
Abstract
Normal and heavy water are solvents most commonly used to study the isotope effect. The isotope effect of a solvent significantly influences the behavior of a single molecule in a solution, especially when there are interactions between the solvent and the solute. The influence of the isotope effect becomes more significant in D2O/H2O since the hydrogen bond in H2O is slightly weaker than its counterpart (deuterium bond) in D2O. Herein, we characterize the isotope effect in a mixture of normal and heavy water on the solvation of a HOD molecule. We show that the HOD molecule affects the proximal solvent molecules, and these disturbances are much more significant in heavy water than in normal water. Moreover, in D2O, we observe the formation of low-density structures indicative of an ordering of the solvent around the HOD molecule. The qualitative differences between HOD interaction with D2O and H2O were consistently confirmed with Raman spectroscopy and NMR diffusometry.
Similar content being viewed by others


Introduction
Hydrophobicity is a well-known and extensively studied phenomenon1,2,3,4. It reflects a commonly observed tendency of non-polar molecules to form aggregates, which reduces the contact surface with an aqueous polar environment, thus increasing solvent entropy. Comprehensive studies of hydrophobic interactions at various scales are essential for understanding numerous chemical and biological processes, including protein folding, membrane formation, or water transfer through pores5,6,7. The solvent rearrangement in the proximity of a solute molecule has a vast impact on solvophobic interactions. The hydrophobic effect is manifested mainly by the unfavorable free energy change upon mixing the hydrophobic substances with water3,8,9. The introduction of any hydrophobic particle induces unfavorable reorganization of the network of water-water hydrogen bonds. From a thermodynamic point of view, when a non-polar molecule is dissolved in water, there is a considerable loss of entropy and an accompanying increase in enthalpy due to the formation of low-energy and relatively rigid water structures surrounding the solute molecule10.
The solute–solvent interactions could be decomposed into two main parts. The first one corresponds to the direct solute–solvent interactions, while the other reflects the solute-induced reorganization of the solvent structure9,11,12. However, the solute-induced structure of the hydration shell is expected to be less ordered than that of ice.
The first qualitative description of an aqueous solvent was proposed in 1933. Water is described as a collection of chaotically oriented molecules forming a relatively regular network with oxygen atoms located in nodes. The hydrogen atoms covalently bonded to them are arranged so that they can form a hydrogen bond with a proximal oxygen atom13. Then, in 1945, Henry S. Frank and Marjorie Woodard Evans formulated the "iceberg" hypothesis, the first microscopic model of the interaction between water and a dissolved molecule. They proposed that non-polar substances make water form frozen patches or microscopic icebergs around each solute molecule. The water around such a molecule becomes frozen-like, showing properties similar much more to ice than liquid. The extent of the iceberg-like structures increases with the size of the solute molecule. The larger the particles, the larger the icebergs produced in the water, leading to a more significant unfavorable change of dissolving entropy9,14.
In 1976, Pierotti presented the scaled particle theory (SPT) based on the statistical analysis of the properties of a solute. The free energy of introducing a hard-sphere solute into a solvent was thus described by scaling up an infinitely small cavity to the desired radius. Therefore, the free energy of solvation is equivalent to the work of forming an empty solute-sized cavity. The SPT combines the microscopic parameters of the solvent with its macroscopic properties, such as temperature dependencies of density or surface tension, so the application of the SPT theory has been limited to relatively small molecules15,16,17,18,19,20. Another theory used in calculating cavity formation in liquids and often compared with the SPT theory is the Sinanoglu theory. Same as SPT theory, it describes the cavity formation process in an aqueous solution which relies heavily on working against the surface tension forces of the solvent. Sinanoglu’s theory is a macroscopic approach based on the thermodynamic properties of pure liquids and diluted solutions. It allows for determining the Gibbs energy and the enthalpy of cavity formation in an aqueous solution. Contrary to the SPT theory, Sinanonglu's theory does not consider an entropic contribution to the cavity formation process21,22,23. Finally, Lum, Chandler, and Weeks proposed the theory of hydrophobic solvation of both small and large non-polar species in water. They assumed that in the case of small molecules, the hydrogen bonding of water is hindered but remains near solutes. In contrast, in the case of large solute molecules, the number of solute–solvent hydrogen bonds is depleted, which leads to the “drying” of extended non-polar surfaces and large forces of solute–solute attraction. According to the LCW model, for small molecules (i.e., with a radius less than 10 Å), the interaction with water is proportional to the volume, while for large molecules to the surface24,25,26.
The structure of water is also studied in silico, and numerous forcefields have been developed27,28,29,30. Due to the low mass of the proton and the significant role of hydrogen bonds, nuclear quantum effects (NQEs), including tunneling and zero-point energy (ZPE), play an essential role in determining the static and dynamic properties of water31. The NQEs can strengthen the hydrogen bond; however, quantum fluctuations facilitate the proton to spread in different directions. Competing nuclear quantum effects are now believed to explain the isotope effects observed in water31,32,33,34,35. In general, various force fields are preferably used to simulate particular properties of water systems36.
Various stationary and time-resolved vibrational spectroscopy techniques37,38,39,40 and theoretical studies also extensively studied water structure and internal dynamics41,42,43. Because of the enormous intensity of the OH stretching band, quantitative analysis of the IR absorption of water is challenging44. This is not the case with the Raman spectrum. Analysis of Raman spectra is often based on applying multivariate curve resolution (MCR)38,45,46,47,48, a technique that allows separating the spectrum into contributions from individual components49,50. Normal water (H2O) and heavy water (D2O) are the solvents most commonly used to study the isotope effect. The isotope replacement in a solvent significantly influences the behavior of a single molecule in solution, especially when there are interactions between the solvent and the solute. This effect becomes more significant in D2O/H2O since the hydrogen bond in H2O is weaker than its counterpart, the deuterium bond in D2O. Moreover, liquid heavy water is more "structured" than normal water51,52.
Using mixtures of normal and heavy water with a significant excess of one isotopologue, e.g., 5% D2O in H2O or vice versa, allows for observing the OH or OD stretching vibrations decoupled from the stretching vibrations of the environment. For the OH oscillator, studies in dilute solutions of H2O in D2O lead to HOD molecules being surrounded by D2O. Naturally, the opposite occurs when small amounts of D2O are added to H2O: now, the HOD is solvated by normal water, and the OD oscillator can be regarded as isolated. Applying MCR, it is possible to separate the spectral features of the bulk (pure solvent) from the vibrational modes affected by the presence of the solute. The latter are contained in the so-called solute correlated (SC) spectrum, which includes both the contribution from the isolated oscillator and those solvent molecules perturbed by the solute (HOD). This perturbation extends well beyond the first solvation shell. The number of H2O solvent molecules coupled to a single OH oscillator of (solute) HOD has been estimated to be as large as 1853.
In our work, we characterize the isotope effect of the solvent, normal water, and heavy water on the solvation of a single HOD molecule. Currently, no experimental data in the literature evidences the partial molar volume of the HOD in D2O and H2O solutions, respectively. In the presented study, we tested our model of hydrophobic solvation, according to which the solvent density in the solvation shell depends on the solute molecule’s structure and polarity12,54,55,56. Additionally, the excess volume, defined as the difference between the experimentally measured partial molar volume and the in silico calculated molecular volume of a solute, seems to be a reasonable estimator of hydrophobic contribution to protein–ligand binding affinity57. We used a simple variant of the MCR technique to assess the disturbance in normal and heavy water structures caused by the HOD molecule. We also determined the diffusion coefficients for proton and deuterium in normal and heavy water.
Results
Partial molar volume and the apparent volumetric thermal expansion coefficient of HOD in H2O and D2O solutions
Our long-term studies have shown that the partial molar volume V20, determined from a series of density measurements, could state a measure of the hydrophobic interactions of the dissolved molecule with the aqueous solvent12. In general, the larger the difference between V20 (the experimental partial molar volume) and the expected molecular volume Vmol (calculated in silico from the solute structure), the more significant the hydrophobic effect54,55. Recently, we have demonstrated that V20 could be used as one of the ADME parameters that describe solute–solvent interactions57,58.
The partial molar volume for the HOD molecule in either H2O or D2O solution was determined from the four dilution series performed at 20–45 °C, analyzed globally according to Eq. (6) (Fig. 1, Supplementary Figs. S1 and S2). The proposed method of data analysis used allowed us to determine the apparent molar volume for both the HOD molecule and the density of pure solvent.

Temperature-dependence of density-molality relationship observed in a single dilution experiment for D2O in H2O (left) and H2O in D2O (right). The density dependence on molality measured at six temperatures of 20 to 45 °C was analyzed according to Eqs. (6–9). All data snapshots obtained for four independent series of dilution experiments for D2O in H2O and H2O in D2O are shown in Supplementary Figs. S1 and S2, respectively.
The temperature-dependence of the bulk solvent density (either H2O or D2O) estimated with Eqs. (6–9) from the density data measured for binary H2O/D2O mixtures is consistent with the literature data (Fig. 2)59,60,61.

The temperature dependence of the density of pure H2O and D2O determined in the range of 20–45 °C. The relationships obtained by us (solid lines) are compared with the literature data for H2O59 (black diamonds) and D2O 60,61 (black circles). The values estimated from binary H2O/D2O solvents are denoted in magenta.
The observed agreement between the density measured directly for the pure solvent and the value extrapolated from diluted binary solutions supports the applicability of the proposed method of data analysis. Therefore, the partial molar volume of the solvent molecule in bulk can be estimated directly from the density according to the following equation
where M0 is the molecular mass of the solvent (18.015 and 20.028 g/mol for H2O and D2O, respectively) and ρ0 is the solvent density. The partial molecular volumes at 25 °C are summarized in Table 1. Interestingly, the volume occupied by a single solvent molecule is almost identical for water and heavy water (18.069 and 18.132 cm3/mol, respectively), indicating that under normal conditions, the general microscopic organization of heavy water resembles that of the normal “light” water. The volumetric thermal expansion coefficient obtained at 25 °C for both H2O (257.30 ± 0.25 × 10–6/K) and D2O (193.06 ± 0.15 × 10–6/K) is consistent with the literature data (α0 = 257.21 × 10–6 K-1)59, and (α0 = 191.65 × 10–6/K)60, respectively. It is worth noting that the volumetric expansion coefficient for heavy water is substantially lower than that for “normal” water, thus reflecting differences in the internal dynamics of these two solvents52,62,63.
The more spectacular differences are observed for the solute, i.e., the HOD molecule (Fig. 3).

Temperature dependence of the partial molar volume determined from density measurements. The data obtained in H2O and D2O are in red and blue, respectively. Triangles represent the HOD molecule, while circles denote bulk solvent (H2O or D2O).
At normal conditions (25 °C), the partial molar volume of HOD in H2O resembles that of H2O in the bulk solvent (18.10 ± 0.01 vs. 18.07 cm3/mol), while in D2O is much higher (20.08 ± 0.05 cm3/mol). Such a difference should be attributed to the solute-induced reorganization of the proximal solvent molecules rather than to the real solvent-induced change in the solute structure. The observed effect demonstrates that HOD only minutely affects the structure of the surrounding H2O molecules, the average organization of which must resemble bulky H2O. Contrary, the same HOD molecule strongly affects proximal D2O molecules. We have already demonstrated that the apparent expansion of a solute molecule in an aqueous solution, which commonly results from the ordering of water molecules in the solvation shell, is indicative of hydrophobic interactions57,58. In this view, the observed excess volume of ~ 2 cm3/mol upon virtual transfer of HOD from water to heavy water (i.e., 10% of the apparent HOD volume in H2O) indicates relevant “solvophobic” interactions experienced by HOD in D2O. Bearing in mind the still proton-deuterium exchange (Eq. 4), one must conclude that the structure of water solvating deuteron resembles that of a bulk solvent. At the same time, the HOD proton substantially affects the structure of the solvating D2O molecules.
Furthermore, the apparent volumetric thermal expansivity of the HOD molecule is higher than that of a bulk D2O. So, an average structure of proximal D2O molecules is more susceptible to temperature-induced changes than a pure solvent (a 2.5-fold increase relative to bulk D2O). Interestingly, no such effect is observed in H2O (expansivity just between the values for pure H2O and D2O). So, the local D2O structure is much more perturbed by HOD (i.e., proton) than the structure of H2O by deuteron.
Raman spectroscopy
We have applied a simple variant of the MCR technique to assess the perturbations in normal and heavy water structures caused by HDO. For that purpose, Raman spectra have been measured for H2O and D2O at four different temperatures: 274, 292, 313, and 331 K. The corresponding spectra were registered for mixtures of water and heavy water containing (a) 5% D2O in H2O; (b) 5% H2O in D2O. The spectra are shown in Supplementary Figs. S3 and S4. It can be readily observed that the differences between bulk D2O and D2O containing HOD are much more significant than for the other case (H2O compared with H2O in the presence of HOD).
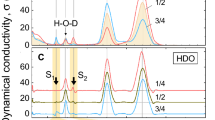
In the next step, the spectra of bulk H2O/D2O were subtracted from those of the mixtures (Fig. 4.). The resulting solute-correlated spectra consist of (1) contribution from isolated OD or OH and (2) the spectrum of the perturbed solvent shell around HOD. Since the first contribution is known from the spectra of isotopically diluted H2O and D2O (see Supplementary Figs. S3, S4), it can be subtracted from the corresponding SC curve, yielding the spectrum assigned to HOD perturbed solvent molecules. The comparison of these spectra (blue curves in Fig. 4) unambiguously shows that the intensity of the spectrum originating from the perturbed solvent is much stronger relative to the spectrum of the isolated OD/OH for the case of HDO in D2O.

Black, solute-correlated spectra for HOD in D2O (left) and H2O (right). Red, the contributions from the isolated OD/OH stretching vibration. Blue, the spectrum assigned to solvent molecules perturbed by HOD.
This result suggests that, in agreement with densimetric data, the OD oscillators in D2O are more strongly perturbed by HOD than the corresponding OH oscillators in H2O. An explanation of this finding can be related to the fact that the OD···O hydrogen bond is considered to be stronger than OH···O34,65.
Most IR/Raman papers dealing with water structure focus on the OH stretching region; it has been recently postulated that the bending mode of water can also be used66. We found that the analysis of the bending region is more challenging because of the much smaller energy spacing between the bands due to different isotopologues, which results in the spectral overlap.
Since hydrogen bonding may be considered the primary mode of interaction between HOD and the aqueous environment, it is natural to expect that the stronger interaction will lead to a more significant solvent perturbation.
IR spectroscopy
The attempts to perform a similar analysis for the IR spectra were unsuccessful. It was necessary to obtain very thin (< 2 µm) films of water squeezed in-between two IR windows to obtain reliable IR absorption values (optical density less than 2.0). Using such a procedure, it was impossible to maintain the same optical path length for each sample. Still, the spectra obtained for samples of various solute content (from 2.5 to 10%) could be compared after normalizing to the absorption maximum (Supplementary Fig. S5). We noticed that the differences between pure and isotopically diluted H2O spectra were minute, more minor than in the case of Raman spectra. A possible explanation may be related to the fact that the OH stretching band of water contains a contribution from the bending overtone67,68. Such contribution is expected to be smaller in the Raman spectrum.
It was also impossible to compare the SC IR spectra in the region of the first overtone of OD and OH. The problem was the overlap, in the OD region, with the combination of the OH stretching and HOH bending modes (from both H2O and HOD).
While the more significant perturbation of D2O than H2O by HOD seems to be well supported by the Raman spectra, the interpretation of temperature dependence requires further, more detailed studies. In agreement with previous reports69, the contribution from intermolecular coupling decreases at elevated temperatures. This decrease is monotonous for HDO in H2O. However, the situation is more complicated for HDO in D2O (Fig. 4). Here, the intermolecular contribution is relatively more significant at 292 K than at 284 K. We checked that this observation is not an experimental artifact caused by laser intensity fluctuation because the intensity of the isolated OH stretch remains constant (within 1%) in the whole temperature range. The origin of this maximum is not clear at present. It may involve modulating hydrogen-bonding strength by thermal activation of torsional and intermolecular stretching modes. To tackle this issue, registration of Raman spectra with small temperature increments is mandatory.
NMR diffusometry
Self-diffusion coefficients for both proton and deuteron are visibly affected by the isotopic composition of the H2O/D2O solvent (Fig. 5). Both cations (deuteron and proton) diffuse faster in H2O than in D2O, and in the same, a solvent deuteron diffuses slightly faster than a proton does. The lowest self-diffusion coefficient is observed for proton in D2O. The temperature dependence of self-diffusion enabled the estimation of proton and deuteron behavior in these two environments. The activation energies, Ea, and the self-diffusion coefficients at 25 °C, D25, are listed in Table 1, The models fitted to the individual data are shown in Fig. S6.

Temperature dependence of self-diffusion coefficients. The activation energies and diffusion coefficients at 25 °C are listed in Table 1. The models fitted to the individual data are shown in Fig. S6.
Interestingly, the deuteron/proton self-diffusion coefficients ratio is higher in D2O than in H2O and increases when the temperature decreases. The latter effect can be directly attributed to the differences in the activation energy for self-diffusion, which is related to the average energy and number of hydrogen bonds formed70. Estimated ratio of self-diffusion coefficients at 25 °C (indicated by the vertical dotted line in Fig. 5) for protons in two solvents (1.26 ± 0.02; H2O vs. D2O) exceeds the value determined in these two solvents for deuteron (1.19 ± 0.01), which indicates that proton is more susceptible to the isotope effect of the aqueous solvent.
Discussion
The volumetric thermal expansion coefficients experimentally determined at 25 °C for H2O and D2O are consistent with the literature data59,60. What is more, the bulk solvent density (either H2O or D2O) estimated from the density data measured for binary (H2O/D2O) mixtures is also consistent with the properties of the pure solvents59,60,61. We identified a qualitative asymmetry between the effect of diluting H2O in D2O and D2O in H2O. The density data indisputably show that the HOD molecule induces low-density structures in D2O, while no such effect could be observed in H2O. Since the partial molar volume of HOD in H2O remains the average of the close values determined for bulk H2O and D2O, the density data indicate that the proton affects the structure of D2O to a much greater extent. At the same time, the effect of the deuteron on the H2O structure can be assessed as virtually negligible. Therefore, HOD in H2O and H2O in H2O have similar apparent volumes, which differ substantially from the value determined for HOD in D2O.
Furthermore, the apparent HOD volume in D2O depends on temperature much stronger than in H2O. Interestingly, the volumetric thermal expansion coefficient for HOD in H2O is close to the average value for bulk H2O and D2O, indicating the absence of solute-specific perturbations in H2O. Contrary to the latter system, the 2.5-fold increase in the volumetric thermal expansion coefficient for HOD in D2O implies that the proton-induced low-density structures in D2O propagate with the temperature.
Such interpretation agrees with the dynamic picture obtained from NMR diffusometry, in which apparent proton diffusion in D2O is slowed down relative to H2O much more than is observed for deuteron. Moreover, the inspection of thermal trends demonstrates that the activation energy for HOD in D2O exceeds values determined for the three complementary systems, HOD in H2O, D2O in D2O, and H2O in H2O. Taken together, it could be thus concluded that the HOD diffusion in D2O causes a more significant reorganization of the bulk solvent than is required in the three other systems. This effect could be naively assigned to the fact that the amplitude of OH stretching vibrations is minutely larger than that for OD. Hence, the HOD movement in D2O (equivalent to H/D location exchange) causes the local pressure increase at a new proton location, rapidly compensated by a local density decrease. Simultaneously, the deuteron in a new location makes a local decrease of pressure, which, however, causes much slower density relaxation due to substantial asymmetry of the Lennard–Jones potential. Consequently, the trajectory of HOD (or proton) diffusing in D2O is denoted by “bubbles” of slowly relaxing low-density regions. Such effect is a close analogy to density perturbation near the wingtips, which forms so-called contrail cousins.
The Raman spectroscopy data also confirm that the HOD molecule disturbs the structure of the proximal solvent, and these disturbances are much more significant for heavy water than for normal water. Ben-Amotz has already assigned the 3200 cm−1 component of the Raman spectra (blue in Fig. 4) to a low-disorder water characteristic of a highly tetrahedral structure38. This supports our interpretation that HOD-induced low-density structures identified in D2O display lower disorder than a bulk D2O.
Methods
Samples for all experiments were prepared using heavy water (Merck, UVASOL, deuteration degree min. 99.9% for NMR spectroscopy) and standard (i.e., non-deuterated) water demineralized and filtered and with an ELIX system (Millipore). The samples were always degassed before measurements.
Density measurements
Partial Molar Volumes (V20) of the analyzed system, i.e., H2O in D2O and D2O in H2O, were estimated directly based on linear interpolation of the density–molality relationship (Supplementary Figs. S1 and S2) using the high-precision density meter Anton Paar DMA 5000 M equipped with the oscillating u-tube. The density measurements were carried out in the temperature range of 20–45 °C. A molal concentration of the minor component varied between 1 and 10 mM kg−1. It never exceeded 0.1%, sufficient to apply a first-order perturbation approach in the data analysis.
The apparent, concentration-dependent molar volume of a solute is defined as:71
where m is the molal concentration of a solute (mol/kg), M is the molar mass of solute (g/mol), and ρ and ρo are the density of the solution and the 'pure solvent', respectively. Consequently, the partial molar volume, \({V}_{2}^{0}\), can be estimated directly from the density data as the volume of a solute at the infinite dilution, according to Eq. (3)57
where ρo and ∂ρ/∂m are the intercept and slope for the linear approximation of the ρ(m) relationship, respectively.
In the mixture of H2O and D2O, hydrogen and deuterium atoms (1H, 2H) are rapidly exchanged between the solvent and solute molecules and, consequently, the following reaction
takes place, leading to the formation of HOD molecules (semiheavy water)72. So, Eqs. (2) and (3) had to be modified upon the exchange process, considering the mass balance associated with Eq. (4). Therefore, the H2O/D2O mixture consists of three components: H2O, D2O, and HDO. However, in highly diluted H2O/D2O or D2O/H2O mixtures, the minor isotopic form (either H2O or D2O) remains negligible. In such conditions, the apparent molar volume attributed to the semiheavy water, \({V}_{\varphi }^{HOD}\), equals:
where m is the molal concentration of the solute (mol/kg), Mo is the molar mass of the solvent, M is the molar mass of the solute, and ρ and ρo are the density of the solution and the bulk solvent, respectively.
Consequently, the partial molar volume of the HOD molecule, \({V}_{2}^{0}(\text{HOD})\), can be estimated as:
where ρo and ∂ρ/∂m state the intercept and slope for the linear approximation of the ρ(m) relationship.
The density measurements were carried out at the temperature range of 20–45 °C. Such an approach allowed the determination of the apparent thermal volumetric expansivity of the HOD molecule. Independently, the thermal volumetric expansivity of the bulk solvent (either H2O or D2O) can be estimated from the density data extrapolated to infinite solute concentration at different temperatures. Because of a limited temperature range sampled, we have used a first-order approximation for the V20(T) relationship, assuming the temperature-independent volumetric thermal expansion coefficient αHOD (Eq. 7).
The algorithm based on Eq. (6) was implemented in Origin (version 9.9; www.originlab.com). The model parameters were fitted globally to all dilution series (either D2O in H2O or H2O in D2O), assuming for HOD molecules the global values of V20(Tref) at Tref = 25 °C and the associated thermal volumetric expansivity coefficient, αHOD.
Finally, the resulting changes in the bulk solvent density, ρ0(T), were analyzed according to the third-order polynomial.
The thermal volumetric expansivity of a bulk solvent, α0(T), was further estimated directly from the temperature-induced variation of the pure solvent density ρ0(T).
We have additionally tested that the apparent coefficient of thermal volumetric compressibility coefficient, \(\frac{1}{{V}_{\varphi }^{HOD}}\cdot \frac{\partial {V}_{\varphi }^{HOD}}{\partial T}\), does not depend on the solute concentration. This observation justifies the global analysis neglecting the second-order correction for solute–solute interactions.
Raman Spectra
Raman measurements were made for pure H2O, pure D2O, and mixtures of 5% D2O in H2O and 5% H2O in D2O. Raman scattering spectra were measured on the InVia Renishaw microspectrometer equipped with a macroscopic adapter. Measurements were made using the 632.8 nm line of a HeNe laser (linear polarization 100:1), a 1200 l/mm grating, and a CCD camera as a detector. The "continuous scan" mode was used, in which the grating rotates synchronously with the shift of charges on the pixels of the CCD camera. Spectra were recorded in the 800–4000 cm−1 range. Accumulation for a single measurement was set to 10 s, and the laser power on the sample was 2 mW. Water mixtures were measured in a 10 mm cuvette using a 30 mm lens in a back-scattering configuration. The cuvette was thermostated to an accuracy of 0.1 K using a NESLAB RTE200 flow thermostat. The measurements were made at 284, 292, 313, and 331 K. Every sample was stabilized for about one hour after any temperature change. Ten spectra were recorded at each temperature for each mixture, then the artifacts due to cosmic rays were manually removed, and all ten spectra were averaged.
Infrared spectroscopy (IR spectra)
IR measurements were carried out for pure H2O and D2O, and 2.5%, 5%, 7.5%, and 10% mixtures of D2O in H2O and, vice versa, H2O in D2O. IR spectra (1 cm−1 spectral resolution) were recorded on a Nicolet Magna-560 FTIR instrument equipped with an MCT/B liquid N2-cooled detector. In order to obtain a thin layer of the investigated mixture, a small drop of solution was placed between two CaF2 windows. Afterward, two windows without a spacer were tightly pressed against each other in a home-built IR cell. The optical path (a thickness of water film between two windows) was calculated based on the molar absorption coefficient of water73. The obtained thickness of the water film varied for different experiments from 1.4 to 2 µm.
NMR diffusometry
The self-diffusion coefficients of D2O in H2O were measured using 2H PGSE Dbppste pulse sequence on Agilent DD2 600 MHz spectrometer (Santa Clara, California, USA) equipped with DOTY DSI-1372 multinuclear probe-head with a maximum magnetic field gradient of 30 T/m (Doty Scientific, Clemson Rd, Columbia, USA). The self-diffusion coefficients of H2O in D2O were measured using 1H PGSE Dbppste74 using Agilent DD2 800 MHz spectrometer. The exact temperatures were calibrated using ethylene glycol by analyzing the chemical shift difference between CH2 (ethylene glycol) and OH groups according to the Bruker VT-Calibration Manual. Gradient calibration constants on both spectrometers were determined using a water sample obtained from Mili-Q using the Agilent gradient calibration procedure at 21 °C.
NMR samples were poured into 5 mm NMR sample tubes (0.01% D2O in H2O and 0.01% H2O in D2O). The spin-echo intensities were fitted according to the Stejskal-Tanner equation75. using MestReNova 14 software (https://mestrelab.com/software/mnova/).
The self-diffusion coefficient at T0 = 25 °C, D25, and the activation energy, Ea, were estimated according to the modified Arrhenius Eq. (11) implemented in Origin (version 9.9; www.originlab.com).
Data availability
The raw density data, Raman spectra, and NMR data collected and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Meyer, E. E., Rosenberg, K. J. & Israelachvili, J. Recent progress in understanding hydrophobic interactions. Proc. Natl. Acad. Sci. USA. 103(43), 15739–15746 (2006).
Baldwin, R. L. Dynamic hydration shell restores Kauzmann’s 1959 explanation of how the hydrophobic factor drives protein folding. Proc. Natl. Acad. Sci. USA. 111(36), 13052–13056 (2014).
Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 437(7059), 640–647 (2005).
Baldwin, R. L. & Rose, G. D. How the hydrophobic factor drives protein folding. Proc. Natl. Acad. Sci. USA. 113(44), 12462–12466 (2016).
Sur, U.K. Behaviour of water at hydrophobic interfaces. J. Mol. Liquids. 348 (2022).
Beckstein, O., Biggin, P. C. & Sansom, M. S. P. A hydrophobic gating mechanism for nanopores. J. Phys. Chem. B 105(51), 12902–12905 (2001).
Kronberg, B. The hydrophobic effect. Curr. Opin. Colloid Interface Sci. 22, 14–22 (2016).
Rodnikova, M. N. Mechanism of solvophobic interactions. Russ. J. Phys. Chem. 80(10), 1605–1607 (2006).
Grabowska, J., Kuffel, A. & Zielkiewicz, J. Revealing the Frank-Evans “iceberg” structures within the solvation layer around hydrophobic solutes. J. Phys. Chem. B 125(6), 1611–1617 (2021).
Grunwald, E. & Steel, C. Solvent reorganization and thermodynamic enthalpy-entropy compensation. J. Am. Chem. Soc. 117(21), 5687–5692 (1995).
Poznanski, J. Partial molar volume as an important thermodynamic parameter. Application for uracil methyl derivatives. J. Mol. Liq. 121(1), 15–20 (2005).
Zielenkiewicz, W. & Poznanski, J. Partial molar volumes of hydrophobic compounds—Insight into the solvation shell? Part I. J. Solution Chem. 27(3), 245–254 (1998).
Bernal, J. D. & Fowler, R. H. A theory of water and ionic solutions, with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1, 513–548 (1933).
Frank, H. S. & Evans, M. W. Free volume and entropy in condensed systems. 3. Entropy in binary liquid mixtures—Partial molal entropy in dilute solutions—Structure and thermodynamics in aqueous electrolytes. J. Chem. Phys. 13(11), 507–532 (1945).
Jackson, R. M. & Sternberg, M. J. E. Application of scaled particle theory to model the hydrophobic effect—Implications for molecular association and protein stability. Protein Eng. 7(3), 371–383 (1994).
Pierotti, R. A. Scaled particle theory of aqueous and non-aqueous solutions. Chem. Rev. 76(6), 717–726 (1976).
Stillinger, F. H. Structure in aqueous solutions of nonpolar solutes from the standpoint of scaled-particle theory. J. Solution Chem. 2(2–3), 141–158 (1973).
Holovko, M., Patsahan, T. & Dong, W. Fluids in random porous media: Scaled particle theory. Pure Appl. Chem. 85(1), 115–133 (2013).
Heying, M. & Corti, D. S. Scaled particle theory revisited: New conditions and improved predictions of the properties of the hard sphere fluid. J. Phys. Chem. B 108(51), 19756–19768 (2004).
Riess, H. Scaled particle methods in the statistical thermodynamics of fluids. Adv. Chem. Phys. 9, 1–84 (1965).
Moreldesrosiers, N. & Morel, J. P. Evaluation of thermodynamic functions relative to cavity formation in liquids—Uses and misuses of Scaled Particle Theory. Can. J. Chem.-Revue Canadienne De Chimie. 59(1), 1–7 (1981).
Zielenkiewicz, W., Zielenkiewicz, P. & Lapshov, P. V. Evaluation of thermodynamic functions relative to cavity formation in aqueous-solutions-comparison of the results calculated from Scaled Particle Theory and Sinanoglu theory for 2-alkyl-9-methyladenines. J. Therm. Anal. 45(4), 775–779 (1995).
Mouraramos, J. J., Dionisio, M. S., Goncalves, R. C. & Diogo, H. P. A further view on the calculation of the enthalpy of cavity formation in liquids—The influence of the cavity size and shape. Can. J. Chem.-Revue Canadienne De Chimie. 66(11), 2894–2902 (1988).
Lum, K., Chandler, D. & Weeks, J. D. Hydrophobicity at small and large length scales. J. Phys. Chem. B. 103(22), 4570–4577 (1999).
Huang, D. M. & Chandler, D. Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc. Natl. Acad. Sci. USA. 97(15), 8324–8327 (2000).
Huang, D. M. & Chandler, D. The hydrophobic effect and the influence of solute-solvent attractions. J. Phys. Chem. B 106(8), 2047–2053 (2002).
Stillinger, F. H. & Rahman, A. Improved simulation of liquid water by molecular-dynamics. J. Chem. Phys. 60(4), 1545–1557 (1974).
Mahoney, M. W. & Jorgensen, W. L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 112(20), 8910–8922 (2000).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91(24), 6269–6271 (1987).
Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. B 105(43), 24A (2001).
Ceriotti, M. et al. Nuclear quantum effects in water and aqueous systems: Experiment, theory, and current challenges. Chem. Rev. 116(13), 7529–7550 (2016).
Berger, A., Ciardi, G., Sidler, D., Hamm, P. & Shalit, A. Impact of nuclear quantum effects on the structural inhomogeneity of liquid water. Proc. Natl. Acad. Sci. USA. 116(7), 2458–2463 (2019).
Morrone, J.A., Car, R. Nuclear quantum effects in water. Phys. Rev. Lett. 101(1) (2008).
Clark, T., Heske, J. & Kuhne, T. D. Opposing electronic and nuclear quantum effects on hydrogen bonds in H2O and D2O. ChemPhysChem 20(19), 2461–2465 (2019).
Chen, B., Ivanov, I., Klein, M.L., Parrinello, M. Hydrogen bonding in water. Phys. Rev. Lett. 91(21) (2003).
Cisneros, G. A. et al. Modeling molecular interactions in water: From pairwise to many body potential energy functions. Chem. Rev. 116(13), 7501–7528 (2016).
Bakker, H. J. & Skinner, J. L. Vibrational spectroscopy as a probe of structure and dynamics in liquid water. Chem. Rev. 110(3), 1498–1517 (2010).
Ben-Amotz, D. Hydration-shell vibrational spectroscopy. J. Am. Chem. Soc. 141(27), 10569–10580 (2019).
Perakis, F. et al. Vibrational spectroscopy and dynamics of water. Chem. Rev. 116(13), 7590–7607 (2016).
Fournier, J. A., Carpenter, W., De Marco, L. & Tokmakoff, A. Interplay of ion-water and water-water interactions within the hydration shells of nitrate and carbonate directly probed with 2D IR spectroscopy. J. Am. Chem. Soc. 138(30), 9634–9645 (2016).
Reddy, G., Lawrence, C. P., Skinner, J. L. & Yethiraj, A. Liquid state theories for the structure of water. J. Chem. Phys. 119(24), 13012–13016 (2003).
Auer, B. M. & Skinner, J. L. IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys. 128(22), 224511 (2008).
Hunter, K. M., Shakib, F. A. & Paesani, F. Disentangling coupling effects in the infrared spectra of liquid water. J. Phys. Chem. B. 122(47), 10754–10761 (2018).
Maréchal, Y. The molecular structure of liquid water delivered by absorption spectroscopy in the whole IR region completed with thermodynamics data. J. Mol. Struct. 1004(1), 146–155 (2011).
Davis, J. G., Gierszal, K. P., Wang, P. & Ben-Amotz, D. Water structural transformation at molecular hydrophobic interfaces. Nature 491(7425), 582–585 (2012).
Davis, J. G., Rankin, B. M., Gierszal, K. P. & Ben-Amotz, D. On the cooperative formation of non-hydrogen-bonded water at molecular hydrophobic interfaces. Nat. Chem. 5(9), 796–802 (2013).
Matt, S. M. & Ben-Amotz, D. Influence of intermolecular coupling on the vibrational spectrum of water. J. Phys. Chem. B. 122(21), 5375–5380 (2018).
Ahmed, M., Singh, A. K. & Mondal, J. A. Hydrogen-bonding and vibrational coupling of water in a hydrophobic hydration shell as observed by Raman-MCR and isotopic dilution spectroscopy. Phys. Chem. Chem. Phys. 18(4), 2767–2775 (2016).
Fega, K. R., Wilcox, A. S. & Ben-Amotz, D. Application of Raman multivariate curve resolution to solvation-shell spectroscopy. Appl. Spectrosc. 66(3), 282–288 (2012).
Ruckebusch, C. & Blanchet, L. Multivariate curve resolution: A review of advanced and tailored applications and challenges. Anal. Chim. Acta. 765, 28–36 (2013).
Bennaim, A., Wilf, J. & Yaacobi, M. Hydrophobic interaction in light and heavy-water. J. Phys. Chem. 77(1), 95–102 (1973).
Marcus, Y. & Bennaim, A. A study of the structure of water and its dependence on solutes, based on the isotope effects on solvation thermodynamics in water. J. Chem. Phys. 83(9), 4744–4759 (1985).
Kananenka, A. A., Hestand, N. J. & Skinner, J. L. OH-stretch Raman multivariate curve resolution spectroscopy of HOD/H2O mixtures. J. Phys. Chem. B. 123(24), 5139–5146 (2019).
Zielenkiewicz, W., Poznanski, J. & Zielenkiewicz, A. Partial molar volumes of alkylated uracils—Insight into the solvation shell? Part II. J. Solution Chem. 27(6), 543–551 (1998).
Zielenkiewicz, W. & Poznanski, J. Partial molar volumes-insights into molecular structure. J. Mol. Liq. 81(1), 37–45 (1999).
Zielenkiewicz, W., Poznanski, J. & Zielenkiewicz, A. Partial molar volumes of aqueous solutions of some halo and amino derivatives of uracil. J. Solution Chem. 29(8), 757–769 (2000).
Szymaniec-Rutkowska, A. et al. Does the partial molar volume of a solute reflect the free energy of hydrophobic solvation? J. Mol. Liq. 293, 111527 (2019).
Czapinska, H. et al. Halogen atoms in the protein-ligand system. Structural and thermodynamic studies of the binding of bromobenzotriazoles by the catalytic subunit of human protein kinase CK2. J. Phys. Chem. B. 125(10), 2491–2503 (2021).
Kell, G. S. Density, thermal expansivity, and compressibility of liquid water from 0 degrees to 150 degrees—Correlations and tables for atmospheric-pressure and saturation reviewed and expressed on 1968 temperature scale. J. Chem. Eng. Data 20(1), 97–105 (1975).
Chang, T. L. & Tung, L. H. Density of heavy water. Nature 163(4149), 737–737 (1949).
Millero, F. J., Dexter, R. & Hoff, E. Density and viscosity of deuterium oxide solutions from 5 to 70 degrees C. J. Chem. Eng. Data 16(1), 85–000 (1971).
Hepler, L. G. Thermal expansion and structure in water and aqueous solutions. Can. J. Chem. 47(24), 4613–5000 (1969).
Brulmans, J., Verdonck, J. & Eschbach, H. L. Thermal-expansion of heavy water between 7 and 26 degrees C. Zeitschrift Fur Naturforschung Section A J. Phys. Sci. 30(1), 107–108 (1975).
Holz, M., Heil, S. R. & Sacco, A. Temperature-dependent self-diffusion coefficients of water and six selected molecular liquids for calibration in accurate H-1 NMR PFG measurements. Phys. Chem. Chem. Phys. 2(20), 4740–4742 (2000).
Scheiner, S. & Cuma, M. Relative stability of hydrogen and deuterium bonds. J. Am. Chem. Soc. 118(6), 1511–1521 (1996).
Seki, T. et al. The bending mode of water: A powerful probe for hydrogen bond structure of aqueous systems. J. Phys. Chem. Lett. 11(19), 8459–8469 (2020).
Auer, B.M., Skinner, J.L. IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys. 128(22) (2008).
Kananenka, A. A., Hestand, N. J. & Skinner, J. L. OH-stretch Raman multivariate curve resolution spectroscopy of HOD/H2O mixtures. J. Phys. Chem. B 123(24), 5139–5146 (2019).
Matt, S. M. & Ben-Amotz, D. Influence of intermolecular coupling on the vibrational spectrum of water. J. Phys. Chem. B 122(21), 5375–5380 (2018).
Wang, J. H. Self-diffusion and structure of liquid water. 1. Measurement of self-diffusion of liquid water with deuterium as tracer. J. Am. Chem. Soc. 73(2), 510–513 (1951).
Jolicoeur, C., Boileau, J., Bazinet, S. & Picker, P. Thermodynamic properties of aqueous organic solutes in relation to their structure. 2. Apparent molal volumes and heat-capacities of C-alkylamine hydrobromides in water. Can. J. Chem.-Revue Canadienne De Chimie. 53(5), 716–722 (1975).
Narten, A. Thermodynamic effects of mixing light + heavy water. J. Chem. Phys. 41(5), 1318–2000 (1964).
Bertie, J. E. & Lan, Z. D. Infrared intensities of liquids XX: The intensity of the OH stretching band of liquid water revisited, and the best current values of the optical constants of H2O(l) at 25°C between 15,000 and 1 cm-1. Appl. Spectrosc. 50(8), 1047–1057 (1996).
Wu, D. H., Chen, A. D. & Johnson, C. S. An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Magn. Reson. Ser. A 115(2), 260–264 (1995).
Stejskal, E. O. & Tanner, J. E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 42(1), 288 (1965).
Acknowledgements
The equipment used was sponsored in part by the Centre for Preclinical Research and Technology (CePT).
Author information
Authors and Affiliations
Contributions
J.P. and J.W. supervised the project; A.S. performed density measurements and analyzed the data; S.G., B.G. and A.G. performed Raman/IR measurements and interpreted the data; K.S. performed NMR and analyzed the data. A.S., S.G., B.G., K.S., J.P. prepared figures; A.S. manuscript drafting. All authors reviewed the manuscript and agreed to publish it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stefaniuk, A., Gawinkowski, S., Golec, B. et al. Isotope effects observed in diluted D2O/H2O mixtures identify HOD-induced low-density structures in D2O but not H2O. Sci Rep 12, 18732 (2022). https://doi.org/10.1038/s41598-022-23551-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23551-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
