
Abstract
Antagonism of the angiotensin II type 1 receptor (AT1) improves neurological function and reduces brain damage after experimental traumatic brain injury (TBI), which may be partly a result of enhanced indirect angiotensin II type 2 receptor (AT2) stimulation. AT2 stimulation was demonstrated to be neuroprotective via anti-inflammatory, vasodilatory, and neuroregenerative mechanisms in experimental cerebral pathology models. We recently demonstrated an upregulation of AT2 after TBI suggesting a protective mechanism. The present study investigated the effect of post-traumatic (5 days after TBI) AT2 activation via high and low doses of a selective AT2 agonist, compound 21 (C21), compared to vehicle-treated controls. No differences in the extent of the TBI-induced lesions were found between both doses of C21 and the controls. We then tested AT2-knockdown animals for secondary brain damage after experimental TBI. Lesion volume and neurological outcomes in AT2-deficient mice were similar to those in wild-type control mice at both 24 h and 5 days post-trauma. Thus, in contrast to AT1 antagonism, AT2 modulation does not influence the initial pathophysiological mechanisms of TBI in the first 5 days after the insult, indicating that AT2 plays only a minor role in the early phase following trauma-induced brain damage.
Similar content being viewed by others


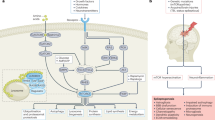
Introduction
Traumatic brain injury (TBI) is the most common cause of trauma-related death and disability in industrialized countries1. Following the primary injury, mechanisms such as cerebral inflammation lead to further (secondary) brain damage2. Increasing evidence indicates that the intrinsic central renin-angiotensin system (RAS) is involved in the pathophysiology of various cerebral pathologies, including TBI3. The RAS effector peptide angiotensin II (AngII) binds predominately to two G protein-coupled transmembrane receptors known as angiotensin II receptor type 1 (AT1) and type 2 (AT2). RAS-mediated vasoconstriction and inflammation are produced by AngII binding to AT14. Recent studies have revealed that blockade of AT1 improved neurological outcomes and reduced secondary brain damage after experimental TBI 5,6,7. Our previous data indicate that there is also a post-traumatic upregulation of AT2 in the first days after TBI in the perilesional tissue 5, which may be related to the initiation of protective repair mechanisms8. Indeed, RAS signaling via AT2 is known to mediate its anti-inflammatory and neuro-regenerative functions9. Upregulated in various cerebral insults10,11,12,13,14,15,16, AT2 activation may counterbalance pathological processes mediated by AT117,18. Because reducing AT1 activity augments AT2 activation 19, the neuroprotective properties of AT1 antagonists may actually be related to enhancement of AT2 function. Iwai et al. reported increased infarct size and neurological impairment in AT2-knockdown mice (AT2−/y) after permanent middle cerebral artery occlusion (MCAO)20. Furthermore, in AT2−/y and under concomitant AT2 antagonism, AT1 blockade was less effective in reducing the infarct size and improving neurological outcomes, suggesting that indirect AT2 stimulation underlies AT1 inhibition-mediated protection17. Signal transduction after AT2 involves the neurotrophic tyrosine kinase receptor type 1 (NTRK1) and the extracellular signal-regulated kinases 1 and 2, as well as protein kinase B and brain-derived neurotrophic factor (BDNF)10,21,22,23.
Long-term treatment of mice with an AT2 agonist peptide was shown to improve anatomical and functional markers of recovery after closed-head TBI24, and post-MCAO treatment of mice with a non-peptide specific and selective AT2 agonist, compound 21 (C21), was found by Schwengel et al. to reduce mortality and neurological impairment25, summarized in26. These results suggest that AT2 activation may be a novel approach to treating TBI. In the present study, we aimed to clarify the significance of AT2 activation in the context of TBI.
To this end, we investigated the role of AT2 in secondary brain damage, cerebral inflammation, and neurologic outcomes after TBI in mice by (1) treating mice with high or low doses of C21 and comparing the results to those in vehicle-treated mice; and (2) analyzing the same outcomes after TBI in AT2-knockdown versus wild-type mice. We also analyzed gene expression of cytokines and neurotrophins to elucidate the putative underlying molecular mechanisms involved.
Materials and methods
Animals
In the first (pharmacology) study, C57Bl6N adult male mice (Charles River Laboratory, Sulzfeld, Germany) were used; in the second (AT2 knockdown) study, AT2−/y (Agtr2tm1a(EUCOMM)Wtsi) and wild-type litter mates (WT; AT2+/y; C57Bl6N background) were investigated. The studies were performed with the approval of the Animal Care and Ethics Committee of Rhineland-Palatinate, Germany in accordance with the institutional guidelines of the Johannes Gutenberg University, Mainz (protocol numbers: 23177–07/G07-1–021 and 23,177–07/G13-1–046) and in compliance with the ARRIVE guidelines. The animals were kept under controlled light and environmental conditions (12-h dark/light cycle, 23 ± 1 °C, 55% ± 5% relative humidity), and had free access to food (Altromin, Germany) and water at all times before and after the experiments.
Experimental traumatic brain injury
The animals were anesthetized with sevoflurane (4% by volume) and an air mixture (40% O2 and 60% N2) via face mask. The rectal temperature was maintained constant at 37 °C by a feedback-controlled heating pad (Hugo Sachs, March-Hugstetten, Germany). Experimental brain trauma was induced by controlled cortical impact (CCI) as described previously 27. Briefly, the animal’s head was fixed in a stereotactic frame (Kopf Instruments, Tujunga, LA, USA) and a large craniotomy (4 × 4 mm2) was performed above the right parietal cortex between the sagittal, lambdoid, and coronal sutures and the insertion of the temporal muscle. A custom-fabricated pneumatic impactor (L. Kopacz, Mainz, Germany) was placed perpendicularly to the brain surface and the impactor tip (diameter, 3 mm) centered in the middle of the craniotomy. The impact parameters were as follows: velocity, 7.5 m/s; duration, 200 ms; brain penetration, 1.5 (study I) and 1.0 mm (study I confirmatory study and study II). Immediately after CCI, the craniotomy was closed with conventional tissue glue (Histoacryl; Braun-Melsungen, Melsungen, Germany) and filament sutures. The animals were randomly assigned to groups, which were followed-up for 15 min (primary injury), 24 h (1 day post injury, 1dpi), or 120 h post-CCI (5 days post injury, 5dpi). Animals in the 15 min group remained anesthetized on the heating pad until their brains were removed; animals in the other groups were placed in their individual cages at the end of surgery and allowed to recover for 6 h in an incubator heated to 33 °C and with 35% humidity (IC8000; Dräger, Germany).
During surgery, the blood pressure was measured 5 min before and after CCI at the tail using a modified NIBP system (RTBP 2000; Kent, USA) as previously described28. Additionally, the blood pressure was determined daily for 8 days before (training phase) and 4 days after CCI. Cuff pressure signals were recorded with a sample rate of 100 Hz (A/D converter: PCI 9112; Adlink Technology, Taiwan; PC software: Dasylab 5.0; measX, Germany) and analyzed offline (Flexpro 6.0, Weisang, Germany) for systolic blood pressure. Perioperative body temperature was measured by a rectal temperature probe (Physitemp; Clifton, NJ, USA).
Experimental groups
Study I: Effect of a 5-day post-CCI treatment with the selective AT2 agonist C21
WT mice were randomized (n = 9/group) to daily intraperitoneal (i.p.) injection with low- (0.03 mg/kg; LD) or high- (0.1 mg/kg; HD) dose C21 or vehicle (saline 0.9%). The injections started 30 min after TBI and were repeated every 24 h until day 4. The cortical cytokine and neurotrophin gene expressions of the CCI groups were compared to those of naïve (non-operated) animals (n = 4). In order to differentiate primary from secondary injury, a group of WT mice (n = 8) was anesthetized, underwent CCI, and then sacrificed 15 min post-TBI, brains were then removed and analyzed for primary injury volume. The protocol of study I was then repeated in a confirmatory study using modified CCI device settings (see above) with eight animals/group.
Study II: TBI in AT2-knockdown mice compared to WT mice
AT2−/y and AT2+/y mice were followed up for either 24 h (n = 12/group) or 5 days after TBI (n = 6/group). Neurological assessment was performed before and after TBI. At the end of the follow-up period, the brains were removed and examined immunohistologically for cerebral lesion volume and activated microglia/macrophages, and cytokine and neurotrophin expressions were quantified via quantitative real-time polymerase chain reaction (qPCR). To differentiate primary from secondary injury, one group of WT mice (n = 6) and one group of AT2-/y mice (n = 5) was anesthetized, underwent CCI, and then sacrificed 15 min post-TBI, at which point the brains were removed and analyzed for primary injury volume.
Neurological assessment
The neurological outcome was determined by neurological severity score (NSS29, in study I), by modified NSS (mNSS7, in study II), and body weight 1 day before and 24, 72, and 120 h after CCI by an investigator blinded toward group allocation.
In study I, to calculate NSS, general behavior, alertness, motor ability, and balance were rated over 10 different tasks. Each task was scored from 0 (normal) to 1 (failed task). One day prior to their surgeries, all animals were tested with the NSS. Healthy animals were required to pass the test with 0 or 1 point to be enrolled in the study.
In study II different and older mice were used. Therefore, a modified NSS (mNSS) was applied to have a more specific differentiation of neurological status7. The neurological outcome was determined by mNSS, modified after Tsenter et al. (2008)29, 1 day before and 24, 72, and 120 h after CCI by an investigator blind toward the group allocation. To calculate mNSS7, general behavior, alertness, motor ability and balance were rated with 10 different tasks. Each task was scored from 0 (normal) up to 3 (failed task). The mNSS ranges from 0 (healthy) to 16 (severely impaired) points7,29,30 (Table 1).
Histologic evaluation of secondary brain damage and immunohistochemistry
For tissue evaluation, the brains were frozen in powdered dry ice after dissection and stored at − 20 °C. They were then cut in the coronal plane with a cryostat (HM 560 Cryo-Star; Thermo Fisher Scientific, Germany). The first slide was defined according to the first section corresponding to bregma + 3.14 mm in the Mouse Brain Library (www.mbl.org). 16 Sects. (10 µm) were collected at 500 µm intervals and placed on Superfrost +™ slides (Thermo Fisher Scientific). In cresyl violet stained sections, total area of both hemispheres and the injured brain tissue area were determined for each section and animal using a computerized image analysis system (Delta Pix Insight; Maalov, Denmark) by an investigator blinded to group allocations. The total hemispheric brain volumes and the lesion volumes were calculated by following formula: 0.5 [mm] × (area of slide 1 [mm2] + area of slide 2 [mm2] + … + area of slide 16 [mm2])5. For immunohistochemical staining of activated microglia/macrophages, cryosections were fixed in paraformaldehyde in phosphate buffered saline, incubated with blocking serum, and then incubated overnight at room temperature with anti-ionized calcium-binding adapter molecule-1 (rabbit anti mouse, anti-Iba-1 antibody; Wako Chemicals, Neuss, Germany, catalogue number: #019–19,741). The sections were washed, incubated with secondary biotin-conjugated antibodies (goat anti-rabbit IgG; Merck; Darmstadt, Germany) and processed according to the manufacturer’s instructions using a Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA, USA). Images were taken at 20 × magnification (Axiovert, Zeiss, Germany). The total number of positive cells ( +) was counted at bregma − 1.28 mm by two investigators blinded to randomization using ImageJ (National Institutes of Health, USA). Iba1-immunolabeled cells with appropriate morphology and appearance31 were identified as activated microglia/macrophages and assessed in a region of interest (ROI) of 0.52 × 0.65 mm2 in the cortical tissue adjacent to the lesion.
Gene expression analysis
Brain tissue samples were excised during histological cryosectioning. Samples from lesion and perilesional tissue (right upper quadrant) and corresponding contralateral cortical tissue (left upper quadrant) were collected, snap frozen in liquid nitrogen, and stored at − 80 °C. qPCR was performed as previously described7,32. Using specific primers7,33,34 (Table 2), absolute copy numbers of BDNF, growth associated protein 43 (GAP43), NTRK1, NTRK2, transforming growth factor beta (TGFβ), interleukin 1β (IL1β), and IL6 were calculated, and were then normalized against the absolute copy numbers of cyclophilin A (PPIA)34.
Data evaluation and statistical analysis
All experiments were randomized and performed by investigators blinded toward the treatment groups (computer-based randomization). In order to determine the required sample size, the a priori power analysis using G ∗ Power35 was performed with the main variable, the primary endpoint, lesion volume data from previously published studies5,36. Therefore, based upon the data of these studies the present a priori power analysis was performed to determine an effect size of d 1.75, with an actual standard statistical power (1 − β) of 0.95, and a significance level (α) of 0.05 and a sample size per group of n = 7. In order to have a sufficient power, we decided to have larger sample sizes (n = 8 – 12, per group). However, due to shortage of AT2 knockout mice, in the 5 days groups, only sample sizes of 6 were possible. Therefore, these results must be carefully considered. Statistical analysis was performed using the GraphPad Prism 8 Statistical Software (GraphPad Software Inc., La Jolla, CA, USA). Data distribution was tested by Shapiro-Wilks test. The comparisons of parametric and non-parametric data between two independent groups were done using the Welch-t test and the Wilcoxon rank sum test, respectively. For the statistical analysis of mNSS we performed ANOVA on ranks with the Kruskal–Wallis test, corrected for multiple comparisons using the Dunn’s test. In this multi-arm parallel group randomized trial, for comparison of multiple independent groups, if the Shapiro–Wilk normality test was passed, one-way analysis of variance (one- way ANOVA) with post-hoc Holm-Šidák comparisons test (comparisons between all groups) was employed. To evaluate group differences in repeated measurements from the same animals (body weight, systolic blood pressure), repeated measures (RM) two-way ANOVA (two-factor repetition) was applied (factors: treatment and time), followed by Šidák’s multiple comparisons test. Whenever there were missing values in the repeated measures dataset and a two-way ANOVA was not possible, repeated measures data were analyzed with the mixed effect model using the restricted maximum likelihood (REML) method with Holm Šidák’s multiple comparison test. Values of p < 0.05 were considered significant. Data are presented as the mean and standard deviation (mean ± SD).
Ethics approval
The studies were performed with the approval of the Animal Care and Ethics Committee of Rhineland-Palatinate, Germany in accordance with the institutional guidelines of the Johannes Gutenberg University, Mainz (protocol numbers: 23177-07/G07-1-021 and 23,177-07/G13-1-046).
Consent for publication
All authors read and approved the final manuscript.
Results
Physiological parameters
Perioperative arterial blood gases were determined 15 min after CCI under general anesthesia in mice (n = 6, respectively) with blood gas analyzer ABL800 Basic (Radiometer Medical ApS, Brønshøj, Denmark) in a parallel investigation, which has been published in 2012 by Timaru-Kast et al.33: pH: 7.32 ± 0.04, paO2: 272 ± 21 mmHg, paCO2: 48 ± 4 mmHg. The data shows that in our standardized anesthesia and operation setting, values are stable and within normal physiological limits33. In the present study in all groups body temperatures during surgery were within physiological levels.
Blood pressure was decreased in high dose C21-treated mice 2 h after CCI
The systolic blood pressure was measured intraoperatively (time point 0), during anesthesia, and postoperatively in awake animals, 2, 6, 24, 48, 72, 96, and 120 h after TBI. While there were no significant changes in the blood pressure in the LD groups, the HD group had significantly lower blood pressure at 2 h after CCI (82 ± 24 mmHg) compared to the vehicle-treated group (116 ± 24 mmHg; p = 0.0353). However, there was no treatment x time interaction for blood pressure and no other differences between the treatment groups were found. The mean systolic blood pressure values were 115 ± 24 mmHg in the vehicle group, 116 ± 28 mmHg in LD, and 107 ± 27 mmHg in the HD group (Fig. 1A).

Effect of AT2 activation with C21 on blood pressure, neurological outcome, lesion volume and cerebral inflammation: Vehicle-treated WT mice (n = 9; white) were compared to WT mice treated with low (0.03 mg/kg; LD C21; n = 9; red), or high dose (0.1 mg/kg; HD C21; n = 9; black) of C21; the data are expressed as mean ± SD; n.s. = not significant. (A) Peritraumatic systolic blood pressure (time point 0 during anesthesia, postoperative in awake animals): while there were no significant changes of blood pressure in the vehicle and LD groups, in the HD group there was a limited drop of blood pressure 2 h after CCI compared to the vehicle-treated group (*p = 0.035). At later time points, the values were within physiological levels, independent of the treatment. (B) NSS: one day (24 h) after TBI in all treatment groups, there was a significant increase in the neurological impairment followed by a significant improvement the following days in all groups. Treatment with C21 in both dosages did not affect neurologic outcome compared to the vehicle. (C) The cerebral lesion volume 5 days after TBI (5 days post-injury, dpi; to rule out effects of edema, data are expressed as % contralateral hemisphere) was elevated compared to the primary lesion (p < 0.05; see results section); however, there was no difference between treatment groups. (D) Five days after TBI (5 dpi), the perilesional gene expression of the cytokine IL1β was not affected by AT2 activation with C21 in both dosages compared to the vehicle treatment.
Neurological outcome and body weight after TBI were not affected by C21 treatment
There was a significant increase in the neurological impairment according to NSS at day 1 post-TBI in all groups, followed by a significant improvement in the following days. However, the neurologic outcomes in the LD and HD groups were not significantly different from those in the vehicle-treated mice (Fig. 1B). Furthermore, the body weight development was similar in all groups. After a decrease at day 1 after CCI body weight increased significantly throughout the observation period (19.7 ± 1.8, 19.7 ± 1.5 and 20.1 ± 1.8 g vs. 20.1 ± 0.7*, 20.7 ± 1.7* and 21.0 ± 1.3* g for vehicle, low and high dose C21 treated mice at day 1 versus day 5, *p < 0.05; Table 3). Increase of body weight was earlier significant compared to day 1 in C21 treated mice (at day 4). However, both dosages C21 did not affect body weight compared to vehicle treated mice at any time point (Table 3).
In a confirmatory investigation in another set of mice with the same treatment regime, but with reduced CCI settings and consecutively 45% smaller lesion volumes, NSS at days 1, 3 and 5 after TBI was without difference between C21-treated groups and vehicle-treated mice (NSS: vehicle: 4 ± 3, 3 ± 2, 1 ± 2; LD: 4 ± 2, 4 ± 2, 2 ± 2; HD: 3 ± 2, 2 ± 2, 1 ± 1 points, respectively).
Lesion volume was not affected by C21 treatment
The primary lesion volume was determined 15 min after CCI (9.2% ± 1.2% contralateral hemisphere). Five days after CCI, the lesion volume was significantly enhanced compared to the primary lesion volume (p < 0.05); however, there was no difference between groups (15.9% ± 4.1%, 15.7% ± 2.0%, and 16.7% ± 2.9% contralateral hemisphere volume for vehicle, LD, and HD, respectively; Fig. 1C). As was found for the initial study, the confirmatory study with altered CCI device settings also demonstrated no significant between-group changes (9.9% ± 1.9%, 9.4% ± 1.9%, and 8.9% ± 1.9% contralateral hemisphere volume for vehicle, LD, and HD, respectively).
Gene expressions of neurotrophin receptors were upregulated, while expressions of neurotrophins and cytokines were unaffected by C21 treatment
Five days after TBI, the perilesional gene expression of the cytokine IL1β, neurotrophin receptors NTRK1 and NTRK2, and neurotrophins GAP43 and BDNF, as assessed by qPCR, were elevated compared to naïve mice (p < 0.05). IL1β was not affected by C21 treatment (Fig. 1D). A dose-dependent upregulation of the neurotrophin receptors NTRK1 (in the HD group only; p = 0.048) and NTRK2 (in both LD and HD groups; p = 0.029 and p = 0.035; Fig. 2A, B) was found. However, gene expression of the neurotrophins GAP43 and BDNF was not significantly different between the vehicle and LD- or HD-group mice. (Fig. 2C, D).

Effect of AT2 activation with C21 on neurotrophin receptor and effector expression: Vehicle-treated WT mice (n = 9; white) were compared to WT mice treated with low (0.03 mg/kg; LD C21; n = 9; red), or high dose (0.1 mg/kg; HD C21; n = 9; black) of C21 at 5 days post-injury (5dpi); the data are expressed as mean ± SD; n.s. = not significant. (A), (B) There were dose-dependent elevated gene expressions of the neurotrophin receptors NTRK1 in HD C21 and of NTRK2 in LD and HD C21 compared to those in the vehicle-treated mice (p < 0.05). (C), (D) The gene expressions of the neurotrophins GAP43 and BDNF were not regulated differently between the vehicle- and C21-treated mice.
AT2 knockdown did not create significant differences from WT mice
The systolic blood pressure was within physiological levels and not significantly different between the AT2-knockdown and WT mice (Fig. 3A).

AT2 deficiency does not affect the blood pressure, neurological outcome and the lesion volume: AT2-deficient mice (AT2−/y; 24 h: n = 12; 5 days: n = 6; blue) were compared to age-matched WT mice (AT2+/y; 24 h: n = 12; 5 days: n = 6; white). The values are presented in mean ± SD; n.s. = not significant. (A) Peritraumatic systolic blood pressure: time point 0 represents the intraoperative measurement immediately after CCI under general anesthesia, and the following postoperative time points measurements were performed in awake animals. The values were within the physiological norms and did not differ between the AT2−/y and AT2+/y animals. (B) The mNSS after CCI was not significantly different between the AT2−/y and AT2+/y animals. (C), (D) Cerebral lesion volume (to rule out effects of edema, data are expressed as % contralateral hemisphere): primary lesion was assessed 15 min after TBI, without difference between AT2+/y and AT2−/y animals. C: The marked secondary increase of lesion volume 24 after TBI (1 day post-injury, 1dpi; p < 0.001) was not different in AT2+/y mice compared to AT2−/y mice. (D): Five days after TBI (5dpi), there was no difference in the lesion volumes between the AT2+/y and AT2−/y mice.
In study II, the neurological status was determined with mNSS. The mNSS scores were not significantly different between the AT2+/y and AT2−/y groups; however, there was a significant increase of neurological impairment 24 h after CCI, followed by a gradual improvement in the following days in all groups (Fig. 3B). Age matched AT2 deficient and wild type litter mate mice were used. Their body weight decreased after CCI continuously from before TBI during 5 days after TBI in both groups. However, AT2 deficiency had no impact on body weight loss after brain trauma (37.8 ± 6.7 vs. 41.2 ± 5.7, 36.3 ± 7.2* vs. 39.3 ± 5.6*, 35.1 ± 6.5*# vs. 37.8 ± 5.5*# and 34.7 ± 7.0*# vs. 37.5 ± 5.1*# g in AT2+/y vs. AT2-/y at time points before CCI, 1 day, 3 days and 5 days after CCI, respectively; *p < 0.05 vs. before TBI, #p < 0.05 vs. day 1 after CCI). The primary injury volumes of AT2−/y (9.0% ± 1.5% contralateral hemisphere) and AT2+/y (12.2 ± 3.2% contralateral hemisphere) were not significantly different (15 min after CCI). Thus, the primary injury volumes of both groups are presented together. There was a significant increase in the lesion volume 24 h after TBI due to secondary damage compared to the primary lesion volume measured 15 min after TBI (primary injury volume, 10.7% ± 3.0% contralateral hemisphere; secondary injury volume, 26.6% ± 5.1% contralateral hemisphere (WT); secondary injury volume, 27.8% ± 3.1% contralateral hemisphere (AT2−/y)). Due to scar tissue formation, compared to 24 h post-CCI, the lesion volumes at day 5 were reduced in AT2+/y and AT2−/y animals (10.4% ± 2.1% and 11.2% ± 1.7% contralateral hemisphere for AT2+/y and AT2−/y, respectively). Neither post-CCI time point demonstrated between-group differences (Fig. 3C, D).
We assessed the number of perilesional microglia/macrophages as a proxy of microglial activation using anti-Iba-1 immunohistochemistry 5 days after CCI in a perilesional ROI (0.52 × 0.65 mm2). The rationale for measuring IBA-1 in an area adjacent to the lesion rather than within the lesioned area was, that inside the lesion, where the tissue is essentially destroyed, microglia/macrophages are almost absent. In the perilesional area, however, there is a robust activation of microglia/macrophages. The numbers of perilesional Iba-1-immunolabelled microglia/macrophages were substantially increased as compared to corresponding sites in the non-injured contralateral hemisphere. However, in the perilesional and in the corresponding contralateral region they were similar between the AT2+/y and AT2−/y groups (perilesional: 183 ± 40** and 198 ± 41** n/ROI, contralateral: 78 ± 7 and 69 ± 7 n/ROI, **p < 0.001 compared to contralateral corresponding region, for AT2+/y and AT2−/y, respectively; Fig. 4A). In addition, the gene expressions IL1β and IL6 (Fig. 4B,C) were not significantly different between AT2+/y and AT2−/y 5 days after TBI. No other measured genes (TGFβ, GAP43, and BDNF; Fig. 4D) demonstrated significant differences between the knockdown and WT groups. Finally, no differences in NTRK1/2 expressions between groups were found.

AT2 deficiency does not affect the neuroinflammation and neurotrophins: AT2-deficient mice (AT2−/y; n = 6; blue) were compared to age-matched WT mice (AT2+/y; n = 6; white) 5 days after TBI (5 dpi). Values are presented in mean ± SD; n.s. = not significant. (A) The number of Iba-1-immunolabelled activated microglia/macrophages 5 days after CCI in the perilesional region of interest (0.52 × 0.65 mm2) was not different between the AT2+/y and AT2−/y groups. (B), (C), (D) The perilesional gene expressions of the cytokines IL1β and IL6 and of the neurotrophin BDNF, determined by qPCR, were not different between the AT2+/y and AT2−/y mice 5 days after TBI (5 dpi).
Discussion
Several studies demonstrated sustained protective effects by blockade of AT1 after TBI5,37, and a neuroprotective role for AT2 in ischemic stroke17,25,38, spinal cord injury10 and nerve regeneration 15. In spite of pathophysiological similarities between brain ischemia and TBI39, the therapeutic potential of the non-peptide selective AT2 agonist C21 after TBI has only been the subject of few studies to date. However, despite the seeming potential for success, in our murine CCI model of TBI, we found that direct AT2 stimulation by repeated application of C21 at high and low dosages did not produce any significant neuroprotection up to 5 days after TBI. Therefore, as proof of concept we investigated the effect of AT2 knockdown on our model system, and again found no differences. The present data surprisingly contradicted our foregoing assumption that AT2 stimulation exerts neuroprotection after TBI. Indeed, there are striking differences between our protocols and TBI models that showed protection by AT2 stimulation. Each of the possible TBI models with inherent advantages and disadvantages, need to be thoroughly considered according to the hypothesis and design of the study when selecting the rodent TBI model40. In a closed-head injury study, Umschweif and colleagues demonstrated reduction of lesion size and improvement in functional recovery by treatment with the peptide AT2 agonist CGP42112A24. The closed-head injury model is designed to replicate the biomechanics and pathobiology of concussive brain injury41,42. The impact of the standardized weight-drop device with dynamic pressure to the closed skull and acceleration and deceleration trauma results predominantly in diffuse brain injury41,43,44,45,46. On the other hand, CCI is a very common model that simulates focal TBI with cortical contusion, brain edema, elevated intracranial pressure, and decreased cerebral blood flow41,47. The murine CCI model offers a very accurate control of the mechanical components of trauma. By exact adjustment of the impact velocity and depth, it is possible to generate a defined, focal cortical lesion with high reproducibility41,48. To rule out the possibility of a too large traumatic brain damage where putative protective effects of AT2 activation may be diminished, we repeated the present study in a confirmatory study with reduced brain penetration of the impactor tip and, apart from that, the same CCI settings and treatment regime. This change of CCI settings lead to a reduced brain damage. However, AT2 activation with C21 in high and low dosages had still no effect on lesion size, compared to vehicle treated mice. Despite equal CCI settings, lesion volumes 5 days after TBI were smaller than 24 h after TBI, due to reorganization of astrocytes, phagocytosis of necrotic and apoptotic cells and debris49,50. Besides expansion of lesion volume, CCI lead to a robust activation of microglia, with increased number of perilesional microglia as compared to corresponding non-injured contralateral regions after CCI51,52, and compared to corresponding brain regions in sham mice53. Besides different TBI models, in contrast to Umschweif et al., the focus of the present study was the early time phase after TBI, and motor impairment and behavior rather than cognitive outcomes by using the NSS29.
One major difference to other related studies was our use of C21. The reason we chose the non-peptide AT2 agonist C21 was, that the peptide alternative CGP42112A has unfavorable pharmacokinetic properties such as very rapid degradation. It has to be applied continuously 24, and therefore in the past it never has been considered to be developed for clinical use54. To model a clinical treatment as possible therapeutic opportunity of AT2 activation after TBI was one aim of the present study14,54,55,56. Therefore, we used C21, the non-peptide AT2 agonist with a more favorable pharmacokinetic profile, as repetitive post-TBI treatment (starting 30 min after CCI) with the potential to be developed in further studies to a possible novel pharmacological concept10. The AT2-specificity of C21 has been proven in several studies10,25,55,57. The decision to choose the two doses of C21 was made, based upon dosages that have shown to be protective in ischemic brain damage, in spinal cord and cardiac injury. The dosage of 0.03 mg/kg/day C21 has been demonstrated to be cardioprotective and to decrease systemic inflammation after left coronary artery occlusion58, higher doses (0.3 mg/kg/day) C21 did not increase cardio-protection58. In a recent rodent brain ischemia study (1.5–3 h after MCAO), it was shown that i.p. treatment with C21 (0.03 mg/kg) at reperfusion downregulated apoptotic and oxidative markers, increased neurotrophin activity, reduced inflammation and infarct size and improved behavioral outcome at 24 h without affecting the blood pressure59. In the present study, our dosage of C21 was chosen based on the murine study of Schwengel et al.25, wherein the effect of post-stroke C21 treatment after MCAO was assessed in AT2−/y and AT2+/y mice. The study demonstrated that 0.03 mg/kg/day C21 significantly improved the survival and neurologic outcomes after MCAO compared to the vehicle treatment, although it had no impact on infarct size 96 h post-stroke in AT2+/y mice25. Expression of BDNF and its receptor NTRK2 and GAP43 were significantly increased in the peri-infarct cortex of C21-treated, compared to vehicle-treated WT mice25. Therefore, based upon these data, we decided to use two doses (0.03 and 0.1 mg/kg/day) to reduce the possibility of under-dosage and investigate the dose–response relationship.
Passage through the blood brain barrier (BBB) and availability of C21 in the cerebral tissue is substantial for the effectivity of C21. Indeed, the lack of measuring the effect site concentration of C21 in the brain tissue after TBI, is a limitation of the present study. In naïve rodents, after i.p.-injection, recent concentration analyses showed only minimal passage of C21 through the intact BBB60,61. After ischemic stroke, however, 30 min following i.p.-injection of 0.03 mg/kg, levels of C21 within the brain cortex and striatum were significant61. Several studies have shown a peak in permeability in the acute and subacute phase followed by a gradual reduction of BBB permeability after ischemic stroke62. Rodent models of TBI have demonstrated a biphasic increase in the BBB permeability to albumin and other high- molecular-weight proteins peaking at 4–6 h and 2–3 days after injury63,64. In our present model, there is a distinct BBB disruption with high permeability peaking at 6–24 h after CCI, as it has been shown earlier by Luh and coworkers32. In human TBI, BBB disturbances can be detected from several days up to years after the trauma65. On the basis of these findings we postulate, that due to post-traumatic increased permeability of the disturbed BBB and repetitive treatment, in the present study there was an adequately high effect site concentration of C21 for receptor stimulation of AT2 in the injured brain tissue55.
In a recent preliminary murine CCI-study with C21 (0.03 mg/kg, i.p.; compared to saline) treatment at 1 h and 3 h post-TBI66, NSS was improved and inflammatory markers, like IL1β reduced at 24 h post-TBI in the pericontusional areas. However, lesion volume was not measured in this preliminary study. In a recent study we demonstrated that IL1β expression was increased in the first 6 h after TBI 5. However, C21 did not affect IL1β expression 5 days after TBI herein.
Moreover, in the present study, the primary endpoint lesion volume was not affected by treatment with C21. This is in accordance with recent studies with spinal cord injury10 and brain ischemia. Schwengel and coworkers noted reduced mortality and neurological impairment by post-stroke treatment with C21 without effect on the infarct size25. Therefore, AT2 activation with C21 may have no effect on the secondary brain damage expansion. Lee and colleagues showed in a cerebral ischemia model (MCAO) in mice the neuroprotective effects of the peptide AT2 agonist CGP42112 with reduced infarct volumes, while treatment with the non-peptide C21 failed to show protective properties and had no effect on the neuronal survival67 in an in vitro glucose deprivation model. The authors found that unlike the peptide AT2 agonist CGP42112, there was no protective effect by a similar range of concentrations of C2168. However, the focus of the present study was to investigate a possible in vivo effect of AT2 activation by C21 after TBI in mice with dosages that have been shown to be protective in earlier rodent models25,43,59. We do not believe under-dosage was a problem in the present study despite the relative lack of previous data to draw on. Possible proof of activity of C21 was found in the elevated expressions of the neurotrophin receptors NTRK1 and NTRK2 and in blood pressure reduction 2 h after TBI in the HD compared to vehicle group. However, blood pressure was not altered at most of the time points. This is in line with several studies that showed no or only slight changes in blood pressure with C21 therapy69,70.
Thus, the different TBI model (closed head TBI versus CCI) and the different AT2 agonists (peptide vs. C21) could have led to the different results. Several issues arose by our negative results. Limitations by the substance or by the application protocol may have contributed to the failure to detect a positive effect on the outcome measures in the model. Pre-treatment with C21 may have had different results. Therefore, after performing the experiments with C21, to rule out underdosage or wrong timing of application, we decided to analyze AT2 knockout mice without additional treatment, as proof-of-concept. We investigated the effect of AT2 deficiency 24 h and 5 days after CCI, however, without differences in brain damage, neuroinflammation and neurology.
A possible reason for our negative results, especially those in the AT2-knockdown mice, is the possibility of a non-protective, proinflammatory role of AT2. Ruiz-Ortega and coworkers demonstrated that AngII increased the proinflammatory Nuclear factor-kappaB (NF-κB) activation via AT1 and AT2 in rat smooth muscle cells71,72. Although NF-κB-mediated transcription occurred mainly through AT1, AT2 activation enhanced NF-κB-DNA-binding and NF-κB-mediated transcription of cytokines71. Furthermore, in AT1-knockdown mice, an increase of NF-κB could be demonstrated by AngII stimulation. Blockade of AT2, however, led to a drop of NF-κB in these mice71,72,73,74. Additionally, Wolf investigated the AngII receptor subtypes involved in NF-κB activation, demonstrating in various cell lines that Ang II induces NF-κB activation through AT2 receptor stimulation75. Sabuhi tested the effects of the AT2 activation with CGP42112A on inflammation and oxidative stress in obese Zucker rats and their lean counterparts. While AT2 activation reduced inflammatory and oxidative stress markers in obese rats, it increased such markers in lean rats. These results suggest that AT2 receptors may have pro- or anti-inflammatory effects according to weight or weight-related factors76. Furthermore, previous studies indicate that neuroprotection by AT2 activation is predominantly mediated by concomitant AT1 antagonism17,67. However, aim of the present study was to investigate the role of AT2, examine the effect of AT2 activation by C21 in the acute phase after TBI. However, the question if AT2 activation is neuroprotective with concomitant AT1 inhibition in TBI pathophysiology has to be addressed in future studies.
In summary, in contrast to studies of AT1 inhibition 5, a selective AT2 activation with C21 did not reduce brain damage in the CCI model. Therefore, the present results suggest that neuroprotective properties of AT1 inhibition in the early phase after TBI are not due to enhanced activation of AT2 by AngII. However, the neuroprotective properties of AT2 that contribute to reparative processes may be present in later phases after TBI.
Conclusion
Several recent studies have shown that inhibition of AT1 improved neurological outcome and reduced secondary brain damage after experimental TBI. There is growing evidence that AT2-mediated protective mechanisms may contribute to the protective potency of AT1 inhibition. In the present study, the effect of AT2 activation on neurologic outcome, cerebral inflammation, and lesion volume was assessed for the first time after CCI. The present data could not confirm a protective role of AT2 activation, nor did AT2 deficiency affect brain damage within 5 days after TBI. This suggests that AT2 may play a minor role in the early phase after TBI. The present results further suggest that neuroprotective properties of AT1 inhibition are not due to enhanced activation of AT2 by AngII within the first five days, in the early phase after TBI. Further investigations are required to assess later effects of AT2 activation by C21 after TBI.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Langlois, J. A., Rutland-Brown, W. & Wald, M. M. The epidemiology and impact of traumatic brain injury: A brief overview. J. Head Trauma Rehabil. 21, 375–378 (2006).
Morganti-Kossmann, M. C., Rancan, M., Otto, V. I., Stahel, P. F. & Kossmann, T. Role of cerebral inflammation after traumatic brain injury: A revisited concept. Shock 16, 165–177 (2001).
Saavedra, J. M. Brain angiotensin II: new developments, unanswered questions and therapeutic opportunities. Cell Mol. Neurobiol. 25, 485–512 (2005).
Saavedra, J. M. Angiotensin II AT(1) receptor blockers ameliorate inflammatory stress: A beneficial effect for the treatment of brain disorders. Cell. Mol. Neurobiol. 32, 667–681. https://doi.org/10.1007/s10571-011-9754-6 (2012).
Timaru-Kast, R. et al. Delayed inhibition of angiotensin II receptor type 1 reduces secondary brain damage and improves functional recovery after experimental brain trauma*. Crit. Care Med. 40, 935–944. https://doi.org/10.1097/CCM.0b013e31822f08b9 (2012).
Villapol, S., Balarezo, M. G., Affram, K., Saavedra, J. M. & Symes, A. J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain J. Neurol. https://doi.org/10.1093/brain/awv172 (2015).
Timaru-Kast, R. et al. Angiotensin II receptor 1 blockage limits brain damage and improves functional outcome after brain injury in aged animals despite age-dependent reduction in AT1 expression. Front. Aging Neurosci. 11, 18. https://doi.org/10.3389/fnagi.2019.00063 (2019).
Loane, D. J. & Faden, A. I. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 31, 596–604. https://doi.org/10.1016/j.tips.2010.09.005 (2010).
Sumners, C. et al. Protective arms of the renin-angiotensin-system in neurological disease. Clin. Exp. Pharmacol. Physiol. 40, 580–588. https://doi.org/10.1111/1440-1681.12137 (2013).
Namsolleck, P. et al. AT2-receptor stimulation enhances axonal plasticity after spinal cord injury by upregulating BDNF expression. Neurobiol. Dis. 51, 177–191. https://doi.org/10.1016/j.nbd.2012.11.008 (2013).
Bottari, S. P., de Gasparo, M., Steckelings, U. M. & Levens, N. R. Angiotensin II receptor subtypes: Characterization, signalling mechanisms, and possible physiological implications. Front. Neuroendocrinol. 14, 123–171. https://doi.org/10.1006/frne.1993.1005 (1993).
Wruck, C. J. et al. Regulation of transport of the angiotensin AT2 receptor by a novel membrane-associated Golgi protein. Arterioscler. Thromb. Vasc. Biol. 25, 57–64. https://doi.org/10.1161/01.ATV.0000150662.51436.14 (2005).
Laflamme, L., Gasparo, M., Gallo, J. M., Payet, M. D. & Gallo-Payet, N. Angiotensin II induction of neurite outgrowth by AT2 receptors in NG108–15 cells. Effect counteracted by the AT1 receptors.J. boil. chem. 271, 22729–22735 (1996).
Gendron, L., Payet, M. D. & Gallo-Payet, N. The angiotensin type 2 receptor of angiotensin II and neuronal differentiation: From observations to mechanisms. J. Mol. Endocrinol. 31, 359–372 (2003).
Reinecke, K. et al. Angiotensin II accelerates functional recovery in the rat sciatic nerve in vivo: Role of the AT2 receptor and the transcription factor NF-kappaB. FASEB J. Off. Publ. Federation Am. Soc. Exp. Biol. 17, 2094–2096. https://doi.org/10.1096/fj.02-1193fje (2003).
Gendron, L., Oligny, J. F., Payet, M. D. & Gallo-Payet, N. Cyclic AMP-independent involvement of Rap1/B-Raf in the angiotensin II AT2 receptor signaling pathway in NG108-15 cells. J. Biol. Chem. 278, 3606–3614. https://doi.org/10.1074/jbc.M202446200 (2003).
Li, J. et al. Angiotensin AT2 receptor protects against cerebral ischemia-induced neuronal injury. FASEB J. 19, 617–619 (2005).
Horiuchi, M., Akishita, M. & Dzau, V. J. Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension 33, 613–621 (1999).
Saavedra, J. M., Benicky, J. & Zhou, J. Mechanisms of the anti-ischemic effect of angiotensin II AT(1) receptor antagonists in the brain. Cell Mol. Neurobiol. 26, 1099–1111 (2006).
Iwai, M. et al. Possible inhibition of focal cerebral ischemia by angiotensin II type 2 receptor stimulation. Circulation 110, 843–848 (2004).
Porrello, E. R., Delbridge, L. M. & Thomas, W. G. The angiotensin II type 2 (AT2) receptor: An enigmatic seven transmembrane receptor. Front Biosci. (Landmark Ed) 14, 958–972 (2009).
Plouffe, B., Guimond, M. O., Beaudry, H. & Gallo-Payet, N. Role of tyrosine kinase receptors in angiotensin II AT2 receptor signaling: Involvement in neurite outgrowth and in p42/p44mapk activation in NG108-15 cells. Endocrinology 147, 4646–4654. https://doi.org/10.1210/en.2005-1315 (2006).
Hashikawa-Hobara, N. et al. Candesartan cilexetil improves angiotensin II type 2 receptor-mediated neurite outgrowth via the PI3K-Akt pathway in fructose-induced insulin-resistant rats. Diabetes 61, 925–932. https://doi.org/10.2337/db11-1468 (2012).
Umschweif, G. et al. Angiotensin receptor type 2 activation induces neuroprotection and neurogenesis after traumatic brain injury. Neurotherapeutics J. Am. Soc. Exp. NeuroTherapeutics 11, 665–678. https://doi.org/10.1007/s13311-014-0286-x (2014).
Schwengel, K. et al. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J. Mol. Med. (Berl.) 94, 957–966. https://doi.org/10.1007/s00109-016-1406-3 (2016).
Bennion, D. M., Steckelings, U. M. & Sumners, C. Neuroprotection via AT2 receptor agonists in ischemic stroke. Clin. Sci. (Lond.) 132, 1055–1067. https://doi.org/10.1042/CS20171549 (2018).
Griemert, E. V. et al. Plasminogen activator inhibitor-1 augments damage by impairing fibrinolysis after traumatic brain injury. Ann. Neurol. 85, 667–680. https://doi.org/10.1002/ana.25458 (2019).
Thal, S. C. & Plesnila, N. Non-invasive intraoperative monitoring of blood pressure and arterial pCO(2) during surgical anesthesia in mice. J.Neurosci.Methods 159, 261–267 (2007).
Tsenter, J. et al. Dynamic changes in the recovery after traumatic brain injury in mice: Effect of injury severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J. Neurotrauma 25, 324–333 (2008).
Thal, S. C. et al. Inhibition of proteasomal glucocorticoid receptor degradation restores dexamethasone-mediated stabilization of the blood-brain barrier after traumatic brain injury. Crit. Care Med. 41, 1305–1315. https://doi.org/10.1097/CCM.0b013e31827ca494 (2013).
Donat, C. K., Scott, G., Gentleman, S. M. & Sastre, M. Microglial activation in traumatic brain injury. Front Aging Neurosci. 9, 208. https://doi.org/10.3389/fnagi.2017.00208 (2017).
Luh, C. et al. Inhibition of myosin light chain kinase reduces brain edema formation after traumatic brain injury. J. Neurochem. 112, 1015–1025. https://doi.org/10.1111/j.1471-4159.2009.06514.x (2010).
Timaru-Kast, R. et al. Influence of age on brain edema formation, secondary brain damage and inflammatory response after brain trauma in mice. PLoS ONE 7, e43829. https://doi.org/10.1371/journal.pone.0043829 (2012).
Timaru-Kast, R., Herbig, E. L., Luh, C., Engelhard, K. & Thal, S. C. Influence of age on cerebral housekeeping gene expression for normalization of quantitative PCR after acute brain injury in mice. J. Neurotrauma https://doi.org/10.1089/neu.2014.3784 (2015).
Faul, F., Erdfelder, E., Buchner, A. & Lang, A. G. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav. Res. Methods 41, 1149-1160, doi:https://doi.org/10.3758/BRM.41.4.1149 (2009)
Timaru-Kast, R. et al. Angiotensin II receptor 1 blockage limits brain damage and improves functional outcome after brain injury in aged animals despite age-dependent reduction in AT1 expression. Front Aging Neurosci. 11, 63. https://doi.org/10.3389/fnagi.2019.00063 (2019).
Villapol, S. et al. Candesartan, an angiotensin II AT(1)-receptor blocker and PPAR-gamma agonist, reduces lesion volume and improves motor and memory function after traumatic brain injury in mice. Neuropsychopharmacol. Off. Publ. Am. College Neuropsychopharmacol. 37, 2817–2829. https://doi.org/10.1038/npp.2012.152 (2012).
McCarthy, C. A. et al. Angiotensin II type 2 receptor stimulation initiated after stroke causes neuroprotection in conscious rats. Hypertension 60, 1531–1537. https://doi.org/10.1161/HYPERTENSIONAHA.112.199646 (2012).
Werner, C. & Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 99, 4–9. https://doi.org/10.1093/bja/aem131 (2007).
Marklund, N. Rodent models of traumatic brain injury: Methods and challenges. Methods Mol. Biol. 1462, 29–46. https://doi.org/10.1007/978-1-4939-3816-2_3 (2016).
Cernak, I. Animal models of head trauma. NeuroRx 2, 410–422. https://doi.org/10.1602/neurorx.2.3.410 (2005).
Umschweif, G. et al. Neuroprotection after traumatic brain injury in heat-acclimated mice involves induced neurogenesis and activation of angiotensin receptor type 2 signaling. J. Cereb. Blood flow Metabolism Off. J. Int. Soc. Cereb. Blood Flow Metabolism 34, 1381–1390. https://doi.org/10.1038/jcbfm.2014.93 (2014).
Flierl, M. A. et al. Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 4, 1328–1337. https://doi.org/10.1038/nprot.2009.148 (2009).
Chen, Y., Constantini, S., Trembovler, V., Weinstock, M. & Shohami, E. An experimental model of closed head injury in mice: Pathophysiology, histopathology, and cognitive deficits. J. Neurotrauma 13, 557–568. https://doi.org/10.1089/neu.1996.13.557 (1996).
Berkner, J., Mannix, R. & Qiu, J. Clinical traumatic brain injury in the preclinical setting. Methods Mol. Biol. 1462, 11–28. https://doi.org/10.1007/978-1-4939-3816-2_2 (2016).
Cernak, I., Stoica, B., Byrnes, K. R., Di, G. S. & Faden, A. I. Role of the cell cycle in the pathobiology of central nervous system trauma. Cell Cycle 4, 1286–1293 (2005).
Smith, D. H. et al. A model of parasagittal controlled cortical impact in the mouse: Cognitive and histopathologic effects. J. Neurotrauma 12, 169–178. https://doi.org/10.1089/neu.1995.12.169 (1995).
Faden, A. I. et al. Neuroprotective and nootropic actions of a novel cyclized dipeptide after controlled cortical impact injury in mice. J. Cereb. Blood flow Metabolism Off. J. Int. Soc. Cereb. Blood Flow Metabolism 23, 355–363. https://doi.org/10.1097/01.WCB.0000046144.31247.33 (2003).
Chen, Y. & Swanson, R. A. Astrocytes and brain injury. J. Cereb. Blood flow Metabolism Off. J. Int. Soc. Cereb. Blood Flow Metabolism 23, 137–149. https://doi.org/10.1097/01.WCB.0000044631.80210.3C (2003).
Fitch, M. T., Doller, C., Combs, C. K., Landreth, G. E. & Silver, J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: In vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J. Neurosci. Off. J. Soc. Neurosci. 19, 8182–8198 (1999).
Kramer, T. J. et al. Depletion of regulatory T cells increases T cell brain infiltration, reactive astrogliosis, and interferon-gamma gene expression in acute experimental traumatic brain injury. J. Neuroinflammation 16, 163. https://doi.org/10.1186/s12974-019-1550-0 (2019).
Kramer, T. J. et al. Correction to: Depletion of regulatory T cells increases T cell brain infiltration, reactive astrogliosis, and interferon-gamma gene expression in acute experimental traumatic brain injury. J. Neuroinflammation 16, 176. https://doi.org/10.1186/s12974-019-1577-2 (2019).
Menzel, L. et al. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury. Glia 65, 278–292. https://doi.org/10.1002/glia.23091 (2017).
Steckelings, U. M. et al. The past, present and future of angiotensin II type 2 receptor stimulation. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 11, 67–73, https://doi.org/10.1177/1470320309347791 (2010).
Steckelings, U. M. et al. Non-peptide AT2-receptor agonists. Curr. Opin. Pharmacol. 11, 187–192. https://doi.org/10.1016/j.coph.2010.11.002 (2011).
Wan, Y. et al. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J. Med. Chem. 47, 5995–6008. https://doi.org/10.1021/jm049715t (2004).
Namsolleck, P., Recarti, C., Foulquier, S., Steckelings, U. M. & Unger, T. AT(2) receptor and tissue injury: Therapeutic implications. Curr. Hypertens. Rep. 16, 416. https://doi.org/10.1007/s11906-013-0416-6 (2014).
Kaschina, E. et al. Angiotensin II type 2 receptor stimulation: A novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction?. Circulation 118, 2523–2532. https://doi.org/10.1161/CIRCULATIONAHA.108.784868 (2008).
Alhusban, A. et al. Compound 21 is pro-angiogenic in the brain and results in sustained recovery after ischemic stroke. J. Hypertens. 33, 170–180. https://doi.org/10.1097/HJH.0000000000000364 (2015).
Shraim, N. et al. Microbore liquid chromatography with UV detection to study the in vivo passage of compound 21, a non-peptidergic AT(2) receptor agonist, to the striatum in rats. J. Neurosci. Meth. 202, 137–142. https://doi.org/10.1016/j.jneumeth.2011.06.009 (2011).
Bennion, D. M. et al. Protective effects of the angiotensin II AT2 receptor agonist compound 21 in ischemic stroke: A nose-to-brain delivery approach. Clin. Sci. (Lond.) 132, 581–593. https://doi.org/10.1042/CS20180100 (2018).
Moisan, A. et al. Microvascular plasticity after experimental stroke: A molecular and MRI study. Cerebrovasc Dis 38, 344–353. https://doi.org/10.1159/000368597 (2014).
Baskaya, M. K., Rao, A. M., Dogan, A., Donaldson, D. & Dempsey, R. J. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 226, 33–36. https://doi.org/10.1016/s0304-3940(97)00239-5 (1997).
Baldwin, B. A. & Sukhchai, S. Intracerebroventricular injection of CCK reduces operant sugar intake in pigs. Physiol. Behav. 60, 231–233. https://doi.org/10.1016/0031-9384(95)02211-2 (1996).
Tomkins, O. et al. Blood-brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry 79, 774–777. https://doi.org/10.1136/jnnp.2007.126425 (2008).
Ismael, S. & Ishrat, T. Compound 21, a direct AT2R agonist, induces IL-10 and inhibits inflammation in mice following traumatic brain injury. Neuromolecular Med. https://doi.org/10.1007/s12017-021-08687-7 (2021).
Lee, S. et al. Neuroprotective effect of an angiotensin receptor type 2 agonist following cerebral ischemia in vitro and in vivo. Exp. Transl. Stroke Med. 4, 16. https://doi.org/10.1186/2040-7378-4-16 (2012).
Bosnyak, S. et al. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin. Sci. (Lond.) 121, 297–303. https://doi.org/10.1042/CS20110036 (2011).
Bosnyak, S. et al. Stimulation of angiotensin AT2 receptors by the non-peptide agonist, compound 21, evokes vasodepressor effects in conscious spontaneously hypertensive rats. Br. J. Pharmacol. 159, 709–716. https://doi.org/10.1111/j.1476-5381.2009.00575.x (2010).
Hilliard, L. M., Mirabito, K. M. & Denton, K. M. Unmasking the potential of the angiotensin AT2 receptor as a therapeutic target in hypertension in men and women: What we know and what we still need to find out. Clin. Exp. Pharmacol. Physiol. 40, 542–550. https://doi.org/10.1111/1440-1681.12067 (2013).
Ruiz-Ortega, M. et al. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: Molecular mechanisms. Circ. Res. 86, 1266–1272 (2000).
Ruiz-Ortega, M., Lorenzo, O., Ruperez, M., Suzuki, Y. & Egido, J. Angiotensin II activates nuclear transcription factor-kappaB in aorta of normal rats and in vascular smooth muscle cells of AT1 knockout mice. Nephrol. Dial. Transplant. 16(Suppl 1), 27–33 (2001).
Suzuki, Y. et al. Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 35, 881–900 (2003).
Ruiz-Ortega, M., Lorenzo, O., Ruperez, M., Blanco, J. & Egido, J. Systemic infusion of angiotensin II into normal rats activates nuclear factor-kappaB and AP-1 in the kidney: Role of AT(1) and AT(2) receptors. Am. J. Pathol. 158, 1743–1756 (2001).
Wolf, G. “The road not taken”: Role of angiotensin II type 2 receptor in pathophysiology. Nephrol. Dial. Transplant. 17, 195–198 (2002).
Sabuhi, R., Ali, Q., Asghar, M., Al-Zamily, N. R. & Hussain, T. Role of the angiotensin II AT2 receptor in inflammation and oxidative stress: Opposing effects in lean and obese Zucker rats. Am. J. Physiol. Renal Physiol. 300, F700-706. https://doi.org/10.1152/ajprenal.00616.2010 (2011).
Acknowledgements
The authors want to thank Dana Pieter and Tobias Hirnet for their excellent technical assistance. Some of the data in this article are from the doctoral thesis of Andreas Garcia Bardon and the professorial dissertation (Habilitation) of Ralph Timaru-Kast, both presented to the Medical Faculty of the Johannes Gutenberg University, Mainz, Germany.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was supported by a grant from the University Medical Center of the Johannes Gutenberg University, Mainz, Germany (Stufe 1).
Author information
Authors and Affiliations
Contributions
R.T.-K.: corresponding author, developed the study conception and design, conducted data acquisition, analysis and interpretation, supervised the experiments, wrote and revised the main manuscript and prepared the figures. A.G.B.: performed experiments, data acquisition, analysis and interpretation and made substantial contribution to conception and design, wrote parts of the manuscript. C.L.: involved in data acquisition, analysis and interpretation, made substantial contribution to conception and design. S.P.C.-C.: performed experiments, data acquisition, analysis and interpretation, made substantial contribution to conception and design, wrote parts of the manuscript. P.S.: performed experiments, conducted data acquisition, analysis and interpretation. E-V.G.: conducted data acquisition, analysis and interpretation, made substantial contribution to conception and design, revised the manuscript critically T.K.: conducted data acquisition, analysis and interpretation, revised the manuscript. A.S.: conducted data acquisition, analysis and interpretation, revised the manuscript. U.M.S.: provided AT2 agonist C21, revised the manuscript critically and contributed important intellectual content. S.C.T.: senior author, planned and supervised the study, conducted data interpretation, made substantial contribution to conception and design, corrected and prepared the figures, revised the manuscript critically. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Timaru-Kast, R., Garcia Bardon, A., Luh, C. et al. AT2 activation does not influence brain damage in the early phase after experimental traumatic brain injury in male mice. Sci Rep 12, 14280 (2022). https://doi.org/10.1038/s41598-022-18338-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18338-x
This article is cited by
-
AT 1 inhibition mediated neuroprotection after experimental traumatic brain injury is dependent on neutrophils in male mice
Scientific Reports (2023)
-
Early DNase-I therapy delays secondary brain damage after traumatic brain injury in adult mice
Scientific Reports (2023)
-
The Renin Angiotensin System as a Therapeutic Target in Traumatic Brain Injury
Neurotherapeutics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
