
Abstract
Withania somnifera owing to its strong and remarkable stress tolerance property is a reliable candidate for the determination of genes involved in mechanism of adaption/tolerance of various stress conditions. 187 AP2/ERF gene related transcripts (GRTs) were identified during comprehensive search in W. somnifera transcriptome repertoire. Major hits in homology search were observed from the model plant Arabidopsis and members of Solanaceae family. Cloning, expression analysis of the gene and genetic transient transformation with the gene (WsAP2) were performed to predict its functional role in planta. Enhanced expression of some of the pathway genes for terpenoid biosynthesis was observed in transformed tissues in comparison to the control tissues. It is speculated that WsAP2 gene crucially regulates the expression of GGPPS gene in addition to the regulation of other important genes of terpenoid pathway via induction of expression of other genes such as HMGR, CAS, DXS and DXR. To the best of our knowledge, this is the first report representing detailed study of AP2/ERF gene family in W. somnifera. It is also suggested from the study that gene might have role in eliciting responses to combat stress and attribute the strong stress tolerant property associated with the plant.
Similar content being viewed by others


Introduction
Plant transcription factors are involved in the regulation of various aspects of plant growth and development including metabolism, ripening of fruits and defense responses etc. APETALA2/ethylene response factor (AP2/ERF) is a large super family of transcription factors (TF) in plant kingdom, which has been reported to be involved in various cellular processes1. AP2/ERF superfamily of transcription factors have been reported to be plant specific however the domain has also been encountered in the proteins associated with cyanobacteria2. Classification and structural analysis of AP2 superfamily revealed that the members of this superfamily have AP2 DNA binding domain of 60 amino acids. On the basis of repetitions and sequence of these AP2 domain, the superfamily has been classified in five major subfamilies i.e. AP2, RAV, ERF (Ethylene response factor), DREB (dehydration-responsive element-binding protein) subfamilies and Soloist3,4. The ERF and DREB subfamily contains a conserved WLG motif and a single AP2 binding domain while AP2 subfamily has two AP2 domains. The RAV subfamily members have a B3 DNA binding domain in addition to AP2 domain. Other members have single AP2 domain but do not have WLG motif, the characteristic of DREB and ERF subfamily5. One twenty one ERF proteins from Arabidopsis thaliana were classified in two different groups of DREB and ERF, based on the sequence similarity of their AP2 domain and these two groups have been divided into six subgroups3. Further analysis of the exon-intron structure and presence of additional motifs in ERF proteins of Arabidopsis thaliana as well as of Oryza sativa, has led to division of these proteins into 12 subgroups based on similar regulatory features6. The 60 amino acid long AP2 domain of AP2 superfamily is divided into two groups. At N terminal end, 20 amino acid long YRG region is rich in basic and hydrophilic residues that is responsible for binding with DNA element, while 40 amino acid long stretch at C terminal end is known as RAYD region that forms an alpha helix with its 118 amino acid residues. This RAYD region is responsible to mediate protein-protein interactions7. The double repeat of AP2 domain in AP2 subfamily is attached by a linker sequence of about 25 amino acids which is highly conserved and important for proper binding of AP2 proteins with their respective DNA elements8.
AP2/ERF superfamily genes and related transcripts were previously identified in various plants and extensively studied with respect to stress tolerance9,10. Various plants have been examined for genome-wide studies related to AP2 gene in plants such as Arabidopsis thaliana11, Ricinus communis L12, Brassica rapa ssp. pekinensis13, Vitis vinifera14, Lotus japonicus15, Medicago truncatula16, Populus trichocarpa17, Musa species18, Glycine max L19 etc. Further, transcriptomic and EST related studies for AP2 have been carried out in Brassica sp20,21., Hevea brasiliensis22, Camellia sinensis23 and Tricitum aestivum24. Here, in this study, we have identified 187 AP2/ERF GRTs in W. somnifera transcriptome. The study presents the first report on identification of AP2/ERF genes in W. somnifera in addition to its classification and characterization. Expression of various transcription factors control and regulate the developmental and cellular processes of plants. Therefore, the study will provide a platform for detailed functional analysis of AP2/ERF family genes to serve as resource for understanding the molecular mechanisms associated with stress responses in W. somnifera.
One hundred forty five, AP2/ERF genes were characterized in Arabidopsis containing 6, 65, 56 and 1 gene representatives belonging to AP2, RAV, ERF, DREB and Soloist respectively. Recent reports on C. annum analysed presence of 175 AP2/ERF related transcripts in the latest genome database which were classified as AP2, RAV, ERF, and Soloist members25. Similarly, 155 tentative ERF genes were determined in Solanum tuberosum using the genome database and the comparison was carried out with A. thaliana26. Isolation of a cold-inducible protein was proposed to contain AP2/ERF domain and reported in S tuberosum previously27. Further five ERF genes were also reported to be cloned from S tuberosum with predictive roles in hormonal and stress regulation of potato28. However, in W. somnifera not much information is available on the identification and characterization of the AP2 transcription factor gene, so far.
Putative GRTs encoding for AP2/ERF in W. somnifera were used for the identification of total 182 transcripts in the respective superfamily. Phylogenetic as well as protein motif structure analysis was also carried out for AP2/ERF genes. Furthermore, the demonstration of length wise transcript expression in different tissues for these transcripts was also performed. Thus, the functional motif identification and classification into various groups will facilitate further studies related to biological functions for the AP2/ERF family genes in W. somnifera.
Materials and Methods
Identification of AP2/ERF GRTs in W. somnifera through transcriptome analysis
The transcriptome datasets (SRA053485) at NCBI database (https:/www.ncbi.nlm.nih.gov/) and illumina sequenced pooled berry tissue data were used to mine out the AP2 GRTs. GRTs related to AP2/ERF were isolated from W. somnifera transcriptome through BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and annotation analysis. To avoid the redundancy of gene sequences the putative GRTs were assembled further through CAP3 assembler (http://doua.prabi.fr/software/cap3). The sequences were processed and assembled appropriately to ensure dataset having significant transcript length and reduced error rate, as done previously29,30. The transcripts screened with appropriate parameters were analyzed using Blast2GO pipeline31. The annotations for the GRTs were also confirmed by datasets from AP2/ERF genes at PlantTFDB (http://planttfdb.cbi.pku.edu.cn/) and transcription factor genes at PlantTFcat (http://plantgrn.noble.org/PlantTFcat/).
Sequence alignment and phylogenetic analysis
The query sequences from W. somnifera transcriptome were subjected to multiple sequence alignment along with the the sequences from S. lycopersicum, C. annum and N. tabacum and other plants using tools available (https://www.ebi.ac.uk/Tools). The dataset was then used for tree construction using MEGA 6.06 (https://www.megasoftware.net/). The extent of closeness among the sequences was determined through maximum likelihood using earlier method32. Highest log likelihood was applied to construct the tree with branch length obtained through calculation of occurrence of substitution at a particular position.
Motif analysis
MEME suite, a motif based sequence analysis tool was used for motif identification with average amino acid sequences of upto 25 (http://meme-suite.org/) with a value of 35 as maximum number of motifs allowed for analysis. MEME was used to predict the probability of amino acid at a particular site in the pattern33.
Plant materials
Withania somnifera (NMITLI-118) plants were grown the experimental farm and glasshouse of the CSIR- Central Institute of Medicinal and Aromatic Plants, Lucknow, India. Young plantlets of five leaf stage (one month old) were taken for stress and elicitation treatments while mature (six months old) plants were harvested for tissue wide expression analysis and gene isolation studies.
Isolation of RNA and cDNA synthesis
Flowers of W. somnifera (NMITLI-118) were collected and frozen in liquid nitrogen immediately after harvesting. For the isolation of total RNA, TRI reagent (Sigma- Aldrich, US) was utilized following the standard manufacturer’s instructions. The quality of RNA was assesed on 0.8% agarose gel and quantity was checked by spectrophotometric analysis (ND- 1000 Nanodrop, NanoDrop Technologies, US). cDNA was synthesized by using the Revert Aid kit for cDNA synthseis (Fermentas, US) following the manufacturer’s methods.
Tissue wide transcript quantification studies of WsAP2
For relative quantification of levels of transcripts of WsAP2, different plant parts (leaf, root, berry and flower) were collected from six months old mature plants of W. somnifera and immediately frozen in liquid nitrogen. Further, RNA was isolated and cDNA was synthesized following standard procedure provided by manufacturers (Fermentas) followed by real time PCR analysis using SYBR green master mix (Applied Biosystems). Gene specific real time primers WST11RTF and WST11RTR were used with keeping β-actin gene as an internal reference control. Reactions were carried out using the cycling conditions as described for qRT-PCR above. The ΔΔCt method was used to calculate the expression levels of WsAP234.
Stress and elicitor treatments
One month old plantlets of five leaf stage were employed for various elicitor and stress treatments such as gibberellic acid (GA), methyl jasmonate (MeJA), salicylic acid (SA), wounding, heat-shock and cold. For GA, SA and MeJA treatments, plantlets were dipped in distilled water supplemented with 0.1 mM and 1 mM SA, GA and MeJA separately for 3 hrs, 6hrs, 9hrs, 12hrs, 24 hrs, and 48 hrs, respectively. For heat shock treatment, plantlets were kept at 65 °C for 30 min and 1 hr, for cold treatment plantlets were kept at 4 °C for 30 min and 1 hr. The wound treatment was given by rubbing and puncturing the leaves by sterile syringe, followed by dipping the plantlets in distilled water for 30 min and 1 hr. RNA isolation was done followed by cDNA synthesis and real time transcript quantification of WsAP2 was carried out by methods described above using same set of primers.
Cloning of APETALA2/ethylene response factor (AP2-ERF) (WsAP2) gene from W. somnifera
Full length clone of WsAP2 (Node_3814) was obtained using cDNA synthesized from RNA of W somnifera flowers with gene specific primers. For amplification, Pfu DNA polymerase (Fermentas, US) was used and the PCR thermocyling program started from denaturation at 94 °C for 3 min, followed by 32–35 cycles at 94 °C for 40 sec, 55 °C for 1 min and 72 °C for 2.0 min, and terminated by a final extension step at 72 °C for 10 min. The purified product was then cloned in the pJET1.2 vector and transformed in E. coli DH5α cells. Positive clones were screened by colony PCR and used for plasmid isolation. Plasmid was digested by appropriate restriction enzymes and cloned into pBI121 vector containing CaMV 35 S promoter. Positive clones were confirmed by PCR and digestion and was further utilized for transformation in A. tumefaciens.
Transient transformation of W. somnifera with WsAP2-pBI121 construct
Transient transformation was performed to transiently overexpress WsAP2 transcription factors in W. somnifera by the method described in our earlier studies35. The cycling parameters were same as described above and β-actin was taken as endogenous control for semi-quantitative PCR. The semi quantitative expression pattern of WsAP2 was visualized on 0.8% agarose gel. Quantitative real time PCR was performed to validate the results obtained from semi-quantitative PCR and to quantify the WsAP2 transcript abundance in transformed tissues. qRT-PCR was performed with an ABI PRISM 7500 Real-Time PCR System (ABI, US) with a thermocycling program of 95 °C for 30 sec, followed by 40 cycles of amplification (95 °C for 5 sec, 60 °C for 20 sec, 72 °C for 20 sec). For normalization of expression values, actin of W. somnifera was used as a reference gene. All the reactions were conducted with three biological replicates in 10 µl reaction volume.
GUS assay of putative transformants
A. tumefaciens infected putative tissue transformants after transient expression were checked for GUS expression36. Briefly, transiently transformed leaves tissues were checked for expression of GUS by utilizing in-situ GUS reaction mixture. Tissues were incubated for overnight at 37 °C followed by washing with sterile double distilled water. Further, the tissues were dipped in 70% ethanol for removal of chlorophylls. The tissues were observed under a microscope (Leica EZ4D, Switzerland), and scored for the presence of GUS foci with photo-documentation.
Transcript abundance studies on secondary metabolite pathway genes in WsAP2 transformedtissues
The selected genes for this study were CAS (cycloartenol synthase), HMGR (3-hydroxy-3-methylglutaryl coenzyme A reductase), DXS (1-deoxy-D-xylulose-5-phosphate synthase), DXR (1-deoxy-D-xylulose 5-phosphate reductoisomerase) and GGPPS (geranylgeranyl diphosphate synthase). RNA was isolated from wild type and transiently transformed tissues of WsAP and cDNA was synthesized. This cDNA was utilized for qRT-PCR analysis as described above. The primer sequences of selected genes are mentioned in Table S1.
Results
Mining and annotation of AP2/ERF GRTs
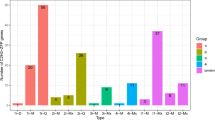
Transcripts with significant range of length for AP2/ERF GRTs were observed in the transcriptome (Pooled berry tissue transcripts represented by red bars as various NODE ids and green and yellow bars denote transcripts named as various contig ids for leaf and root tissues respectively, as in our previous reports). Overall 187 AP2/ERF GRTs were found to be present in W. somnifera transcriptomes. Significant number of AP2/ERF GRTs of considerable lengths were observed in berry (89), leaf (36) and root (62) tissues during the study which included Blastx analysis for homology search, GO analysis, Interproscan assignment analysis etc. The transcripts having length between 500 to 3000 bp were plotted together for the three tissues (Fig. 1). Top hits were observed for genes reported from A. thaliana, various members from Solanaceae family (S.lycopersicum, C. annum, N. tabacum, S. tuberosum etc.), G. max and various other plants (Fig. 2). The major descriptions for the hits observed in AP2/GRTs were AINTEGUMENTA-like 6, AP2 family protein, ERF family protein, cytokinin response factor, 4, AP2 6 l, ethylene responsive element binding factor2 and so on. 27 transcripts out of total input transcripts were observed to be annotated with Arabidopsis AP2/ERF gene sequences at PLANTTFDB containing important gene definitions (Table 1).


Top hit species distribution for the whole repertoire (combined transcripts from all the transcriptomes) of AP2 GRTs in W. somnifera based on annotations obtained through Blast2GO analysis.
Gene ontology (GO) assignment
Gene ontology search assigned various terms to the gene transcripts such as cell growth, transport, response to stress, response to stimulus (abiotic, biotic and endogenous), signal transduction, flower and embryo development, secondary metabolic process and so on. Similarly, for metabolic function DNA-binding transcription activity, DNA binding, protein binding and so on were the major terms in addition to the transporter activity. Additionally, for cellular component the major expressed terms were nucleus, cytoplasm, plasma membrane, cytosol and some other terms (Fig. 3).

Gene Ontology annotation by assignment of terms for (a) Biological process (BP), (b) Cellular component (CC) and (c) Molecular function (MF) to the whole repertoire (combined transcripts from all the transcriptomes) of AP2 GRTs in W. somnifera.
Phylogenetic reconstruction of AP2/ERF GRTs in W. somnifera
Multiple sequence alignment was done using the amino acid sequences of putative AP2/ERF GRTs against proteins. MEGA 6.06 software was used for analysis of the phylogeny and molecular evolutionary pattern (Fig. 4). On analyzing the closeness of AP2/ERF GRTs from W. somnifera with C. annum, S. lycopersicum and N. tabacum, 26 transcripts branched as separate groups in which starting from the first group anticlockwise 7 transcripts were classified as separate clades and 18 other transcripts together clubbed in other clade. Root contig 13458 was classified as a separate branch. In another group 21 W. somnifera AP2/ERF GRTs were grouped separately but lying in the same clade having 3 AP2/ERF genes from S. tuberosum, and 1 related gene from C. annum which were closely placed with root contigs 57460, 18800, 18921 and 6893. Leaf contig 28320, Node 26035 and root contig 45742 were branched separately. Similarly, some other transcripts from W. somnifera in the same group were observed to form different clades. 12 AP2/ERF GRTs were aligned together with 11 AP2 genes from Solanaceae species containing 6 sequences from C. annum, 1 from N. tabacum and 4 from S. tuberosum (Fig. 4). Node 7315, 95324 and leaf contig 7075, 14770 branched as additional separate groups,. Further, the next two groups mostly were populated with the reference gene sequences from Solanaceae species. Importantly, the AP2/ERF GRTs importantly that aligned with the above mentioned groups were Node 69997, 57223, 8963, 22774, 38413, and leaf contig 37257. Node 38413 was observed to be closely related to S. tuberosum, N. tabacum and C. annum assigned to the same group but branched differently (Fig. 4).

Phylogenetic tree construction for the whole repertoire (combined transcripts from all the transcriptomes) of AP2 GRTs in W. somnifera and genes from members of family Solanaceae (S. tuberosum, C. annum and N. tabacum). Node_3814 which was used for further analysis has been separately highlighted by orange color arrow.
Conserved motif analysis
The annotated sequences contain enriched motifs for AP2/ERF related transcription factors. The analysis suggested that various AP2/ERF genes contain conserved motifs. Three motifs which were of considerable importance, have been shown for the selected sequences (Fig. 5). Major proportions of W. somnifera GRTs showed full AP2/ERF domains while some contained partial domains. The 3 majorly observed motifs were SKKLYRGVRQRPWGKWVAEIRLP as motif 1, AARAYDAAALKLRGKKA as motif 2, and KLNFPENRP as motif 3. The domain importantly present in all the sequences was IPR001471 which has been defined as AP2/ERF domain. The motifs are closely located in some of the sequences while distantly apart in various other sequences (Fig. 5).

Identification of motifs for the selected AP2 GRTs in W. somnifera using MEME suite. (a) Sequence logo for three major motifs (b) Block diagrams to show the location of the motifs in representative sequences.
Cloning of WsAP2
The complete ORF of WsAP2 was cloned in pJET 1.2 blunt end vector and sequenced (Fig. 6a). The full length (1098 bp) amplicon of WsAP2 from W. somnifera codes for the predicted protein of 366 amino acids (Fig. 6a–e). The calculated mass of WsAP2 protein was 40.2 kDa. Homology matching of WsAP2 sequence with transcriptomic dataset (Node_3814) confirmed existence of 99 percent sequence identity. The phylogenetic tree constructed for WsAP2 sequence with the top hit matching sequences during homology search also revealed the closeness of the WsAP2 gene sequence with other members of family Solanaceae such as S. tuberosum (XP_006341423.1), N. tabacum (XP_016457719.1), N. attenuata (XP_019244135.1), C. annum (XP_016562990.1) and C. chinense (PHU24180.1) (Fig. 7).

Cloning, restriction digestion, and confirmation of positive clones by colony PCR of full length WsAP2 (a). Recombinant construct map of pBI121-WsAP2 (b).Total RNA isolation from flower of W. somnifera (c) Amplification of WsAP2 (1098 bp). (d) Restriction digestion of pJET::WsAP2 clone by respective restriction enzymes for expression in pBI121 binary vector. (e) Colony PCR confirmation of cloned WsAP2.

Phylogenetic tree for full length cloned WsAP2 in W. somnifera.
Tissue specific abundance of WsAP2 transcripts
Since expression level of a gene varies across parts of a plant under various biotic and abiotic stresses37,38, we analyzed the expression levels of WsAP2 in different tissues of the W somnifera . Our investigations suggested that WsAP2 was expressed ubiquitously in all parts of W. somnifera. Comparative analysis of expression revealed that it was highest in flower followed by leaf. Lowest expression level were recorded in root tissue (Fig. 8a).

(a) Transcript abundance in different tissues of W. somnifera (L = Leaf; R = Root; B = Berry and F = Flower). Induced relative expression of WsAP2 gene after elicitor’s treatment (b) 1 mM Gibberellic acid (c) 1 mM Salicylic acid (d) 1 mM Methyl Jasmonate (e) Stress (Wounding, Cold and Heat) treatment for 0.5 and 1 hour.
Modulation of WsAP2 gene expression by signaling molecules and stress treatments
The analysis revealed that the increased expression of the gene was noticed after elicitor (salicylic acid, methyl jasmonate and gibberellic acid), wounding, heat and cold treatments (Fig. 8a–e). In the case of 1 mM gibberellic acid treatment, increment in expression level was recorded after 9 hrs (95 folds) (Fig. 8b).The expression of the gene was upregulated upto 245 folds during the first 24 hrs after treatment with salicylic acid after which down regulation of gene occurred (Fig. 8c). The most profound effect was observed with MeJA treatment with 1 mM concentration where the elevated expression was recorded after 9 hrs of treatment (upto 155 folds) followed by a decreased expression at 12 hrs intervals (Fig. 8d). Wounding increased the transcripts level of WsAP2 after one hour of treatment (282 folds) while cold treatment reduced the expression of WsAP2 from 30 min (32 folds) to one hour (26 folds) post treatment. Heat treatment reduced the expression level of transcripts in one hour (16 folds) as compared to 30 min (38 folds) (Fig. 8e).
Transcript abundance of WsAP2 and expression level of pathway genes in transiently transformed leaf tissues
The transient transformation assay was chosen for assessment of the functional role of WsAP2 gene. WsAP2 was successfully cloned in pBI121 (Fig. 6a,b) and overexpressed in W. somnifera leaf explant as indicated by the GUS histochemical analysis (Fig. 9a). Semi-quantitative as well as quantitative real time PCR analysis was done to assess the expression level of WsAP2 in transformed tissues of W. somnifera in comparison with wild type and empty vector control (Fig. 9b,c). Expression levels of transcripts of WsAP2 in transiently transformed tissues was 6 to 8 times higher in comparison to wild type tissues and empty vector transformed tissues. The data revealed that the elevated expression in transcripts level of WsAP2 gene in the transgenic tissues, with variation in transcript abundance in different transformed lines.

Transient expression of WsAP2 gene in W somnifera leaves with pBI::WsAP2 construct. (a) GUS Histochemical assay in WsAP2 transformed tissues for confirmation of successful transformation event (b) Semi-quantitative gene expression analysis in wild type, vector control and transformed leaves. (c) Real time qRT PCR analysis in wild type, vector control and transformed leaves. (d) Expression profiling of key genes of MVA and MEP pathway.
By using qRT-PCR analysis the expression level of secondary metabolic pathway (terpenoids) related genes like CAS, HMGR, DXS, DXR and GGPPS in transiently transformed tissues of WsAP2 was also monitored. It was found that all the transcripts showed increased expression in transformed tissues as compared to wild type and empty vector control but the expression of GGPPS was more pronounced (more than 80 folds) than the remaining gene. This result suggested that WsAP2 may be a master regulator of GGPPS gene and also regulated the MVA and MEP pathway of terpenoid biosynthesis via inducing the expression of HMGR, CAS, DXS and DXR gene (Fig. 9d).
Disscussion
Increasing interest of medicinal plants for therapeutic, neutraceuticals and various other purposes have resulted in the attention of a number of related sectors towards more research on these plants. However, there are many biotic and abiotic+ stresses which these plants have to tolerate posing serious threats to the growth and development of plants. For the exploitation of medicinal plants in different applications and better sustainability, these have to be pest and disease free with improved yield and fewer requirements of fertilizers and water39,40. Environmental factors and pathogens have strong impact on plants during its development. Since AP2/ERF genes play significant role in plant development and various stress responses, whether biotic or abiotic, these present ideal candidature for investigating the regulatory mechanism of related process. The improved tolerance for biotic and abiotic stress requires special focus on plants under the physiological and molecular mechanism towards these stresses as for example in W. somnifera. The significance of the objective to identify and isolate AP2/ERF GRTs is towards understanding the molecular genetic basis which would facilitate the improvement of W. somnifera and provide the functional genetic resource meant for transgenic research. Transcription factors are very important in modulation of acclimatization of plant responses towards different external or internal cues. These significantly govern downstream gene expression in response to stress exposure via gene activation/repression in case of stress and signal transduction pathways. These are present in the plant genome in large numbers. In our previous study, we have demonstrated the presence of significant proportion of transcription factors in W. somnifera29. On the basis of conserved AP2-related domains AP2/GRTs were identified to be grouped as AP2/ERF superfamily members in W. somnifera transcriptome in this analysis. The availability of datasets for various plants at Plant (TFDB) facilitated the identification and comparison of families and groups at default parameters. The annotation as predicted, based on comparison with various databases (db) such as Arabidopsis genome db, Interpro db and TFCAT db confirmed the presence AP2/ERF domains in the sequences and thus suggests the correctness of annotation via these databases. The results of selected AP2/ERF genes through phylogenetic analysis were observed to be in confirmation with the results of whole transcripts phylogenetic comparison with other Solanaceae species members, as the selected Node_3814 was aligned close to the Nicotiana sp., S. tuberosum, Capsicum sp. Not every W. somnifera AP2/ERF gene has counterpart in N. tabacum, S. tuberosum, C. annnum depicting the probability that W. somnifera had undergone differential expansion separately. The candidate transcripts for AP2/ERF proteins were used in MEME motif analysis for identification of conserved motifs in families and subfamilies. The results from conserved motif analysis may provide leads to further classify the putative candidates, as identical motifs are likely to have similar function41.
Various transcription factors were cloned and characterized from W. somnifera but the role of AP2-ERF TF was still not understood. For the assesment of the role of AP2-ERF transcription factor in W. somnifera via in-silico methods followed by in-planta validation, this study was carried out.The transient overexpression study of WsAP2 showed that this gene was successfully transformed in W. somnifera. It was already known that AP2 transcription factor was involved in growth and development of plant as well as in the regulation of biotic and abiotic stress response3,42. It was reported that salicylic acid and jasmonic acid are important activators of defence related genes in plant system. In addition to this plant hormone gibberellic acid regulates the crosstalk of different signaling cascades occurring during different abiotic stress responses43, such as drought, salt, and cold. In our current study, the transient overexpression of WsAP2 was found to be induced not only by salicylic acid, methyl jasmonate and gibberellic acid but also by abiotic stresses like wound, heat and cold treatments. This has led to the conclusion that WsAP2 might act as a connecting link between different signalling pathways abiotic stress responses in plant. Jasmonic acid is involved in the rearrangement of gene expression of secondary metabolism in response to various types of environmental and developmental stimuli. Thus, jasmonic acid is a strong inducer of secondary metabolism44. Several jasmonate (methyl jasmonate) inducible AP2 transcription factors were studied to be involved in the regulation of secondary metabolic pathway related enzymes. For example overexpression of AaERF1 or AaERF2 were associated with increased accumulation of artemisinin and artemisinic acids45,46. In our study, it was found that after transient overexpression of WsAP2 in plant, the expression levels of secondary metabolism related genes like CAS, HMGR, DXS and DXR was increased and GGPPS were maximally induced after WsAP2 overexpression confirming the involvement of WsAP2 in terpenoid metabolism.
References
Gu, C. et al. Multiple regulatory roles of AP2/ERF transcription factor in angiosperm. Botanical Studies 58, 6 (2017).
Wessler, S. R. Homing into the origin of the AP2 DNA binding domain. Trends Plant Sci. 10, 54–56 (2005).
Sakuma, Y. et al. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration-and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 290, 998–1009 (2002).
Saleh, A. & Pagés, M. Plant AP2/ERF transcription factors. Genetika 35, 37–50 (2003).
Magnani, E., Sjölander, K. & Hake, S. From endonucleases to transcription factors: evolution of the AP2 DNA binding domain in plants. Plant Cell. 16, 2265–2277 (2004).
Nakano, T., Fujisawa, M., Shima, Y. & Ito, Y. The AP2/ERF transcription factor SlERF52 functions in flower pedicel abscission in tomato. J.Exp. Bot. 65, 3111–3119 (2014).
Okamuro, J. K., Caster, B., Villarroel, R., Van Montagu, M. & Jofuku, K. D. The AP2 domain of APETALA2 defines a large new family of DNA binding proteins in Arabidopsis. Proc. Natl. Acad. Sci. USA 94, 7076–7081 (1997).
Klucher, K. M., Chow, H., Reiser, L. & Fischer, R. L. The AINTEGUMENTA gene of Arabidopsis required for ovule and female gametophyte development is related to the floral homeotic gene APETALA2. Plant Cell. 8, 137–153 (1996).
Xu, Z. S., Chen, M., Li, L. C. & Ma, Y. Z. Functions and application of the AP2/ERF transcription factor family in crop improvement F. J. Integr. Plant Biol. 53, 570–585 (2011).
Mizoi, J., Shinozaki, K. & Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta Gene Regul. Mech. 1819, 86–96 (2012).
Czechowski, T., Stitt, M., Altmann, T., Udvardi, M. K. & Scheible, W. R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 139, 5–17 (2005).
Xu, W., Li, F., Ling, L. & Liu, A. Genome-wide survey and expression profiles of the AP2/ERF family in castor bean (Ricinus communis L.). BMC Genomics 14, 785 (2013).
Song, X., Li, Y. & Hou, X. Genome-wide analysis of the AP2/ERF transcription factor superfamily in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genomics 14, 573 (2013).
Licausi, F. et al. Genomic and transcriptomic analysis of the AP2/ERF superfamily in Vitis vinifera. BMC Genomics 11, 719 (2010).
Sun, Z. M., Zhou, M. L., Xiao, X. G., Tang, Y. X. & Wu, Y. M. Genome-wide analysis of AP2/ERF family genes from Lotus corniculatus shows LcERF054 enhances salt tolerance. Funct. Integr. Genomics 14, 453–466 (2014).
Shu, Y., Liu, Y., Zhang, J., Song, L. & Guo, C. Genome-wide analysis of the AP2/ERF superfamily genes and their responses to abiotic stress in Medicago truncatula. Front. Plant Sci. 6, 1247 (2016).
Zhuang, J. et al. Genome-wide analysis of the AP2/ERF gene family in Populus trichocarpa. Biochem. Biophys. Res. Commun. 371, 468–474 (2008).
Lakhwani, D. et al. Genome-wide analysis of the AP2/ERF family in Musa species reveals divergence and neofunctionalisation during evolution. Sci. Rep. 6, 18878 (2016).
Zhang, G. et al. Phylogeny, gene structures, and expression patterns of the ERF gene family in soybean (Glycine max L.). J. Exp. Bot. 59, 4095–4107 (2008).
Zhuang, J. et al. Analysis of Brassica rapa ESTs: gene discovery and expression patterns of AP2/ERF family genes. Mol. Biol. Rep. 37, 2485–2492 (2010).
Zhuang, J. & Zhu, B. Analysis of Brassica napus ESTs: gene discovery and expression patterns of AP2/ERF-family transcription factors. Mol. Biol. Rep. 41, 45–56 (2014).
Duan, C. et al. Identification of the Hevea brasiliensis AP2/ERF superfamily by RNA sequencing. BMC Genomics 14, 30 (2013).
Wu, Z. J. et al. Transcriptome-based discovery of AP2/ERF transcription factors related to temperature stress in tea plant (Camellia sinensis). Funct. Integr. Genomics. 15, 741–752 (2015).
Zhuang, J. et al. Discovery and expression profile analysis of AP2/ERF family genes from Triticum aestivum. Mol. Biol.Rep. 38, 745–753 (2011).
Jin, J. H. et al. Genome-wide identification of the AP2/ERF transcription factor family in pepper (Capsicum annuum L.). Genome 61, 663–674 (2018).
Charfeddine, M., Saïdi, M. N., Charfeddine, S., Hammami, A. & Bouzid, R. G. Genome-wide analysis and expression profiling of the ERF transcription factor family in potato (Solanum tuberosum L.). Mol.Biotechnol. 57, 348–358 (2015).
Mine, T., Hiyoshi, T., Kasaoka, K. & Ohyama, A. CIP353 encodes an AP2/ERF-domain protein in potato (Solanum tuberosum L.) and responds slowly to cold stress. Plant Cell Physiol. 44, 10–15 (2003).
Wang, Z. et al. Isolation and characterization of StERF transcription factor genes from potato (Solanum tuberosum L.). C.R. Biol. 338, 219–226 (2015).
Tripathi, S. et al. Transcription factor repertoire in Ashwagandha (Withania somnifera) through analytics of transcriptomic resources: Insights into regulation of development and withanolide metabolism. Sci. Rep. 7, 16649 (2017).
Srivastava, S. et al. Light and auxin responsive cytochrome P450s from Withania somnifera Dunal: cloning, expression and molecular modelling of two pairs of homologue genes with differential regulation. Protoplasma 252, 1421–1437 (2015).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Bailey et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25, 402–408 (2001).
Mishra, S., Sangwan, R. S., Bansal, S. & Sangwan, N. S. Efficient genetic transformation of Withania coagulans (Stocks) Dunal mediated by Agrobacterium tumefaciens from leaf explants of in vitro multiple shoot culture. Protoplasma 250, 451–458 (2013).
Jefferson, R. A. Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol. Biol. Rep. 5, 387–405 (1987).
Pichersky, E. & Gang, D. R. Genetics and biochemistry of secondary metabolites in plants: an evolutionary perspective. Trends Plant Sci. 5, 439–445 (2000).
Sabir, F., Mishra, S., Sangwan, R. S., Jadaun, J. S. & Sangwan, N. S. Qualitative and quantitative variations in withanolides and expression of some pathway genes during different stages of morphogenesis in Withania somnifera Dunal. Protoplasma 250, 539–549 (2013).
Karp, A. et al. Genetic improvement of willow for bioenergy and biofuels free access. J. Integr. Plant Biol. 53, 151–165 (2011).
Miao et al. De novo transcriptome analysis of Medicago falcata reveals novel insights about the mechanisms underlying abiotic stress-responsive pathway. BMC Genomics 16, 818 (2015).
Xie, X. L. et al. Isolation, classification and transcription profiles of the AP2/ERF transcription factor superfamily in citrus. Mol. Biol. Rep. 41, 4261–4271 (2014).
Eckert, D., Buhl, S., Weber, S., Jäger, R. & Schorle, H. The AP-2 family of transcription factors. Genome Biol. 6, 246 (2005).
Shinozaki, K., Yamaguchi-Shinozaki, K. & Seki, M. Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 6, 410–417 (2003).
Kim, J., Chang, C. & Tucker, M. L. To grow old: regulatory role of ethylene and jasmonic acid in senescence. Front. Plant Sci. 6, 20 (2015).
Yu, Z. X. et al. The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol. Plant 5, 353–365 (2012).
Lu, X. et al. A a ORA, a trichome‐specific AP 2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol. 198, 1191–1202 (2013).
Tripathi, S., Sangwan, R. S., Mishra, B., Jadaun, J. S. & Sangwan, N. S. Berry transcriptome: insights into a novel resource to understand development dependent secondary metabolism in (Ashwagandha) . Physiol. Plant 168, 148–173 (2019).
Acknowledgements
The authors express sincere thanks to New Millennium Indian Technology Leadership Initiative (NMITLI). NSS is thankful to UGC for financial support. ST is thankful to WOS-A fellowship by DST and AcSIR PhD program for the award of the degree (Enrolment no.10BB13J10007).
Author information
Authors and Affiliations
Contributions
N.S.S. conceptualized and generated the experimental design. S.T. and Y.S. implemented the design through experiments. S.T. and Y.S. wrote the manuscript which was improved by N.S.S. and R.S.S. All the authors were involved in preparation of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tripathi, S., Srivastava, Y., Sangwan, R.S. et al. In silico mining and functional analysis of AP2/ERF gene in Withania somnifera. Sci Rep 10, 4877 (2020). https://doi.org/10.1038/s41598-020-60090-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60090-7
This article is cited by
-
Pharmaceutical, food potential, and molecular data of Hancornia speciosa Gomes: a systematic review
Genetic Resources and Crop Evolution (2022)
-
Protein Elicitor EsxA Induces Resistance to Seedling Blight and PR Genes Differential Transcription in Rice
Rice (2021)
-
WRKY1-mediated regulation of tryptophan decarboxylase in tryptamine generation for withanamide production in Withania somnifera (Ashwagandha)
Plant Cell Reports (2020)
-
Comparative transcriptome analysis to identify putative genes related to trichome development in Ocimum species
Molecular Biology Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
