
Abstract
The bacterial flagellar protein export machinery consists of a transmembrane export gate complex and a cytoplasmic ATPase complex. The gate complex has two intrinsic and distinct H+-driven and Na+-driven engines to drive the export of flagellar structural proteins. Salmonella wild-type cells preferentially use the H+-driven engine under a variety of environmental conditions. To address how the Na+-driven engine is activated, we analyzed the fliJ(Δ13–24) fliH(Δ96–97) mutant and found that the interaction of the FlgN chaperone with FlhA activates the Na+-driven engine when the ATPase complex becomes non-functional. A similar activation can be observed with either of two single-residue substitutions in FlhA. Thus, it is likely that the FlgN-FlhA interaction generates a conformational change in FlhA that allows it to function as a Na+ channel. We propose that this type of activation would be useful for flagellar construction under conditions in which the proton motive force is severely restricted.
Similar content being viewed by others
Introduction
The bacterial flagellum is a macromolecular protein complex responsible for rapid and efficient movement of bacterial cells towards more suitable environments. The flagellum is composed of the basal body, which acts as a rotary motor, the hook, which serves as a universal joint, and the filament, which forms a helical propeller1,2. To construct the flagellum on the cell surface, a specialized protein export machinery located at the flagellar base transports flagellar structural subunits from the cytoplasm to the distal end of the growing flagellar structure. The flagellar export machinery is composed of a transmembrane export gate complex powered by a proton motive force (PMF) across the cytoplasmic membrane and a cytoplasmic ATPase ring complex3,4. This export machinery is structurally and functionally similar to virulence-related type III secretion systems of pathogenic bacteria, which directly inject virulence effector proteins into eukaryotic host cells5.
The transmembrane export gate complex is located inside the basal body MS ring and acts as a proton/protein antiporter to drive H+-coupled protein translocation across the cytoplasmic membrane3,4. FliP, FliQ, and FliR form a polypeptide channel complex for the translocation of export substrates across the cytoplasmic membrane6,7. FlhB associates with the FliP/FliQ/FliR complex and is postulated to coordinate opening of the polypeptide channel8. FlhA associates not only with the FliP/FliQ/FliR complex but also with the MS ring9. Because FlhA promotes the transit of both H+ and Na+ across the cytoplasmic membrane, it seems to act as the export engine of the export gate complex10,11. The C-terminal cytoplasmic domains of FlhA (FlhAC) and FlhB (FlhBC) project into the central cavity of the basal body C ring and form a docking platform for the cytoplasmic ATPase complex (FliH, FliI, FliJ), flagellar chaperones (FlgN, FliS, FliT) and export substrates12,13. This docking platform coordinates the order of flagellar protein export with assembly in a highly organized and well-controlled manner14.
FliH, FliI, and FliJ form the cytoplasmic ATPase ring complex at the flagellar base15. This structure is not essential for flagellar protein export in Salmonella enterica serovar Typhimurium (hereafter referred to as Salmonella)16,17,18, but it ensures robust and efficient coupling of energy to flagellar protein export19,20. The FliI ATPase hydrolyses ATP and activates the export gate complex through an interaction between FlhAC and FliJ, which is located at the center of the FliI hexamer ring, thereby allowing the transmembrane export gate complex to become active in coupling influx of H+ through the FlhA ion channel with the translocation of export substrates through the FliP/FliQ/FliR polypeptide channel21,22,23.
The export gate complex can also use a sodium motive force (SMF) across the cytoplasmic membrane to drive Na+-coupled protein export when the cytoplasmic ATPase ring complex is absent or nonfunctional10. Because the FlhA ion channel conducts both H+ and Na+, it is plausible that the ATPase complex may switch the ion channel properties of FlhA from a dual ion mode to a highly efficient H+ channel through an interaction between FlhAC and FliJ10. However, it remains unknown how this might happen.
In planktonic Salmonella wild-type cells, the transmembrane export gate complex preferentially uses the PMF to transport flagellar structural subunits to the cell exterior under a variety of environmental conditions16,17,18,21. However, when the cytoplasmic ATPase ring complex becomes nonfunctional, as during biofilm formation24, the export gate preferentially uses the SMF over a wide range of external pH10. A subpopulation of planktonic cells can rapidly move in the biofilm structure by rotating flagella to keep cells in the biofilm alive and healthy25. The second messenger molecule 3′–5′ cyclic diguanylate monophosphate, which induces biofilm formation, not only inhibits the transcription of flagellar genes but also binds to the FliI ATPase to suppress flagellar assembly of the cells in the biofilm24. The total PMF seems to be quite low in the cells living in the biofilm because the membrane voltage is quite small26. These observations suggest that the flagellar protein export apparatus would evolve to retain the Na+-driven export engine so that flagellated cells could arise in biofilms.
To clarify how the Na+-driven export engine is activated, we analyzed the export properties of the Salmonella MM104H-3 [fliJ(Δ13–24) fliH(Δ96–97)] strain (hereafter referred to as J(Δ13–24) H*, Table 1), in which the extragenic fliH suppressor mutation partially rescues the interaction of FliJ(Δ13–24) with FlhAC, thereby restoring flagellar formation in the presence of the fliJ(Δ13–24) mutation21. We show that the J(Δ13–24) H* cells use the SMF to produce flagella and that an interaction of FlhAC with FlgN is essential for this Na+-coupled protein export.
Results
Effect of the SMF on flagellar protein export by the J(Δ13–24) H* strain
To clarify whether an altered interaction of FliJ with FlhAC induces opening of a Na+ channel in FlhA, we analyzed the effect of the SMF on flagellar formation by the J(Δ13–24) H* strain, in which FliJ has a decreased affinity for FlhAC21. We set the external pH at 7.5 to diminish the chemical potential gradient of H+ 27 and confirmed that was the case by using a pH indicator protein, pHluorin(M153R)28,29 to show that the intracellular pH of Salmonella cells was 7.41 ± 0.05. The results for all strains we used in this study are qualitatively summarized in Table 1. Motility of the J(Δ13–24) H* strain was better in the presence of 100 mM NaCl than in its absence (Fig. 1a and Supplementary Fig. 1). The amount of FlgD and FliC secreted by these cells was also higher in the presence of 100 mM NaCl than in its absence (Fig. 1b). Because the growth rate of Salmonella cells is slower under no-salt conditions compared to in the presence of 100 mM NaCl or 100 mM KCl (Supplementary Fig. 2), we also analyzed the motility of the J(Δ13–24) H* cells in the presence of 100 mM KCl. These cells were less motile in the presence of 100 mM KCl than in the presence of 100 mM NaCl (Supplementary Fig. 3a). Also, unlike NaCl, KCl did not enhance the secretion level of FliC (Supplementary Fig. 3b). These results demonstrate that Na+ facilitates protein export by the J(Δ13–24) H* cells, whereas neither motility nor flagellar protein export by wild-type cells showed any Na+ dependence (Fig. 1 and Supplementary Fig. 3), in agreement with a previous report10.

a Motility of SJW1103 (wild-type, indicated as WT), MM104H-3 [fliJ(Δ13–24) fliH(Δ96–97), indicated as J(Δ13–24) H*], MMHI0117 [ΔfliH-fliI flhB(P28T), indicated as ΔHI B*] and MMHIJ0117 [ΔfliH-fliI-fliJ flhB(P28T), indicated as ΔHIJ B*] in 0.35% soft agar plates in the absence and presence of 100 mM NaCl. The diameter of the motility ring of 10 colonies of each strain was measured in the presence and absence of 100 mM NaCl. The average diameter of the motility ring of each strain grown in the presence of 100 mM NaCl was set to 1.0, and then relative diameter of the motility ring of cells grown in the absence of NaCl was calculated (mean ± SD, n = 10). Scar bar, 1.0 cm. b Effect of Na+ on flagellar protein export at external pH 7.5. Immunoblotting, using polyclonal anti-FlgD or anti-FliC antibody, of whole-cell proteins (Cell) and culture supernatant fractions (Sup) prepared from the above strains grown exponentially at 30 °C in T-broth (pH 7.5) with or without 100 mM NaCl. The regions of interest were cropped from original immunoblots shown in Supplementary Fig. 10.
To quantify the efficiency of flagellar assembly, we labelled the filaments with a fluorescent dye (Fig. 2a) and measured the number and length of the filaments (Supplementary Table 1). Wild-type cells produced an average of 2.7 ± 1.1 filaments per cell (mean ± SD, n = 152) in the absence of NaCl and 3.3 ± 1.5 filaments per cell (n = 153) in the presence of 100 mM NaCl (Fig. 2b). The average filament length was 11.3 ± 2.1 μm (n = 50) in the absence of NaCl and 12.9 ± 2.5 μm (n = 50) in the presence of 100 mM NaCl (Fig. 2c). Because the transcription levels of flagellar genes were not increased by adding 100 mM NaCl10, we suggest that the wild-type protein export apparatus may also utilize the SMF to some extent. In the absence of NaCl, 38.5% of the J(Δ13–24) H* cells had no visible filaments. The remaining population produced an average of 1.3 ± 0.5 filaments per cell (n = 118) (Fig. 2b). The average filament length was 7.0 ± 2.9 μm (n = 50), which is about 1.6-fold shorter than the length of the wild-type filaments in the absence of NaCl (Fig. 2c), indicating that the growth rate of filaments in these cells is slower than in the wild-type. In contrast, 87.3% of the J(Δ13–24) H* cells produced the filaments in the presence of 100 mM NaCl, with an average number of 2.0 ± 1.0 per cell (n = 145) (Fig. 2b). The average filament length was 10.7 ± 3.1 μm (n = 50), which is about 1.5-fold longer than the filament length of cells grown without NaCl (Fig. 2c). These results suggest that the transmembrane export gate complex of this strain uses the SMF in addition to the PMF to transport flagellar structural proteins during flagellar assembly. Based on these results, we propose that an altered interaction between FliJ and FlhAC activates a Na+-driven export engine to promote Na+-coupled flagellar protein export.

a Fluorescent images of the SJW1103 (WT) and MM104H-3 (J(Δ13–24) H*) cells. The cells were grown in T-broth (pH 7.5) with or without 100 mM NaCl until the cells reached the stationary phase, and then flagellar filaments were labelled with a fluorescent dye, Alexa Fluor 594. The fluorescence images of the filaments labelled with Alexa Fluor 594 (magenta) were merged with the bright field images of the cell bodies. Scale bar, 5.0 μm. b Average number of flagellar filaments. Box plots show the number of the flagellar filaments in the WT and J(Δ13–24) H* cells. Lower and upper box boundaries are 25th and 75th percentiles, respectively. The line in the middle of the box is median. Lower and upper error lines are the smallest and largest values, respectively. More than 110 cells were counted. c Scatter plots of flagellar filament length. Filament length is the average of 50 filaments, and vertical lines are standard deviations. Comparisons between datasets were performed using a two-tailed Student’s t-test. A P-value of <0.05 was considered to be statistically significant difference. **P < 0.01; ***P < 0.001. (Also see Supplementary Table 1).
Effect of FlgN on flagellar protein export by J(Δ13–24) H* cells
The Salmonella MMHI0117 [ΔfliH-fliI flhB(P28T)] strain (hereafter referred to as ΔHI B*, Table 1) is a pseudorevertant isolated from a mutant with a deletion of the two genes that form the cytoplasmic ATPase complex16. The ΔHI B* cells also preferentially use the SMF rather than the PMF to produce flagella and support some motility (Fig. 1a)10. Under no-salt conditions, FliC is not expressed in the ΔHI B* strain (Fig. 1b) because FlgM, the transcription repressor of class 3 flagellar proteins such as FliC until hook assembly is complete30,31, is also not secreted out of the cytoplasm (Supplementary Fig. 4). In agreement with a previous report10, addition of 100 mM KCl did not enhance motility or flagellar protein export by the ΔHI B* cells (Supplementary Fig. 3). It has been shown that a nonfunctional variant of FliJ, GST-FliJ, binds to FlhA and inhibits flagellar protein export by the ΔHI B* cells32. Therefore, to identify the flagellar protein required for activation of the Na+-driven export engine, GST-FliJ was overexpressed in ΔHI B* cells, and whole-cell lysates were subjected to GST affinity chromatography. In addition to FlhA, FlgN also co-purified with GST-FliJ, but not with GST alone (Supplementary Fig. 5). FlgN is a flagellar export chaperone specific for two hook-filament junction proteins, FlgK and FlgL33. The FlgN chaperone protects these two proteins from proteolysis in the cytoplasm34 and also facilitates the docking of FlgK and FlgL to FlhAC to expedite rapid and efficient export of these proteins35,36,37. Unlike the FliS and FliT chaperones, which require their cognate export substrates, FliC and FliD, respectively, to bind to FlhAC, FlgN binds to FlhAC with nanomolar affinity even in the absence of FlgK and FlgL36. This property of FlgN raises the question of whether its interaction with FlhAC activates the Na+-driven export engine.
To answer this question, we introduced a ΔflgN::tetRA allele into the wild-type and J(Δ13–24) H* strains by P22-mediated transduction to produce the ΔflgN and J(Δ13–24) H* ΔflgN strains (hereafter referred to as ΔN and J(Δ13–24) H* ΔN, respectively, Table 1) and analyzed motility of these two transductants in soft agar. About 35.2% of the ΔN cells produced a single flagellar filament (Supplementary Fig. 6a), and this level of flagellar synthesis was sufficient to generate a small motility ring on soft agar plates (Fig. 3a). In contrast, the J(Δ13–24) H* ΔN cells were completely nonmotile (Fig. 3a), and no filaments were seen on these cells (Supplementary Fig. 6a).

a Motility of SJW1103 (WT), MM104H-3 (J(Δ13–24) H*), MM9001 (ΔN) and MM9003 (J(Δ13–24) H* ∆N) in 0.35% soft agar plates containing 100 mM NaCl. Scale bar, 1.0 cm. b Immunoblotting, using polyclonal anti-FlgD (1st row), anti-FlgE (2nd row), anti-FlgK (3rd row), anti-FlgL (4th row) or anti-FliC (5th row) antibody, of whole-cell proteins (Cell) and culture supernatant fractions (Sup) prepared from the above strains. The regions of interest were cropped from original immunoblots shown in Supplementary Fig. 11a. c Electron micrograms of hook-basal bodies isolated from the above stains. Scale bar, 100 nm. d Motility of MM9003, MM9003-2 (J(Δ13–24) H* ∆N A1*) and MM9003-3 (J(Δ13–24) H* ∆N A2*) in 0.35% soft agar plates containing 100 mM NaCl. Scale bar, 1.0 cm. e Immunoblotting, using polyclonal anti-FlgD (1st row), anti-FlgE (2nd row), anti-FlgK (3rd row), or anti-FlgL (4th row) antibody, of whole-cell proteins (Cell) and culture supernatant fractions (Sup) prepared from the above strains. The regions of interest were cropped from original immunoblots shown in Supplementary Fig. 11b.
To discover why the J(Δ13–24) H* ΔN cells do not produce flagellar filaments, we next analyzed the impact of ΔflgN::tetRA on flagellar protein export. The loss of FlgN considerably reduced the levels of FlgK and FlgL secreted by wild-type cells but had little effect on secretion of the hook-capping protein, FlgD or the hook protein, FlgE (Fig. 3b). The ΔN cells produced visible hook-basal bodies (HBBs) (Fig. 3c), in agreement with a previous report38. In contrast, the presence of ΔflgN::tetRA inhibited the secretion of FlgD and FlgE by the J(Δ13–24) H* cells (Fig. 3b). Whereas the J(Δ13–24) H* cells produced HBBs, the J(Δ13–24) H* cells containing ΔflgN::tetRA produced only the MS-C ring structures (Fig. 3c). Also, the intracellular levels of FliC, FlgK and FlgL were much lower in the absence of FlgN than in its presence in the J(Δ13–24) H* mutant background (Fig. 3b), suggesting that FlgN is also required for FlgM secretion. When FlgN was expressed from a pTrc99A based plasmid in the J(Δ13–24) H* ΔN cells, the motility was restored to a level comparable to that of the J(Δ13–24) H* strain (Supplementary Fig. 6b). These results indicate that FlgN becomes essential for the export of all flagellar structural proteins by the J(Δ13–24) H* cells.
The ΔHI B* and MMHIJ0117 [ΔfliH-fliI-fliJ flhB(P28T)] (hereafter referred to as ΔHIJ B*, Table 1) cells showed a clear Na+ dependence on flagellar protein export (Fig.1 and Supplementary Fig. 3). To confirm our observations described above, we also introduced the ΔflgN::tetRA allele into these two strains and found that a loss of FlgN results in a completely nonmotile phenotype (Fig. 4a and Supplementary Fig. 6b). Also, neither FlgD, FliC nor FlgM were seen in the culture supernatants of these two strains containing ΔflgN::tetRA (Fig. 4b and Supplementary Figs 7 and 8). Therefore, we conclude that FlgN is essential for Na+-coupled protein export by the transmembrane export gate complex when the functional cytoplasmic ATPase complex is absent.

a Fluorescent images of MMHI0117 (ΔHI B*), MMHIJ0117 (ΔHIJ B*), MMHIJ0117-2 (ΔHIJ B* A1*), MMHIJ0117-3 (ΔHIJ B* A2*), MM9002 (ΔHI B* ΔN), MM9004 (ΔHIJ B* ΔN), MM9004-2 (ΔHIJ B* ΔN A1*), and MM9004-3 (ΔHIJ B* ΔN A2*). The cells were grown in T-broth (pH 7.5) containing 100 mM NaCl until the cells reached the stationary phase. Flagellar filaments were labelled with Alexa Fluor 594. The fluorescence images of the filaments labelled with Alexa Fluor 594 (magenta) were merged with the bright field images of the cell bodies. Scale bar, 5.0 μm. b Immunoblotting, using polyclonal anti-FlgD (1st row) or anti-FliC (2nd row) antibody, of whole-cell proteins (Cell) and culture supernatant fractions (Sup) prepared from the MMHI0117, MMHIJ0117, MMHIJ0117-2, and MMHIJ0117-3 cells. The regions of interest were cropped from original immunoblots shown in Supplementary Fig. 12. c, d Average number and length of flagellar filaments in the MMHI0117, MMHIJ0117, MMHIJ0117-2, and MMHIJ0117-3 cells. Filament length is the average of 50 filaments, and vertical lines are standard deviations. Comparisons between datasets were performed using a two-tailed Student’s t-test. A P-value of <0.05 was considered to be statistically significant difference. ***P < 0.001; n.s. no statistical significance. (See Supplementary Table 2).
Pseudorevertants of the J(Δ13–24) H* ΔN cells
The flhA(D456V) or flhA(T490M) mutation partially restores motility to the ΔHI B* strain lacking FlgN35 (Supplementary Fig. 7). Therefore, we introduced these alleles into the J(Δ13–24) H* ΔN cells by P22-mediated transduction to see whether motility was rescued. As expected, either of these flhA mutations improved motility and flagellar protein export (Fig. 3d and e), indicating that these flhA mutations can also activate the Na+-driven export engine in the absence of FlgN. Therefore, we suggest that these single-residue substitutions in FlhAC allow FlhAC to adopt a conformation mimicking a FlgN-bound state of FlhAC, thereby allowing the export gate complex to utilize the SMF to transport flagellar structural subunits to the cell exterior even in the absence of FlgN.
Effect of deletion of FliJ residues 13–24 on the interactions of FlgN and FlhAC
The interaction of FliJ with the linker region of FlhAC (FlhAL) is required for activation of the transmembrane export gate complex, and FliH and FliI are required for efficient interaction between FliJ and FlhAL21,32. In addition to binding with high affinity to FlhAC, FlgN binds to FliJ with a KD of 22 μM39. Thus, FlgN may be important in the docking of FliJ to FlhAC, especially when the functions of FliH and FliI are compromised. Therefore, we first investigated whether the fliJ(Δ13–24) deletion mutation affects the interaction of FliJ with FlgN. FlgN co-purified with GST-FliJ(Δ13–24) (Fig. 5a), indicating that the deletion does not abolish interaction with FlgN. To assess the strength of the FliJ-FlgN interaction, we examined binding of FlgN to immobilized GST-FliJ or GST-FliJ(Δ13–24) by surface plasmon resonance (SPR). Steady-state analysis of the SPR data with a 1 to 1 binding model indicated that the KD values for the FliJ-FlgN and FliJ(Δ13–24)-FlgN interactions were 27.4 ± 13.0 μM and 7.74 ± 0.12 μM, respectively (Fig. 5b). Thus, the deletion actually increases the binding affinity by about 3.5-fold.

a Interaction between FliJ and FlgN. Cell lysates prepared from Salmonella SJW1368 (∆flhDC-cheW) cells expressing either GST-FliJ (indicated as GST-J) or GST-FliJ(∆13–24) (indicated as GST-J(∆13–24)) were mixed with those from E. coli BL21(DE3) Star cells producing His-FlgN (indicated as N), and then each mixture (indicates as L) was loaded onto a GST column. After extensive washing, proteins were eluted with a buffer containing 10 mM reduced glutathione. Eluted fractions were analyzed by SDS-PAGE with CBB staining. The regions of interest were cropped from original CBB-stained gels shown in Supplementary Fig. 13a. b Measurements of the binding affinities of FliJ and FliJ(∆13–24) for FlgN by SPR. His-FlgN of various concentrations was flowed over the sensor surface with immobilized GST-FliJ or GST-FliJ(∆13–24) in 10 mM HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% Surfactant P20 at a flow rate of 20 μl min−1. All experiments were performed at 25 °C. The steady-state resonance units (RU) were plotted against FlgN concentrations. c, d Effect of FlgN on the FliJ-FlhA interaction. Purified His-FlhAC was mixed with purified c GST-FliJ or d GST-FliJ(∆13–24) in the absence (upper panel) and presence (lower panel) of purified His-FlgN, and dialyzed overnight against PBS. Each mixture (L) was loaded onto a GST column. After washing with 10 ml PBS at a flow rate of about 0.5 ml min−1, proteins were eluted with 10 mM reduced glutathione. Flow through fraction (F.T.), wash fractions (W), and elution fractions (E) were analyzed by SDS-PAGE with CBB staining. The regions of interest were cropped from original CBB-stained gels shown in Supplementary Fig. 13b.
We next investigated whether FlgN participates in the interaction of FliJ(Δ13–24) with FlhAC. The amount of FlhAC that co-purified with GST-FliJ(Δ13–24) was much lower than that with GST-FliJ (Fig. 5c and d, upper panels), in agreement with a previous report21. FlgN did not improve the interaction of FliJ(Δ13–24) with FlhAC (lower panels). These results suggest that a direct interaction between FlgN and FlhAC turns on Na+-coupled protein export independently of FliJ when the FliJ-FlhAC interaction is compromised.
Effect of the flhA(D456V) and flhA(T490M) mutations on Na+-coupled protein export by cells lacking FliH, FliI, and FliJ
FliJ is apparently more important for Na+-coupled flagellar protein export than FliH and FliI21 because the protein export activity of the ΔHIJ B* strain is much lower than that of the ΔHI B* strain (Fig. 1 and Supplementary Fig. 1). To confirm this, we first analyzed the number and length of flagellar filaments produced by the ΔHI B* and ΔHIJ B* cells (Fig. 4a and Supplementary Table 2). About 78.2% of the ΔHI B* cells produced the filaments with an average of 1.6 ± 0.7 filaments per cell (n = 266) and an average length of 7.8 ± 2.5 µm (n = 50) (Fig. 4c, d). In contrast, only 13.5% of the ΔHIJ B* cells produced the filaments with an average of 1.1 ± 0.2 filaments per cell (n = 57) and an average length of 5.1 ± 2.2 µm (n = 50) (Fig. 4c, d). Therefore, we conclude that FliJ is required for efficient activation of the export gate complex even in the absence of FliH and FliI.
Since FliJ is not critical for activation of the Na+-driven export engine, we hypothesized that the interaction of FliJ with FlhAC may be required for efficient opening of the polypeptide channel for the substrate entry into the channel. Because it has been shown that the flhA(D456V) or flhA(T490M) mutation significantly increases the probability of entry of export substrates such as FlgD and FlgE into the polypeptide channel in the ΔHI B* cells20, we investigated whether either of these two flhA mutations might overcome the effects of the loss of both FliJ and FlgN. To clarify this, we introduced these flhA alleles into the ΔHIJ B* strain and found that either mutation increased the secretion levels of FlgD and FliC by more than 10 fold (Fig. 4b). Consistently, more than 95% of ΔHIJ B* cells containing either flhA(D456V) or flhA(T490M) mutation had several flagellar filaments (Fig. 4a, c and Supplementary Table 2) although the average length of those filaments was almost the same as that of the much fewer filaments by the ΔHIJ B* strain (Fig. 4d). The loss of FlgN did not significantly reduce the secretion levels of FlgD and FliC by the ΔHIJ B* cells with either of the flhA mutation (Fig. 4b). Furthermore, of the ΔHIJ B* cells containing ΔflgN::tetRA, 20.8% with flhA(D456V) and 8.5% with flhA(T490M) cells produced a single flagellar filament (Fig. 4a) in a way similar to the ΔN mutant strain (Supplementary Fig. 6a). Therefore, we suggest that the flhA(D456V) and flhA(T490M) mutations can activate the export gate complex to become a highly efficient Na+-driven engine in the absence of FliH, FliI, FliJ, and FlgN.
Discussion
The flagellar protein export machinery maintains its activity despite various internal and external perturbations. To do so, this export machinery has evolved to become a dual-fuel export machine to exploit both H+ and Na+ as the coupling ion10. The wild-type export engine predominantly uses H+ as a coupling ion21,23. However, when the ATPase complex does not work properly, the export engine uses its Na+ channel to continue flagellar assembly10, but the mechanism of the switching of the coupling ion was unknown. Here, we show that an impaired interaction between FliJ and FlhAC caused by diminished ATPase activity activates Na+-coupled protein export (Figs. 1 and 2). We also found that an interaction between FlgN, an export chaperone specific for FlgK and FlgL33, and FlhAC becomes essential for Na+-coupled protein export (Figs. 3 and 4). FlgN promotes the docking of FlgK and FlgL to the FlhAC platform of the export gate complex to facilitate rapid and efficient export of these proteins35,36,37. Therefore, the loss of FlgN reduces the secretion levels of FlgK and FlgL, resulting in a considerable reduction in the probability of filament formation at the tip of the HBB38. We found here that in the J(Δ13–24) H* cells, deletion of FlgN inhibits the export of FlgD and FlgE (Fig. 3b). This result suggests that FlgN acts not only as a substrate-specific export chaperone but also as a switch to activate a backup mechanism that in the absence of the FliHIJ ATPase complex, turns on the Na+-driven export engine. FlgN interacts directly and with high affinity with FlhAC to accomplish this activation (Fig. 6).

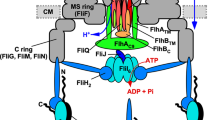
The flagellar protein export machinery is composed of a transmembrane export gate complex made of FlhA, FlhB, FliP, FliQ, and FliR and a cytoplasmic ATPase complex consisting of FliH, FliI, and FliJ. The export gate complex is located inside the MS ring and utilizes proton motive force (PMF) across the cytoplasmic membrane (CM) to drive proton (H+)-coupled flagellar protein export. FliP, FliQ and FliR form a polypeptide channel. FlhB associates with the FliP/FliQ/FliR complex and controls opening of the polypeptide channel. The C-terminal cytoplasmic domain of FlhA (FlhAC) projects into the central cavity of the C ring. The N-terminal transmembrane domain (FlhATM) forms an ion channel for the translocation of H+ and sodium ion (Na+) from the periplasm to the cytoplasm. The cytoplasmic ATPase ring complex associates with the C ring through an interaction between FliH and a C ring protein, FliN. ATP hydrolysis by the FliI ATPase activates the export gate complex through an interaction between FliJ and FlhAL connecting FlhAC to FlhATM, becoming an active protein transporter to couple the H+ flow through the FlhA channel to the translocation of export substrates into the polypeptide channel. When the cytoplasmic ATPase complex does not function properly, FlgN binds to FlhAC to open the Na+ channel of FlhATM, allowing the export gate complex to utilize sodium motive force (SMF) across the cytoplasmic membrane to drive Na+-coupled protein export.
The flhA(D456V) or flhA(T490M) mutation has been isolated as a bypass mutation of the motility defect of the ΔHI B* ΔN strain35. Here, we found that these flhA mutations overcome the effects of loss of both FlgN and FliJ (Fig. 4), suggesting that these two mutations allow FlhAC to adopt a conformation mimicking its conformation in the active FlgN/FliJ/FlhAC trimeric complex. Because the interaction between FliJ and FlhAC is not directly involved in activation of the Na+-driven export engine, we propose that the interactions of FlhAC with FlgN and FliJ activate the Na+ channel of FlhA and the polypeptide channel formed by FliP, FliQ, and FliR, respectively, so that the export gate complex efficiently couples the Na+ flow through the FlhA channel with substrate entry into the polypeptide channel (Fig. 6).
FlgN binds to a well-conserved hydrophobic dimple of FlhAC formed by Asp-456, Phe-459, and Thr-49035,36,40. When the ATPase complex is functional, FliJ binds to the flexible linker region of FlhA (FlhAL) connecting FlhAC to the N-terminal transmembrane domain that forms an ion channel21,41. This interaction activates the export gate complex to become an active H+-driven export engine21,42 (Fig. 6).This conclusion is supported by the observation that deletion of residues 328–351 of FlhAL significantly weakens the FliJ-FlhAC interaction (Supplementary Fig. 9a) but not the FlgN-FlhAC interaction (Supplementary Fig. 9b). FlgN bound to FliJ(∆13–24) but did not restore the impaired interaction between FliJ(∆13–24) and FlhAC (Fig. 5). FliJ not only binds to FlgN39 but also to the FlgN/FlgK complex35. The FlgN/FlgK/FliJ trimeric complex docks to the FlhAC platform (Supplementary Fig. 9c)35. When the GST-FlgN/FlgK/FliJ complex was mixed with FlhAC lacking residues 328–351 of FlhAL, only a very small amount of FliJ co-purified with this complex (Supplementary Fig. 9c), indicating that FliJ dissociates from FlgN upon binding of the FlgN/FlgK/FliJ complex to FlhAC lacking residues 328–351 of FlhAL. Because protein transport activity was higher in the presence of FliJ than in its absence (Fig. 1), we propose that the cytoplasmic FlgN/FliJ complex docks to the FlhAC platform through an interaction between FlgN and FlhAC, which then induces the dissociation of the FlgN/FliJ complex into FlgN and FliJ subunits to bind to the hydrophobic dimple of FlhAC and FlhAL, respectively. These interactions then fully activate the Na+-driven engine of the export gate complex in the absence of an active ATPase complex (Fig. 6). This conclusion is supported by the crystal structure of a FliJ homologue, CdsO, in complex with CdsVC, which is a FlhAC homologue43. It remains unknown how FliJ binds to FlhAL because CdsO does not bind to the linker region of CdsVC in the crystal structure.
The 3′-5′ cyclic diguanylate monophosphate molecule binds to the FliI ATPase to inhibit the FliI ATPase activity24. This event might be expected to inhibit the H+-coupled activity of the protein export channel. A subpopulation of planktonic cells is generated during biofilm development, perhaps as a “hedge-betting ploy” for cells to escape the biofilm25. Because the total PMF is quite low in the cells living in the biofilm structure26, we propose that activation of the Na+-driven export engine would provide a selective advantage for cells living in the biofilm.
Methods
Bacterial strains, plasmids, transductional crosses, and DNA manipulations
Wild-type and mutant strains of S. enterica serovar Typhimurium and plasmids used in this study are listed in Supplementary Table 3. P22-mediated transductional crosses were carried out with p22HTint. DNA manipulations were performed using standard protocols. DNA sequencing reactions were carried out using BigDye v3.1 (Applied Biosystems) and then the reaction mixtures were analyzed by a 3130 Genetic Analyzer (Applied Biosystems).
Motility assays in soft agar
Fresh colonies were inoculated onto soft agar plates [1% (w/v) tryptone, 10 mM potassium phosphate pH 7.5, 0.35%(w/v) Bacto agar] or soft agar plates containing 100 mM NaCl or 100 mM KCl and incubated at 30 °C. At least ten independent measurements were performed. A diameter of the motility ring of each Salmonella strain was measured using ImageJ software version 1.52 (National Institutes of Health).
Secretion assay
Wild-type and mutant cells of S. enterica serovar Typhimurium were grown overnight in T-broth [1%(w/v) Bacto tryptone, 10 mM potassium phosphate pH 7.5] without 100 mM NaCl. A 50 μl of the overnight culture was inoculated into a 5 ml of fresh T-broth (pH 7.5) or T-broth (pH 7.5) containing 100 mM NaCl or 100 mM KCl and incubated at 30 °C with shaking until the cell density had reached an OD600 of ca. 1.4–1.6. Cultures were centrifuged to obtain cell pellets and culture supernatants. The cell pellets were resuspended in a sample buffer solution [62.5 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate (SDS), 10% glycerol, 0.001% bromophenol blue] containing 1 μl of 2-mercaptoethanol. Proteins in the culture supernatants were precipitated by 10% trichloroacetic acid and suspended in a Tris/SDS loading buffer (one volume of 1 M Tris, nine volumes of 1× sample buffer solution)44 containing 1 μl of 2-mercaptoethanol. After boiling proteins in both whole cellular and culture supernatant fractions at 95 °C for 3 min, these protein samples were separated by SDS–polyacrylamide gel (normally 12.5% acrylamide) electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Bio-Rad) using a transblotting apparatus (Hoefer). Then, immunoblotting with polyclonal anti-FlgD, anti-FlgE, anti-FlgK, anti-FlgL, or anti-FliC antibody was carried out using iBand Flex Western Device as described in the manufacturer’s instructions (Thermo Fisher Scientific). Detection was performed with Amersham ECL Prime western blotting detection reagent (Cytiva). Chemiluminescence signals were captured by a Luminoimage analyzer LAS-3000 (GE Healthcare). All image data were processed with Photoshop software CS6 (Adobe). At least three independent experiments were performed.
Observation of flagellar filaments with a fluorescent dye
Wild-type and mutant cells of S. enterica serovar Typhimurium were grown at 30 °C in T-broth (pH 7.5) with or without 100 mM NaCl until the cells reached a stationary phase. The cells were attached to a cover slip (Matsunami glass, Japan), and unattached cells were washed away with motility buffer (10 mM potassium phosphate pH 7.0, 0.1 mM EDTA, 10 mM L-sodium lactate). A 1 μl aliquot of polyclonal anti-FliC serum was mixed with 50 μl of motility buffer and then 50 μl of the mixture was applied to the cells attached to the cover slip. After washing with the motility buffer, 1 μl of anti-rabbit IgG conjugated with Alexa Fluor 594 (Invitrogen) was added to 50 μl of motility medium, and then the mixture was applied. After washing with the motility buffer, the cells were observed by fluorescence microscopy45. Fluorescence images were analyzed using ImageJ software version 1.52 (National Institutes of Health).
Preparation of HBBs
Wild-type and mutant cells of S. enterica serovar Typhimurium were grown at 30 °C in 500 ml of L-broth until the cell density had reached an OD600 of ca. 1.0. The cells were harvested by centrifugation (10,000 × g, 10 min, 4 °C) and suspended in 20 ml of ice-cold 0.1 M Tris-HCl pH 8.0, 0.5 M sucrose, followed by adding EDTA and lysozyme at final concentrations of 10 mM and 0.1 mg ml−1, respectively. The cell suspensions were stirred for 30 min at 4 °C. Then, the cells were solubilized on ice for 1 h by adding Triton X-100 and MgSO4 at final concentrations of 1%(w/v) and 10 mM, respectively. The cell lysates were adjusted to pH 10.5 with 5 M NaOH and centrifuged (10,000 × g, 20 min, 4 °C) to remove cell debris. After ultracentrifugation (45,000 × g, 60 min, 4 °C), pellets were resuspended in 10 mM Tris-HCl, pH 8.0, 5 mM EDTA, 1%(w/v) Triton X-100, and the solution was loaded a 20–50%(w/w) sucrose density gradient in 10 mM Tris-HCl, pH 8.0, 5 mM EDTA, 1%(w/v) Triton X-100. After ultracentrifugation (49,100 × g, 13 h, 4 °C), intact flagella, HBBs or MS-C rings were collected and ultracentrifuged (60,000 × g, 60 min, 4 °C). For intact flagella, pellets were resuspended in 50 mM glycine, pH 2.5, 0.1%(w/v) Triton X-100, and were incubated at room temperature for 30 min to depolymerize the filaments. After ultracentrifugation (60,000 × g, 60 min, 4 °C), pellets were resuspended in 50 μl of 10 mM Tris-HCl, pH 8.0, 5 mM EDTA, 0.1%(w/v) Triton X-100. Samples were applied to carbon-coated copper grids, followed by negative staining with 2%(w/v) uranyl acetate. Electron micrographs were recorded with a JEM-1011 transmission electron microscope (JEOL, Tokyo, Japan) operated at 100 kV and equipped with a F415 CCD camera (TVIPS, Gauting, Germany) at a magnification of ×5500, which corresponds to 2.75 nm per pixel.
Pull-down assays by GST chromatography
To identify the flagellar protein required for activation of the Na+-driven export engine, GST-FliJ was over-produced in the Salmonella MMHI0117 strain, and then the cells were suspended in PBS (8 g of NaCl, 0.2 g of KCl, 3.63 g of Na2HPO4•12H2O, 0.24 g of KH2PO4, pH 7.4 per liter) and sonicated. After centrifugation of cell lysates to remove undisrupted cells and insoluble membrane fractions, the soluble fractions were loaded onto a glutathione Sepharose 4B column (bed volume, 1 ml) pre-equilibrated with 20 ml of PBS. After extensive washing of the column with PBS, proteins were eluted with 50 mM Tris-HCl, pH 8.0, 10 mM reduced glutathione. Fractions containing GST or GST-FliJ were identified by SDS-PAGE with Coomassie Brilliant blue (CBB) staining. Then, these fractions were analyzed by immunoblotting with polyclonal anti-FlhAC, anti-FliM, anti-FlgN, or anti-FliT antibody.
To analyze the FlgN-FliJ interaction by GST affinity chromatography, cell lysates prepared from SJW1368 cells expressing GST-FliJ or GST-FliJ(Δ13–24), were mixed with those from the Escherichia coli BL21 (DE3) Star strain transformed with pMMGN130 (His-FlgN). To effect of deletion of residues 328–351 of FlhAL on the interactions of FlhAC with FlgN and FliJ, cell lysates prepared from SJW1368 expressing GST-FlgN was mixed with purified FlgK, purified His-FliJ and the soluble fraction isolated from E. coli BL21 (DE3) Star cells over-expressing either His-FlhAC or His-FlhAC lacking residues 328–351 of FlhAL. Then each mixture was loaded onto a Glutathione Sepharose 4B column. After extensive wash of the column with PBS, bound proteins were eluted with 50 mM Tris-HCl, pH 8.0, 10 mM reduced glutathione.
His-FlhAC, His-FlhAC38K, and His-FlgN were overexpressed in E. coli BL21 (DE3) Star cells, and then these proteins were purified from the cell lysates by Ni affinity chromatography with a nickel-nitriloacetic acid (Ni-NTA) agarose column (QIAGEN). GST-FliJ, GST-FliJ(Δ13–24), and GST-FlgN were overexpressed in SJW1368 cells, and then these proteins were purified from cell lysates by GST affinity chromatography. To investigate the effect of deletion of residue 13–24 of FliJ on the FliJ-FlhAC interaction, purified His-FlhAC was mixed with purified GST-FliJ or GST-FliJ(Δ13–24) in the presence and absence of purified His-FlgN. To clarify the role of FlhAL on the interactions of FlhAC with FliJ and FlgN, purified His-FlhAC or His-FlhAC38K was mixed with purified GST-FliJ or GST-FlgN. Each mixture was dialyzed overnight against PBS at 4 °C with three changes of PBS. A 5 ml of each mixture was loaded onto a glutathione Sepharose 4B column and washed with 10 ml of PBS at a flow rate of ca. 0.5 ml min−1. Bound proteins were eluted with 5 ml of 50 mM Tris-HCl, pH 8.0, 10 mM reduced glutathione. At least three independent experiments were carried out.
Surface plasmon resonance (SPR)
Anti-GST antibody was immobilized on a CM5 chip using a GST capture kit as described in the manufacturer’s instructions (GE Healthcare). 40 µl of 10 µg ml−1 GST-FliJ or 10 µg ml−1 of GST-FliJ(Δ13–24) were injected over the chip pre-equilibrated with a binding buffer (10 mM HEPES pH 7.4, 0.15 M NaCl, 3 m M EDTA, 0.005% Surfactant P20) at a flow rate of 20 µl min−1 and immobilized on the sensor chip via the anti-GST antibody. Forty microliter of His-FlgN of various concentrations in the binding buffer to monitor association was passed over the sensor surface and then washed with the buffer to monitor dissociation at a flow rate of 20 µl min−1. An acidic buffer (10 mM Glycine-HCl, pH 2.2) was used for regeneration of the surface of the sensor chip by removal of the captured proteins and any associates. All experiments were done at 25 °C. To obtain the KD value, we analyzed SPR profiles using BIAevaluation software version 4.1 as described in the manufacturer’s instructions (GE Healthcare). At least three independent SPR measurements were carried out.
Statistics and reproducibility
Statistical tests, sample size, and number of biological replicates are reported in the figure legends. Statistical analyses were done using Prism 7.0c software (GraphPad). Comparisons were performed using a two-tailed Student’s t-test. A P-value of < 0.05 was considered to be statistically significant difference. *P < 0.05; **P < 0.01; ***P < 0.001.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated during this study are included in this published article and its Supplementary Information files. Strains, plasmids, polyclonal antibodies and all other data are available from the corresponding author on reasonable request.
References
Morimoto, Y. V. & Minamino, T. Structure and function of the bi-directional bacterial flagellar motor. Biomolecules 4, 217–234 (2014).
Nakamura, S. & Minamino, T. Flagella-driven motility of bacteria. Biomolecules 9, 279 (2019).
Minamino, T. Protein export through the bacterial flagellar type III export pathway. Biochim. Biophys. Acta 1843, 1642–1648 (2014).
Minamino, T. Hierarchical protein export mechanism of the bacterial flagellar type III protein export apparatus. FEMS Microbiol. Lett. 365, fny117 (2018).
Galán, J. E., Lara-Tejero, M., Marlovits, T. C. & Wagner, S. Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 68, 415–438 (2014).
Kuhlen, L. et al. Structure of the core of the type III secretion system export apparatus. Nat. Struct. Mol. Biol. 25, 583–590 (2018).
Ward, E. et al. Type-III secretion pore formed by flagellar protein FliP. Mol. Microbiol. 107, 94–103 (2018).
Kuhlen, L. et al. The substrate specificity switch FlhB assembles onto the export gate to regulate type three secretion. Nat. Commun. 11, 1296 (2020).
Fukumura, T. et al. Assembly and stoichiometry of the core structure of the bacterial flagellar type III export gate complex. PLoS Biol. 15, e2002281 (2017).
Minamino, T., Morimoto, Y. V., Hara, N., Aldridge, P. D. & Namba, K. The bacterial flagellar type III export gate complex is a dual fuel engine that can use both H+ and Na+ for flagellar protein export. PLoS Pathog. 12, e1005495 (2016).
Erhardt, M. et al. Mechanism of type-III protein secretion: regulation of FlhA conformation by a functionally critical charged-residue cluster. Mol. Microbiol. 104, 234–249 (2017).
Minamino, T. & Macnab, R. M. Interactions among components of the Salmonella flagellar export apparatus and its substrates. Mol. Microbiol. 35, 1052–1064 (2000).
Abrusci, P. et al. Architecture of the major component of the type III secretion system export apparatus. Nat. Struct. Mol. Biol. 20, 99–104 (2013).
Minamino, T., Inoue, Y., Kinoshita, M. & Namba, K. FliK-driven conformational rearrangements of FlhA and FlhB are required for export switching of the flagellar potein export apparatus. J. Bacteriol. 202, e00637–19 (2020).
Imada, K., Minamino, T., Uchida, Y., Kinoshita, M. & Namba, K. Insight into the flagella type III export revealed by the complex structure of the type III ATPase and its regulator. Proc. Natl Acad. Sci. USA 113, 3633–3638 (2016).
Minamino, T. & Namba, K. Distinct roles of the FliI ATPase and proton motive force in bacterial flagellar protein export. Nature 451, 485–488 (2008).
Paul, K., Erhardt, M., Hirano, T., Blair, D. F. & Hughes, K. T. Energy source of flagellar type III secretion. Nature 451, 489–492 (2008).
Erhardt, M., Mertens, M. E., Fabiani, F. D. & Hughes, K. T. ATPase-independent type-III protein secretion in Salmonella enterica. PLoS Genet. 10, e1004800 (2014).
Minamino, T. et al. FliH and FliI ensure efficient energy coupling of flagellar type III protein export in Salmonella. Microbiologyopen 5, 424–435 (2016).
Inoue, Y., Morimoto, Y. V., Namba, K. & Minamino, T. Novel insights into the mechanism of well-ordered assembly of bacterial flagellar proteins in Salmonella. Sci. Rep. 8, 1787 (2018).
Minamino, T., Morimoto, Y. V., Hara, N. & Namba, K. An energy transduction mechanism used in bacterial type III protein export. Nat. Commun. 2, 475 (2011).
Minamino, T., Morimoto, Y. V., Kinoshita, M., Aldridge, P. D. & Namba, K. The bacterial flagellar protein export apparatus processively transports flagellar proteins even with extremely infrequent ATP hydrolysis. Sci. Rep. 4, 7579 (2014).
Morimoto, Y. V. et al. High-resolution pH imaging of living bacterial cell to detect local pH differences. mBio 7, 01911–16 (2016).
Trampari et al. Bacterial rotary export ATPases are allosterically regulated by the nucleotide second messenger cyclic-di-GMP. J. Biol. Chem. 290, 24470–24483 (2015).
Houry, A. et al. Bacterial swimmers that infiltrate and take over the biofilm matrix. Proc. Natl Acad. Sci. USA 109, 13088–13093 (2012).
Prindle, A. et al. Ion channels enable electrical communication in bacterial communities. Nature 572, 59–63 (2015).
Nakamura, S. et al. Effect of intracellular pH on the torque-speed relationship of bacterial proton-driven flagellar motor. J. Mol. Biol. 386, 332–338 (2009).
Miesenböck, G., Angelis, D. A. & Rothman, J. E. Visualization secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195 (1998).
Morimoto, Y. V., Kojima, S., Namba, K. & Minamino, T. M153R mutation in a pH-sensitive green fluorescent protein stabilizes its fusion proteins. PLoS ONE 6, e19598 (2011).
Hughes, K. T., Gillen, K. L., Semon, M. J. & Karlinsey, J. E. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science 262, 1277–1280 (1993).
Kutsukake, K. Excretion of the anti-sigma factor through a flagellar substructure couples flagellar gene expression with flagellar assembly in Salmonella typhimurium. Mol. Gen. Genet. 243, 605–612 (1994).
Ibuki, T. et al. Interaction between FliJ and FlhA, components of the bacterial flagellar type III export apparatus. J. Bacteriol. 195, 466–473 (2013).
Fraser, G. M., Bennett, J. C. Q. & Hughes, C. Substrate-specific binding of hook-associated proteins by FlgN and FliT, putative chaperones for flagellum assembly. Mol. Microbiol. 32, 569–580 (1999).
Aldridge, P., Karlinsey, J. E. & Hughes, K. T. The type III secretion chaperone FlgN regulates flagellar assembly via a negative feedback loop containing its chaperone substrates FlgK and FlgL. Mol. Microbiol. 49, 1333–1345 (2003).
Minamino, T. et al. Interaction of a bacterial flagellar chaperone FlgN with FlhA is required for efficient export of its cognate substrates. Mol. Microbiol 83, 775–788 (2012).
Kinoshita, M., Hara, N., Imada, K., Namba, K. & Minamino, T. Interactions of bacterial chaperone-substrate complexes with FlhA contribute to co-ordinating assembly of the flagellar filament. Mol. Microbiol. 90, 1249–1261 (2013).
Kinoshita, M. et al. Rearrangements of α-helical structures of FlgN chaperone control the binding affinity for its cognate substrates during flagellar type III export. Mol. Microbiol. 101, 656–670 (2016).
Kutsukake, K., Okada, T., Yokoseki, T. & Iino, T. Sequence analysis of the flgA gene and its adjacent region in Salmonella typhimurium, and identification of another flagellar gene, flgN. Gene 143, 49–54 (1994).
Evans, L. D. B., Stafford, G. P., Ahmed, S., Fraser, G. M. & Hughes, C. An escort mechanism for cycling of export chaperones during flagellum assembly. Proc. Natl Acad. Sci. USA 103, 17474–17479 (2006).
Inoue, Y. et al. Structural insight into the substrate specificity switch mechanism of the type III protein export apparatus. Structure 27, 965–976 (2019).
Bange, G. et al. FlhA provides the adaptor for coordinated delivery of late flagella building blocks to the type III secretion system. Proc. Natl Acad. Sci. USA 107, 11295–11300 (2010).
Saijo-Hamano, Y., Minamino, T., Macnab, R. M. & Namba, K. Structural and functional analysis of the C-terminal cytoplasmic domain of FlhA, an integral membrane component of the type III flagellar protein export apparatus in Salmonella. J. Mol. Biol. 343, 457–466 (2004).
Jensen, J., Yamini, S., Rietsch, A. R. & Spiller, B. W. The structure of the type III secretion system export gate with CdsO, an ATPase lever arm. PLoS Pathog. 16, e1008923 (2020).
Minamino, T. & Macnab, R. M. Components of the Salmonella flagellar export apparatus and classification of export substrates. J. Bacteriol. 181, 1388–1394 (1999).
Morimoto, Y. V., Nakamura, S., Kami-ike, N., Namba, K. & Minamino, T. Charged residues in the cytoplasmic loop of MotA are required for stator assembly into the bacterial flagellar motor. Mol. Microbiol. 78, 1117–1129 (2010).
Acknowledgements
We acknowledge Kelly T. Hughes for his kind gift of the flgN::tetRA allele, Kouhei Ohnishi for his kind gift of polyclonal anti-FlgM antibody, Yumi Inoue and Yasuyo Abe for technical assistance and Michael D. Manson for critical reading of the manuscript and helpful discussions. This work was supported in part by JSPS KAKENHI Grant Numbers JP26293097 and JP19H03182 (to T.M.), JP18K14638 and JP20K15749 (to M.K.), JP15H05593 and JP18K06159 (to Y.V.M.) and JP25000013 (to K.N.) and MEXT KAKENHI Grant Numbers JP15H01640 and JP20H05532 (to T.M.) and JP26115720 and JP15H01335 (to Y.V.M). This work has also been partially supported by JEOL YOKOGUSHI Research Alliance Laboratories of Osaka University to K.N.
Author information
Authors and Affiliations
Contributions
T.M. and K.N. conceived and designed research; T.M., M.K., and Y.V.M. performed experiments; T.M., M.K., and Y.V.M. analyzed the data, and T.M. and K.N. wrote the paper based on discussion with other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Minamino, T., Kinoshita, M., Morimoto, Y.V. et al. The FlgN chaperone activates the Na+-driven engine of the Salmonella flagellar protein export apparatus. Commun Biol 4, 335 (2021). https://doi.org/10.1038/s42003-021-01865-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-01865-0
This article is cited by
-
FliH and FliI help FlhA bring strict order to flagellar protein export in Salmonella
Communications Biology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
