no differences in the frequencies of males/females between Aβ groups (p > 0.05), and MCI and AD groups were more likely to be Aβ+ than Aβ− (p < 0.0001).
In preliminary analysis containing all the markers, the strongest associations (p < 1e−8) with amyloid status corresponded to a set of 5 SNPs (rs429358, rs769449, rs6857, rs157581, rs2075650) located within a 20 kb region containing the apolipoprotein E (APOE, 3.6 kb) and the translocase of outer mitochondrial membrane 40 (TOMM40, 12.4 kb) genes.
To enhance the identification of additional Aβ specific variants, we conducted the same association analysis including the presence/absence of the ε4 allele as a covariate. A total of 10 SNPs, located within 6 loci, showed nominal evidence for association (p < 5e−3). Results were clumped to keep only the most representative SNP per region of linkage disequilibrium, resulting in 6 independent SNPs (Table 2). Evidence of associations with previously established AD-specific loci include AC103796.1-BDNF (rs2049048; p = 3.62e−04; OR, 0.45 [95% CI, 0.29–0.7]), PPP3R1 (rs7593613; p = 2.04e−03; OR, 1.59 [95% CI, 1.18–2.13]), NGFR (rs9908234; p = 2.45e−03; OR, 2.47 [95% CI, 1.38–4.43]), KL (rs648202; p = 2.90e−03; OR, 1.84 [95% CI, 1.23–2.75]), ABCA7 (rs3764650; p = 3.07e−03; OR, 2.01 [95% CI, 1.27–3.2]), CALHM1 (rs2986018; p = 4.16e−03; OR, 1.59 [95% CI, 1.16–2.18]), Table 2. Details of the genotyped markers and their associated p values are presented in Supplementary Table 1.
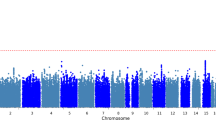
Comparing SNPs at nominal significance for Aβ in this study with the IGAP results, all but one marker (NGFR, rs9908234) was also found significantly associated with AD in the IGAP study. This limited overlap is explained by the use of different proxy SNPs between this study and the IGAP study. For example, the variant rs3764650 in the ABCA7 gene was typed in this study and reached nominal significance. Although this specific variant is not present in IGAP, it is located in the same LD block (r2 > 0.9) as four other markers (rs73505217, rs4147911, rs4147910, rs76348507) that reached genome-wide significance in the IGAP meta-analysis. Figure 1 shows a Manhattan plot of all SNPs tested in the current study, with the -log10 of the p-value on the y-axis, chromosome on the x-axis, and dot colour representing the presence/absence of markers in the IGAP study.

Simplified Manhattan plot for association results. The P values (−log 10) are plotted against their relative positions on each chromosome. To improve the visibility of the figure, positions correspond to the order in which the markers are located on the chromosomes. The threshold for significance was set to 5e10−3.
Lastly, we assessed the gene ontologies and functional interactions amongst the genes that reached nominal significance using the GeneMANIA resource16. Through the gene-gene interaction network, we were able to demonstrate the presence of physical and genetic interactions between the identified genes and several other genes with similar biological functions (Fig. 2).

Gene-gene interaction network. The graph represents possible interactions between 6 genes in which variants associated with Aβ deposition were identified (black nodes). Indirect interactions are facilitated by the introduction of 4 external genes (gray nodes): Neurotrophin 4 (NTF4), Neurotrophic Receptor Tyrosine Kinase 2 (NTRK2), Sortilin 1 (SORT1) and Calcineurin A Alpha (PPP3CA). Edges represent the type of interactions: physical interaction69,70,71) are colored in red and genetic interaction72 in green. The network was generated using GeneMANIA16.
Discussion
As with most complex traits, multiple genetic variants with small and cumulative effects are likely to explain the heritability of AD. Consistent with this assumption, we identified robust associations with several previously established loci in a sample of cognitively normal and AD subjects. First, by identifying several markers located in the TOMM40-APOE region, our analysis supports the hypothesis that APOE on chromosome 19 is a major susceptibility gene for AD. The APOE ε4 allele has been associated with an increased risk of developing AD in a number of independent studies. Therefore, these results were expected but can be considered as a validation of our dataset. Aside from SNPs within the TOMM40-APOE locus, we identified a set of 6 variants at the nominal significance level showing evidence of association with Aβ status. A gene-gene interaction network revealed direct and indirect interactions amongst the 6 genes in which the genetic markers are located, suggesting a collective influence of genetic polymorphisms (Fig. 2).
The top-ranked SNP, rs7593613, was located in the regulatory subunit of the protein phosphatase B gene (PPP3R1), also known as calcineurin B. This marker was in high linkage disequilibrium (LD) with 2 other SNPs located on the same locus and showing significant associations with AD (rs28694054, p = 3.4e−03; rs11692815, p = 4.3e−03). Variants in PPP3R1 have been previously reported as potential modulators of tau and phosphorylated tau levels in the presence of amyloid deposition. These changes are suspected to result in an accelerated progression of AD17,18. Calcineurin is involved in a number of pathways that regulate synaptic activity and neuronal excitability. Thereby, any impairment in this complex could have substantial effects and lead to pathological synaptic loss19.
The second top-ranked SNP identified, rs2049048, is located in the AC103796.1 gene and less than 7 kb from the brain-derived neurotrophic factor (BDNF). Although the role of AC103796.1 remains unclear, the gene overlaps BDNF over 20 kb and therefeore, may contibute to its function. BDNF is a neurotrophin involved in synaptic plasticity, neurogenesis, neuronal survival, and cognitive health20. Changes in BDNF levels are not specific to AD and have been reported in a number of neuropsychiatric disorders. It remains a key target for therapeutic treatment due to its pivotal role in the central nervous system21. Increasing evidence suggests that BDNF could modulate Aβ accumulation by decreasing Aβ formation22, limiting Aβ-mediated cell death23 and repairing Aβ-related damages24. Our findings indicate that a specific polymorphism in the AC103796.1-BDNF gene region (rs2049048; p = 3.62e−04) is indeed over represented in those who were Aβ+. Another variant, rs6265, has been more widely investigated and found to be associated with reduced hippocampal volume25 and cognitive decline26,27,28,29. However, contradictory results have also been reported30,31, suggesting a more complex relationship between AC103796.1-BDNF and cognition. In our analyses, rs6265, did not show a significant association with Aβ accumulation (p = 0.7).
Another notable association was found with the variant rs9908234 (p = 2.45e−03), located in the nerve growth factor receptor (NGFR) gene which encodes for a cell surface receptor for neurotrophins. A gene-gene interaction network indicated that NGFR has multiple indirect interaction with other genes identified in this study, including AC103796.1-BNDF, ABCA7 and CALHM1 (Fig. 2). In a recent meta-analysis of genome-wide association for migraine, rs9908234, was the most significantly associated marker with the disorder32. Although the link between the two conditions remains unclear, migraines are known to cause micro brain lesions33 which are promoting the development of MCI and AD34,35. In addition to the interaction of NGFR with the aforementioned genes, it also binds one of the major receptors for NGF and has also been reported to bind directly to APP36,37. These studies and others38 postulate a relationship between APP processing/Aβ accumulation and NGF/NGF receptor mediated signaling pathways that warrants further investigation. This relationship is further supported by the association of genetic variation in NGFR with Aβ accumulation in the current study.
We also report an association between the rs648202 marker and Aβ accumulation (p = 2.90e−03). The marker is located in the klotho (KL) gene, which codes for a single-pass transmembrane protein involved in cellular metabolism and has been associated with several age-related diseases. Recent studies have shown that mutations of KL caused systemic aging and reduced longevity in mice34,35. Conversely, overexpression of the gene resulted in healthier aging and prolongation of life39,40. Thus, as a key modulator of the aging process, klotho has become a candidate of interest for the development of novel therapeutic treatment for AD41. However, the role of KL in the development of AD remains to be defined, as a recent study showed that a functional variant in KL, namely KL-VS, had no influence on cognitive decline in preclinical AD42. The rs648202 variant, associated with Aβ in this study, is in linkage disequilibrium (D’ = 1.0) with the KL-VS variants (rs9527025/rs9536314), however they are not highly correlated (r2 = 0.03).
The SNP, rs3764650, located in an intron of the ABCA7 gene showed a moderate association with Aβ accumulation (p = 3.07e−03). This specific variant was identified as one of the main susceptibility loci for late-onset AD in two independent cohorts6,43. Furthermore, recent studies have shown that rs3764650 was associated with cortical and hippocampal atrophy in cognitively normal and mild cognitive impairment (MCI) subjects44 as well as with memory decline in MCI and late-onset AD patients45. In addition, ABCA7 has been identified as a major mediator of phagocytic clearance of Aβ46, which supports the reported association.
In CALHM1, the polymorphism rs2986018 showed marginal evidence for association (p = 4.16e−03). Several genetic epidemiological studies have suggested that rs2986017, a marker located in CALHM1 and within the same LD block as rs2986018, could influence age at onset of AD47,48,49. Although the underlying mechanisms by which CALHM1, which codes for a calcium channel, modulates AD’s pathogenesis remain unclear, it has recently been identified as a repressor of Aβ accumulation, in cell lines and in vivo50. These findings indicate that CALHM1 is potentially involved in Aβ degradation in the brain, a molecular mechanism highly relevant to AD’s pathogenesis.
In summary, although the present study was subject to a lack of power due to the limited number of cases and controls available, it provides suggestive evidence for the implication of several genes previously hypothesised to have a role in the development of AD, through the a priori evidence based approach employed for marker selection. Therefore, replication analyses in independent samples is warranted to confirm our findings and increase the significance of true associations. Whilst the nature of the marker selection employed in this study is a potential strength, the biased nature of this selection may have resulted in the exclusion of, as yet unknown, Aβ-associated genetic variants. As such further unbiased approaches in larger sample sizes is also warranted. Finally, the approach undertaken here used a cross-sectional analysis of a dichotomised sample. The use of a continuous variable of Aβ-burden or longitudinal rates of accumulation may identify other genetic variants of interest.
Conclusion
With the increasing numbers of traits examined through genetic association analyses, it has become increasingly clear that individual genetic components are insufficient to explain complex phenotypes such as Alzheimer’s disease. Instead, such traits are most likely modulated by the collective influence of tens or even hundreds of genetic loci with small individual effects. In this study we identified 6 variants associated with the accumulation of Aβ, a key process in the pathogenesis of AD. The identified variants are located in the intronic regions of 6 distinct loci that are involved in major neurological and neurocognitive functions. Further studies are needed to fully understand the role of these variants in the AD’s pathogenesis. However, this study opens doors to the investigation of novel biological targets for AD treatment to be considered in future studies.
Methods
Participants
Data from the AIBL study, a prospective longitudinal study of ageing, is presented here. The AIBL study design, enrolment process, neuropsychological assessments and diagnostic criteria have been previously described51. Of the 1572 participants enrolled into the AIBL study 1416 of these underwent genetic analysis using the methodologies described below. Participants were classified as MCI52 or AD53 when the clinical criteria for diagnosis of was met. In the absence of these diagnoses a classification of cognitively normal (CN) was given by a clinical review panel, blinded to Amyloid-β status. Ethics approval for the AIBL study and all experimental protocols was provided by the ethics committees of Austin Health, St Vincent’s Health, Hollywood Private Hospital and Edith Cowan University. All experiments and methods were carried out in accordance with the approved guidelines and regulations and all volunteers gave written informed consent before participating in the study.
SNP selection, genotyping and quality control
A thorough literature review was conducted in PubMed to identify genes with an a priori evidence of association with AD risk, cognitive performance, pathological characteristics (i.e. Aβ/tau, atrophy), candidate peripheral/CSF biomarkers, hypothesised pathomechanisms (e.g. Aβ clearance/metabolism) and other AD related biological pathways or comorbidities (e.g. endocytosis, cholesterol metabolism, steroidogenesis, diabetes/insulin resistance, cardiovascular disease). This resulted in the selection of an a priori candidate list of 270 genes. The final selection of 2088 genetic markers across these loci was based on prior phenotypic association and/or extended coverage of each loci. The list of genetic variants is available in Supplementary Table 2.
Genotype data was obtained from 1416 samples from the AIBL cohort using using a combination of an Illumina GoldenGate array containing 1536 markers and multiple TaqMan® OpenArray™ assays.
The GoldenGate array was performed by the Beijing Genomics Institute (BGI, Shenzhen, China) as per manufacturer’s protocols. OpenArray™ assays were developed using inventoried or custom designed TaqMan® genotyping assays, whilst TaqMan® assays were used for APOE genotyping (rs7412, assay ID: C____904973_10; rs429358, assay ID: C___3084793_20; Life Technologies, Carlsbad, CA) using the TaqMan® GTXpress® Master Mix (Life Technologies). All TaqMan® and OpenArray™ assays were performed on an Applied Biosystems™ QuantStudio™ 12 K Flex Real-Time PCR system using the manufacturer’s instructions.
Genotype data was prepared by removing markers with a genotyping call rate below 95% and a minor allele frequency (MAF) of at least 0.05. In addition, markers not in Hardy-Weinberg equilibrium (p < 10−4) were removed. Samples identified with discordant sex information and samples with a call rate below 95% were discarded. Approximately 65% of samples (N = 918) and over 77% of genetic markers assayed (N = 1610) reached QC procedures.
This study limited its analyses to a subset of 505 participants who had previously undergone positron emission tomography (PET) to assess neocortical Aβ burden. PET imaging was performed with three different Aβ-imaging radiotracers 11C-Pittsburgh Compound B (PiB), 18F-florbetapir (FLUTE) or 18F-flutemetamol (FBP). Methodology for each tracer has been previously described in54. Briefly, standardised uptake values (SUVs) were calculated via summing spatially normalised PET images sampled using a narrow cortical regions of interest template using CapAIBL®, a web-based, freely available software55,56. The SUVs were then scaled to each tracer’s recommended reference regions to define the SUV ratio (SUVR). Reference region for PiB was the cerebellar cortex57,58, for FLUTE the pons59 and for FBP the whole cerebellum60. All participants were then classified to a dichotomous Aβ deposition phenotype, being either high (Aβ+; n = 258) or low (Aβ−; n = 247), based on each tracer-specific neuropathology established thresholds (PiB: 1.4 SUVR, FLUTE: 0.62 SUVR and FBP: 1.05 SUVR)58,59,60. Despite displaying different dynamic ranges and subtle differences in the uptake and selectivity of the probes61,62, head to head comparisons of the three radiotracers have previously shown >98% concordance in their classification as PET Aβ+ and PET Aβ−63,64,65.
Association analyses
To identify associations between genetic marker and the dichotomous Aβ status, we performed a logistic regression using PLINK266. The analyses incorporated age, gender and the presence/absence of the ε4 allele as covariates. Considering the limited sample size of the data set and the targeted nature of the study, we used the following approach to identify an adequate threshold for significance. To estimate an empirical threshold for significance, we measured the distributions of the P-values of the variants and defined a threshold, Psig, as the 99th percentile (1-α) at a significance level of α = 0.01. We calculated Psig using the Harrell–Davis distribution-free quantile estimator67.
Data availability
All data and samples used in this study are derived from the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study68. All AIBL data, and that specific to this study, is publicly accessible to all interested parties through an Expression of Interest procedure and is governed by the AIBL Data Use Agreement, for more information please see https://aibl.csiro.au/awd/.
References
Villemagne, V. L. et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. The Lancet. Neurology 12, 357–367, https://doi.org/10.1016/S1474-4422(13)70044-9 (2013).
Jack, C. R. et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. The Lancet. Neurology 12, 207–216, https://doi.org/10.1016/S1474-4422(12)70291-0 (2013).
Nelson, P. T., Braak, H. & Markesbery, W. R. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. Journal of Neuropathology and Experimental Neurology 68, 1–14, https://doi.org/10.1097/NEN.0b013e3181919a48 (2009).
Ma, Y. et al. Predictive accuracy of amyloid imaging for progression from mild cognitive impairment to Alzheimer disease with different lengths of follow-up: a meta-analysis. [Corrected]. Medicine 93, e150, https://doi.org/10.1097/MD.0000000000000150 (2014).
Vos, S. J. B. et al. Prevalence and prognosis of Alzheimer’s disease at the mild cognitive impairment stage. Brain: A Journal of Neurology 138, 1327–1338, https://doi.org/10.1093/brain/awv029 (2015).
Hollingworth, P. et al. Common variants at ABCA7, MS4a6a/MS4a4e, EPHA1, CD33 and CD2ap are associated with Alzheimer’s disease. Nature Genetics 43, 429–435, https://doi.org/10.1038/ng.803 (2011).
Seshadri, S. et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840, https://doi.org/10.1001/jama.2010.574 (2010).
Naj, A. C. et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature Genetics 43, 436–441, https://doi.org/10.1038/ng.801 (2011).
Lambert, J.-C. et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics 41, 1094–1099, https://doi.org/10.1038/ng.439 (2009).
Harold, D. et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics 41, 1088–1093, https://doi.org/10.1038/ng.440 (2009).
Lambert, J.-C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature genetics 45, 1452–1458, https://doi.org/10.1038/ng.2802 (2013).
Lim, Y. Y. et al. APOE ε4 moderates amyloid-related memory decline in preclinical Alzheimer’s disease. Neurobiology of Aging 36, 1239–1244, https://doi.org/10.1016/j.neurobiolaging.2014.12.008 (2015).
Lim, Y. Y. et al. APOE and BDNF polymorphisms moderate amyloid β-related cognitive decline in preclinical Alzheimer’s disease. Molecular Psychiatry 20, 1322–1328, https://doi.org/10.1038/mp.2014.123 (2015).
Porter, T. et al. KIBRA is associated with accelerated cognitive decline and hippocampal atrophy in APOE ε4-positive cognitively normal adults with high Aβ-amyloid burden. Scientific Reports 8, 2034, https://doi.org/10.1038/s41598-018-20513-y (2018).
Porter, T. et al. Cognitive gene risk profile for the prediction of cognitive decline in presymptomatic Alzheimer’s disease. Personalized Medicine in Psychiatry 7, 14–20, https://doi.org/10.1016/j.pmip.2018.03.001 (2018).
Montojo, J., Zuberi, K., Rodriguez, H., Bader, G. D. & Morris, Q. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Research 3, https://doi.org/10.12688/f1000research.4572.1 (2014).
Cruchaga, C. et al. SNPs Associated with Cerebrospinal Fluid Phospho-Tau Levels Influence Rate of Decline in Alzheimer’s Disease. PLoS Genetics 6, e1001101, https://doi.org/10.1371/journal.pgen.1001101 (2010).
Peterson, D. et al. Variants in PPP3r1 and MAPT are associated with more rapid functional decline in Alzheimer’s disease: the Cache County Dementia Progression Study. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association 10, 366–371, https://doi.org/10.1016/j.jalz.2013.02.010 (2014).
Reese, L. C. & Taglialatela, G. A Role for Calcineurin in Alzheimer’s Disease. Current Neuropharmacology 9, 685–692, https://doi.org/10.2174/157015911798376316 (2011).
Diniz, B. S. & Teixeira, A. L. Brain-derived neurotrophic factor and Alzheimer’s disease: physiopathology and beyond. Neuromolecular Medicine 13, 217–222, https://doi.org/10.1007/s12017-011-8154-x (2011).
Nagahara, A. H. & Tuszynski, M. H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nature Reviews. Drug Discovery 10, 209–219, https://doi.org/10.1038/nrd3366 (2011).
Rohe, M. et al. Brain-derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 29, 15472–15478, https://doi.org/10.1523/JNEUROSCI.3960-09.2009 (2009).
Nagahara, A. H. et al. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 33, 15596–15602, https://doi.org/10.1523/JNEUROSCI.5195-12.2013 (2013).
Kimura, N., Takahashi, M., Tashiro, T. & Terao, K. Amyloid beta up-regulates brain-derived neurotrophic factor production from astrocytes: rescue from amyloid beta-related neuritic degeneration. Journal of Neuroscience Research 84, 782–789, https://doi.org/10.1002/jnr.20984 (2006).
Honea, R. A. et al. Characterizing the role of brain derived neurotrophic factor genetic variation in Alzheimer’s disease neurodegeneration. PloS One 8, e76001, https://doi.org/10.1371/journal.pone.0076001 (2013).
Hariri, A. R. et al. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 23, 6690–6694 (2003).
Kennedy, K. M. et al. BDNF val66met polymorphism affects aging of multiple types of memory. Brain Research 1612, 104–117, https://doi.org/10.1016/j.brainres.2014.09.044 (2015).
Lim, Y. Y. et al. BDNF Val66met, Aβ amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiology of Aging 34, 2457–2464, https://doi.org/10.1016/j.neurobiolaging.2013.05.006 (2013).
Miyajima, F. et al. Brain-derived neurotrophic factor polymorphism Val66met influences cognitive abilities in the elderly. Genes, Brain, and Behavior 7, 411–417, https://doi.org/10.1111/j.1601-183X.2007.00363.x (2008).
Fehér, A., Juhász, A., Rimanóczy, A., Kálmán, J. & Janka, Z. Association between BDNF Val66met polymorphism and Alzheimer disease, dementia with Lewy bodies, and Pick disease. Alzheimer Disease and Associated Disorders 23, 224–228, https://doi.org/10.1097/WAD.0b013e318199dd7d (2009).
Nagata, T., Shinagawa, S., Nukariya, K., Yamada, H. & Nakayama, K. Association between BDNF polymorphism (Val66met) and executive function in patients with amnestic mild cognitive impairment or mild Alzheimer disease. Dementia and Geriatric Cognitive Disorders 33, 266–272, https://doi.org/10.1159/000339358 (2012).
Ligthart, L. et al. Meta-analysis of genome-wide association for migraine in six population-based European cohorts. European Journal of Human Genetics 19, 901, https://doi.org/10.1038/ejhg.2011.48 (2011).
Chuang, C.-S., Lin, C.-L., Lin, M.-C., Sung, F.-C. & Kao, C.-H. Migraine and risk of dementia: a nationwide retrospective cohort study. Neuroepidemiology 41, 139–145, https://doi.org/10.1159/000353559 (2013).
Provenzano, F. A. et al. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA neurology 70, 455–461, https://doi.org/10.1001/jamaneurol.2013.1321 (2013).
Rodrigue, K. M. Contribution of Cerebrovascular Health to the Diagnosis of Alzheimer Disease. JAMA neurology 70, 438–439, https://doi.org/10.1001/jamaneurol.2013.1862 (2013).
Fombonne, J., Rabizadeh, S., Banwait, S., Mehlen, P. & Bredesen, D. E. Selective vulnerability in Alzheimer’s disease: amyloid precursor protein and p75(NTR) interaction. Annals of Neurology 65, 294–303, https://doi.org/10.1002/ana.21578 (2009).
Zhang, Y.-W. et al. APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PloS One 8, e80571, https://doi.org/10.1371/journal.pone.0080571 (2013).
Matrone, C. et al. APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 31, 11756–11761, https://doi.org/10.1523/JNEUROSCI.1960-11.2011 (2011).
Dubal, D. B. et al. Life extension factor klotho enhances cognition. Cell Reports 7, 1065–1076, https://doi.org/10.1016/j.celrep.2014.03.076 (2014).
Shiozaki, M. et al. Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience 152, 924–941, https://doi.org/10.1016/j.neuroscience.2008.01.032 (2008).
Paroni, G. et al. Klotho at the Edge of Alzheimer’s Disease and Senile Depression. Molecular Neurobiology, https://doi.org/10.1007/s12035-018-1200-z (2018).
Porter, T. et al. Klotho allele status is not associated with Aβ and APOE ε4 related cognitive decline in preclinical Alzheimer’s disease. Neurobiology of Aging, https://doi.org/10.1016/j.neurobiolaging.2018.12.014 (2019).
Reitz, C. et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ε4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492, https://doi.org/10.1001/jama.2013.2973 (2013).
Ramirez, L. M. et al. Common variants in ABCA7 and MS4a6a are associated with cortical and hippocampal atrophy. Neurobiology of Aging 39, 82–89, https://doi.org/10.1016/j.neurobiolaging.2015.10.037 (2016).
Carrasquillo, M. M. et al. Late-onset Alzheimer’s risk variants in memory decline, incident mild cognitive impairment, and Alzheimer’s disease. Neurobiology of Aging 36, 60–67, https://doi.org/10.1016/j.neurobiolaging.2014.07.042 (2015).
Fu, Y., Hsiao, J.-H. T., Paxinos, G., Halliday, G. M. & Kim, W. S. ABCA7 Mediates Phagocytic Clearance of Amyloid-β in the Brain. Journal of Alzheimer’s disease: JAD 54, 569–584, https://doi.org/10.3233/JAD-160456 (2016).
Boada, M. et al. CALHM1 P86l polymorphism is associated with late-onset Alzheimer’s disease in a recessive model. Journal of Alzheimer’s disease: JAD 20, 247–251, https://doi.org/10.3233/JAD-2010-1357 (2010).
Dreses-Werringloer, U. et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell 133, 1149–1161, https://doi.org/10.1016/j.cell.2008.05.048 (2008).
Lambert, J.-C. et al. The CALHM1 P86l polymorphism is a genetic modifier of age at onset in Alzheimer’s disease: a meta-analysis study. Journal of Alzheimer’s disease: JAD 22, 247–255, https://doi.org/10.3233/JAD-2010-100933 (2010).
Vingtdeux, V. et al. CALHM1 ion channel elicits amyloid-β clearance by insulin-degrading enzyme in cell lines and in vivo in the mouse brain. J Cell Sci 128, 2330–2338, https://doi.org/10.1242/jcs.167270 (2015).
Ellis, K. A. et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. International Psychogeriatrics 21, 672–687, https://doi.org/10.1017/S1041610209009405 (2009).
Winblad, B. et al. Mild cognitive impairment-beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. Journal of Internal Medicine 256, 240–246, https://doi.org/10.1111/j.1365-2796.2004.01380.x (2004).
McKhann, G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944 (1984).
Bourgeat, P. et al. Implementing the centiloid transformation for 11c-PiB and β-amyloid 18f-PET tracers using CapAIBL. NeuroImage 183, 387–393, https://doi.org/10.1016/j.neuroimage.2018.08.044 (2018).
Bourgeat, P. et al. Comparison of MR-less PiB SUVR quantification methods. Neurobiology of Aging 36(Suppl 1), S159–166, https://doi.org/10.1016/j.neurobiolaging.2014.04.033 (2015).
Dore, V. et al. CapAIBL: Automated Reporting of Cortical PET Quantification Without Need of MRI on Brain Surface Using a Patch-Based Method. In Wu, G., Coupé, P., Zhan, Y., Munsell, B. C. & Rueckert, D. (eds) Patch-Based Techniques in Medical Imaging, Lecture Notes in Computer Science, 109–116 (Springer International Publishing, 2016).
Rowe, C. C. et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of Aging 31, 1275–1283, https://doi.org/10.1016/j.neurobiolaging.2010.04.007 (2010).
Rowe, C. C. et al. Head-to-head comparison of 11c-PiB and 18f-AZD4694 (NAV4694) for β-amyloid imaging in aging and dementia. Journal of Nuclear Medicine: Official Publication, Society of Nuclear Medicine 54, 880–886, https://doi.org/10.2967/jnumed.112.114785 (2013).
Thurfjell, L. et al. Automated quantification of 18f-flutemetamol PET activity for categorizing scans as negative or positive for brain amyloid: concordance with visual image reads. Journal of Nuclear Medicine: Official Publication, Society of Nuclear Medicine 55, 1623–1628, https://doi.org/10.2967/jnumed.114.142109 (2014).
Clark, C. M. et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 305, 275–283, https://doi.org/10.1001/jama.2010.2008 (2011).
Lowe, V. J. et al. Comparison of [18f]Flutemetamol and [11c]Pittsburgh Compound-B in cognitively normal young, cognitively normal elderly, and Alzheimer’s disease dementia individuals. NeuroImage: Clinical 16, 295–302, https://doi.org/10.1016/j.nicl.2017.08.011 (2017).
Vandenberghe, R. et al. 18f-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Annals of Neurology 68, 319–329, https://doi.org/10.1002/ana.22068 (2010).
Curtis, C. et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA neurology 72, 287–294, https://doi.org/10.1001/jamaneurol.2014.4144 (2015).
Hatashita, S. et al. [18f]Flutemetamol amyloid-beta PET imaging compared with [11c]PIB across the spectrum of Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging 41, 290–300, https://doi.org/10.1007/s00259-013-2564-y (2014).
Wolk, D. A. et al. Amyloid imaging in Alzheimer’s disease: comparison of florbetapir and Pittsburgh compound-B positron emission tomography. Journal of Neurology, Neurosurgery, and Psychiatry 83, 923–926, https://doi.org/10.1136/jnnp-2012-302548 (2012).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7, https://doi.org/10.1186/s13742-015-0047-8 (2015).
Harrell, F. E. & Davis, C. E. A new distribution-free quantile estimator. Biometrika 69, 635–640, https://doi.org/10.1093/biomet/69.3.635 (1982).
the AIBL Research Group et al. Alzheimer’s disease cerebrospinal fluid biomarkers are not influenced by gravity drip or aspiration extraction methodology. Alzheimer’s Research & Therapy 7, https://doi.org/10.1186/s13195-015-0157-7 (2015).
Yu, H. et al. Next-generation sequencing to generate interactome datasets. Nature Methods 8, 478–480, https://doi.org/10.1038/nmeth.1597 (2011).
Wang, J. et al. Toward an understanding of the protein interaction network of the human liver. Molecular Systems Biology 7, 536, https://doi.org/10.1038/msb.2011.67 (2011).
Chatr-Aryamontri, A. et al. The BioGRID interaction database: 2017 update. Nucleic Acids Research 45, D369–D379, https://doi.org/10.1093/nar/gkw1102 (2017).
Lin, A., Wang, R. T., Ahn, S., Park, C. C. & Smith, D. J. A genome-wide map of human genetic interactions inferred from radiation hybrid genotypes. Genome Research 20, 1122–1132, https://doi.org/10.1101/gr.104216.109 (2010).
Acknowledgements
Funding for the AIBL study was provided in part by the study partners [Commonwealth Scientific Industrial and research Organisation (CSIRO), Edith Cowan University (ECU), Mental Health Research institute (MHRI), National Ageing Research Institute (NARI), Austin Health, CogState Ltd.]. The AIBL study has also received support from the National Health and Medical Research Council (NHMRC) and the Dementia Collaborative Research Centres program (DCRC2), as well as funding from the Science and Industry Endowment Fund (SIEF) and the Cooperative Research Centre (CRC) for Mental Health – funded through the CRC Program (Grant ID:20100104), an Australian Government Initiative. Data used in preparation of this article was obtained from the AIBL68 (https://aibl.csiro.au/). We thank all the investigators within the AIBL who contributed to the design and implementation of the resource and/or provided data but did not participate in the analysis or writing of this report. A complete listing of AIBL investigators can be found at https://aibl.csiro.au/about/aibl-research-team/.
Author information
Authors and Affiliations
Contributions
M.V. developed the computational pipeline, performed the data analysis and wrote the initial manuscript. T.P., L.M. and M.P. contributed to the acquisition of genetic data, V.V. contributed to acquisition of imaging data and revising the manuscript. J.D. and M.V. verified the analytical methods. S.L. devised the project and the main conceptual ideas, supervised the findings of this work and obtained funding. M.V., T.P., J.D. and S.L. discussed the results and contributed to the manuscript. D.A., C.M., R.M. and C.R. contributed to AIBL study design and obtaining funding. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vacher, M., Porter, T., Villemagne, V.L. et al. Validation of a priori candidate Alzheimer’s disease SNPs with brain amyloid-beta deposition. Sci Rep 9, 17069 (2019). https://doi.org/10.1038/s41598-019-53604-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53604-5
This article is cited by
-
Large multi-ethnic genetic analyses of amyloid imaging identify new genes for Alzheimer disease
Acta Neuropathologica Communications (2023)
-
Early onset senescence and cognitive impairment in a murine model of repeated mTBI
Acta Neuropathologica Communications (2021)
-
Lower brain-derived neurotrophic factor levels are associated with age-related memory impairment in community-dwelling older adults: the Sefuri study
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
