
Abstract
Deficiency of the immune checkpoint lymphocyte activation gene-3 (LAG3) protein is significantly associated with both elevated HDL-cholesterol (HDL-C) and myocardial infarction risk. We determined the association of genetic variants within ±500 kb of LAG3 with plasma LAG3 and defined LAG3-associated plasma proteins with HDL-C and clinical outcomes. Whole genome sequencing and plasma proteomics were obtained from the Multi-Ethnic Study of Atherosclerosis (MESA) and the Framingham Heart Study (FHS) cohorts as part of the Trans-Omics for Precision Medicine program. In situ Hi-C chromatin capture was performed in EBV-transformed cell lines isolated from four MESA participants. Genetic association analyses were performed in MESA using multivariate regression models, with validation in FHS. A LAG3-associated protein network was tested for association with HDL-C, coronary heart disease, and all-cause mortality. We identify an association between the LAG3 rs3782735 variant and plasma LAG3 protein. Proteomics analysis reveals 183 proteins significantly associated with LAG3 with four proteins associated with HDL-C. Four proteins discovered for association with all-cause mortality in FHS shows nominal associations in MESA. Chromatin capture analysis reveals significant cis interactions between LAG3 and C1S, LRIG3, TNFRSF1A, and trans interactions between LAG3 and B2M. A LAG3-associated protein network has significant associations with HDL-C and mortality.
Similar content being viewed by others
Introduction
Cardiovascular disease (CVD) remains the leading cause of mortality despite effective low-density lipoprotein cholesterol (LDL-C) lowering therapies1,2,3. In addition to LDL-C, other lipid CVD risk factors include high-density lipoprotein cholesterol (HDL-C) and triglycerides4,5,6. The causal role of LDL-C in atherosclerosis is now established while that for HDL-C per se is in doubt7,8,9,10,11,12.
A recent review on the topic of high HDL-C paradox examined findings from two large observational studies (Cardiovascular Health in Ambulatory Care Research Team [CANHEART] and Copenhagen City Heart Study and the Copenhagen General Population Study [Copenhagen Heart Studies]) that identified a U-shaped relationship between mortality and HDL-C showing increased mortality risk with low HDL-C but also with high HDL-C levels13. Voight et al12. showed that in a mendelian randomization study, using either a single nucleotide polymorphism (SNP) for the endothelial lipase gene (LIPG) or a genetic score encompassing fourteen common SNPs, high HDL-C levels did not predict lower myocardial infarction (MI) risk. The study concluded that selected genetic variants associated with higher HDL-C levels were not causally associated with MI risk. There have now been a number of randomized clinical trials showing that pharmacologic inhibition of cholesteryl ester transfer protein did not reduce or minimally reduced MI risk despite the effect of significantly raising HDL-C levels from baseline values7,10,14.
Candidate gene approaches have shown that common and rare genetic variants within the HDL receptor, scavenger receptor class B type I (SCARB1) gene, are significantly associated with increased CVD risk, contributing to the concept of high HDL-C paradox15,16,17,18,19. In CARDIoGRAM, a common variant within SCARB1, rs10846744 with effect allele C that resides within an enhancer region in the first intron of the gene, is significantly associated with prevalent CVD20. A number of experimental approaches have been used to examine the effects of rs10846744 on distally regulating neighboring genes on chromosome 12 (wherein SCARB1 is located) with a novel physical interaction between SCARB1 and the immune checkpoint molecule lymphocyte activation gene-3 (LAG3 gene) and effects on the LAG3 protein having been shown15.
LAG3 is a member of the IgG superfamily and is an important immune checkpoint molecule in regulating further activation of T effector cells21. The prevailing paradigm is that the extracellular domain of LAG3 on T cells binds with high avidity to a select region on MHC-II molecules on antigen presenting cells to suppress further activation of T cells and regulate T cell homeostasis22. In humans, the LAG3 gene resides on the short arm of chromosome 12 (12p13.32) and is within 8.4 kB of CD423. LAG3 is expressed in B cells, T cells, NK lymphocytes, monocytes, and dendritic cells and its distribution is approximately 50% intracellular and 50% on the cell surface24,25,26. Activation of these cells promotes transit of intracellular LAG3 to the cell surface, where extracellular LAG3 is then subject to cleavage by ADAM10 and ADAM17 metalloproteases, resulting in soluble LAG3 (sLAG3)27. In addition to transmembrane LAG3 binding to MHC class II to limit effector T cell expansion, in vitro studies have demonstrated that sLAG3 also binds to MHC class II and regulates CD4-driven signaling pathways28. A subset of Tregs (alternative Tr1 Tregs) has been characterized using flow cytometric LAG3 expression, with these cells being a major source of secretion of the immunosuppressive interleukin 10 (IL-10) cytokine29,30,31,32. Zhu et al.33 observed that in patients with documented coronary artery disease there was a significantly lower expression by flow cytometry of these CD49b+ LAG3+ Tr1 Tregs cells compared with control subjects. Our results and that of Zhu et al33. are consistent that humans with LAG3 deficiency are at increased risk for CVD and have lower circulating IL-10 levels.
In participants of the Multi-Ethnic Study of Atherosclerosis (MESA), we observed that deficiency of plasma or soluble LAG3 protein was significantly associated with elevated HDL-C levels, and paradoxically with increased risk of CVD15. We also reported that age, smoking, lipid medications, and the SCARB1 rs10846744 were independent predictors of plasma LAG3 levels15. In MESA and the Framingham Heart Study (FHS), we have now directly examined the region surrounding the LAG3 locus to identify common and rare variants associated with plasma LAG3 and HDL-C concentrations, and with clinical outcomes. Additionally, in adjusted models, we examined the association of LAG3 co-expressed proteins and their association with HDL-C, coronary heart disease (CHD) and all-cause mortality. We have now identified a common noncoding variant within the LAG3 gene that is significantly associated with plasma LAG3, and a number of LAG3 co-expressed proteins that are significantly associated with LAG3 protein, HDL-C, and all-cause mortality.
Results
Characteristics of the MESA and FHS participants
The median baseline age of participants from MESA (Exam 1) was 61 years (with an interquartile range of 53.0–69.0 years); 50.7 % were women; and self-reported race/ethnic distribution was 41.5% White, 23.2% Hispanic, 22.8% African-American, and 12.5% Chinese-American (Table 1). In FHS (Offspring Exam 5), the median age of participants was 55 years (with an interquartile range of 47–63 years); 53.5% were women; and were 100% White participants (Table 2).
Genetic associations with phenotypic traits
Discovery study in MESA
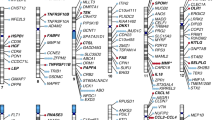
Meta-analysis identified one significant genetic variant within the LAG3 region at FDR < 0.05 (based on a Z-test from meta-analysis for the regression coefficient; Table 3, Fig. 1). The common LAG3 variant rs3782735 (chr12:6775910) effect allele G was positively associated with LAG3 protein levels (Beta = 0.24 for the additive genetic effect on inverse normal transformed protein level; 95% confidence interval [CI] = 0.14–0.34; Z-stat = 4.8; P-value = 4.28 × 10−6; FDR = 0.014) at Exam 1. In race/ethnic-stratified analysis, rs3782735 showed statistically significant association in the larger White group only (FDR = 0.003) (associations of rs3782735 with other phenotypic traits shown in Supplementary Data 1). The common C1S variant rs7970720 (chr12:7048232) effect allele G was negatively associated with peripheral vascular disease as measured by ABI at Exam 5 (FDR = 0.016) (Supplementary Fig. 1). In race/ethnic-stratified analysis, rs7970720 was statistically significant in the larger White group only (FDR = 0.0004).
Validation study in FHS
In FHS, rs3782735 was significantly associated with LAG3 protein levels (Beta = 0.105 for the additive genetic effect on log-transformed protein level; 95% CI = 0.018-0.192; t-stat = 2.38; P-value = 0.018 based on a two-sided t-test for the coefficient from regression with n = 1007), but rs7970720 did not demonstrate association with ABI (P-value > 0.05).
Discovery study in FHS
In the FHS cohort, we did not identify any SNPs that were significantly associated with LAG3 protein levels after adjusting for multiple testing (FDR < 0.05 based on a two-sided t-test for each coefficient from regression with n = 1007). Thus, there was no follow-up of FHS-based findings in MESA (Fig. 2, validation study 2).

The plot presents results for the index variant rs3782735 at chr12:6775910 +/- 250 kb, with linkage disequilibrium determined using the multi-ethnic TOPMed WGS data from MESA.

Abbreviations: Afr. Amer. = African American; HC = heterozygosity count; FDR = False discovery rate.
LAG3 protein association analysis
Discovery study in MESA and validation in FHS
Next, we examined the extent of plasma LAG3-associated protein networks by performing a multivariate linear regression analysis with LAG3 as the outcome. The model examined association of each protein measure with LAG3 under covariate adjustment for age, sex, and PCs of ancestry. LAG3 protein levels in MESA were associated with 669 other protein measures at Exam 1 and 968 proteins at Exam 5 (FDR < 0.05 based on a two-sided t-test for the coefficients from regression with n = 938 and n = 929 at MESA Exams 1 and 5, respectively). Among these, 603 proteins were identified at FDR < 0.05 at both MESA Exams 1 and 5 (Supplementary Data 2), and these proteins were carried forward for validation in FHS. Among these 603 protein measures, 254 demonstrated significant association with LAG3 protein levels in FHS after adjusting for multiple testing (P < 0.05/603 = 8.3 × 10−5 based on a two-sided t-test for the coefficient from regression with n = 1913; Supplementary Data 3).
Discovery study in FHS and validation in MESA
We identified 657 protein measures that were associated with LAG3 protein levels in the FHS proteomics data (FDR < 0.05 based on a two-sided t-test for the regression coefficient; Supplementary Data 4). Among them, 287 and 431 demonstrated significant associations with LAG3 protein levels in MESA Exams 1 and 5, respectively, after adjusting for multiple testing (P < 0.05/657 = 7.6 × 10−5 based on a two-sided t-test for the regression coefficient). Among the associations identified for Exams 1 or 5, 261 of the identified protein measures overlapped with Bonferroni-corrected validation evidence at both MESA Exams 1 and 5 (Supplementary Data 5). Of these, 131 proteins were negatively associated with LAG3 while 130 were positively associated with LAG3.
Overlap of proteins discovered and validated in both MESA and FHS
We identified 183 LAG3 associated proteins that overlapped among the validated lists of associated proteins in FHS, MESA Exam 1, and MESA Exam 5 (Supplementary Data 6). Pathway analysis of the 183 overlapped proteins using GeneAnalyticsTM algorithms revealed sixty-four significant high disease score matches with malignancy, autoimmune disease, neurological disorders, and vascular disease and one hundred fifty-one high scores in the innate immune system pathway (Supplementary Data 7).
LAG3 protein network and association with HDL-C
Since we previously observed a significant inverse association between plasma LAG3 and HDL-C15, we next examined the association of the LAG3 protein network with HDL-C. Of the overlapped 183 LAG3-associated proteins (Supplementary Data 6), thirteen demonstrated associations with HDL-C in MESA with FDR < 0.05 at both MESA Exams 1 and 5 (based on a two-sided t-test for the coefficients from regression with n = 787 and n = 927 at MESA Exams 1 and 5, respectively; Supplementary Data 8). Among these thirteen HDL-C associated proteins in MESA, six of them demonstrated Bonferroni significant validation in FHS (P < 0.05/13 based on a two-sided t-test for the coefficient from regression with n = 1907, Supplementary Data 9). In FHS, 88 of the overlapped 183 LAG3 associated proteins showed associations with HDL-C at FDR < 0.05 based on a two-sided t-test for each coefficient from regression (Supplementary Data 10), and six of these further showed Bonferroni corrected validation in MESA (P < 0.05/88) at both Exams 1 and 5 (Supplementary Data 11). Among the 183 proteins examined for association with HDL-C in both MESA and FHS, four of them were discovered and validated in both cohorts (leucine rich repeats and immunoglobulin like domains 3 [LRIG3], DNV family receptor alpha 1 [GFRA1], insulin like growth factor 1 receptor [IGF1R] and DCTP pyrophosphatase 1 [DCTPP1]; Fig. 3a).

Plots show estimated effects and 95% confidence limits. Analyses were performed with inverse normal transformed protein levels in MESA and log-transformed protein levels in FHS. a Association of HDL-C with protein levels was examined by linear regression with covariate adjustment for age, sex, study site (in MESA), race/ethnicity (in MESA), PCs of ancestry, BMI, triglycerides, pack-years of smoking (in MESA), current smoking (in FHS), current alcohol use, LAG3 protein level, plate ID and batch (in FHS). Among the 183 proteins examined for association with HDL-C in both MESA and FHS, four of them were discovered and validated in both cohorts (leucine rich repeats and immunoglobulin like domains 3 [LRIG3], DNV family receptor alpha 1 [GFRA1], insulin like growth factor 1 receptor [IGF1R] and DCTP pyrophosphatase 1 [DCTPP1]). b Of the 183 proteins examined for association with CHD and all-cause mortality in MESA, we did not observe any results at FDR < 0.05. In FHS, while there were no associations at FDR < 0.05 for CHD, we observed that 18 of the 183 LAG3-associated proteins demonstrating FDR < 0.05 were significantly associated with all-cause mortality based on two-sided Z-tests for the coefficients from Cox regression with a total sample of n = 1913, including 650 events (Supplementary Data 12). In MESA, none of these 18 proteins reached Bonferroni-corrected statistical significance, but four of the 18 proteins showed nominal associations with all-cause mortality (tumor necrosis factor receptor super family 1 A [TNFRSF1A], beta-2-microglobulin [B2M], tumor necrosis factor receptor super family 1B [TNFRSF1B] and cystatin C [CST3]; all P < 0.05 based on two-sided Z-tests for the coefficients from Cox regression with n = 935 including 95 events. Association of baseline protein levels with all-cause mortality was examined under a Cox proportional hazards model with covariate adjustment for age, sex, race/ethnicity (in MESA), PCs of ancestry, BMI, total cholesterol, HDL-C, SBP, DBP, pack-years of smoking (in MESA), current smoking status (in FHS) and batch (in FHS).
LAG3 protein network and association with clinical outcomes (coronary heart disease and all-cause mortality)
Of the 183 proteins examined for association with CHD and all-cause mortality in MESA, we did not observe any results at FDR < 0.05. In FHS, while there were no associations at FDR < 0.05 for CHD, we observed that 18 of the 183 LAG3-associated proteins demonstrating FDR < 0.05 were significantly associated with all-cause mortality based on two-sided Z-tests for the coefficients from Cox regression with a total sample of n = 1913, including 650 events (Supplementary Data 12). In MESA, none of these 18 proteins reached Bonferroni-corrected statistical significance, but four of the 18 proteins showed nominal associations with all-cause mortality (tumor necrosis factor receptor super family 1 A [TNFRSF1A], beta-2-microglobulin [B2M], tumor necrosis factor receptor super family 1B [TNFRSF1B] and cystatin C [CST3]; all P < 0.05 based on two-sided Z-tests for the coefficients from Cox regression with n = 935 including 95 events; Fig. 3b and Supplementary Data 13).
Chromatin capture interactions between LAG3 and loci associated with HDL-C and all-cause mortality
In a hypothesis seeking effort, we performed in situ Hi-C chromatin capture to examine possible chromatin high frequency interactions between LAG3 (chr12: 6772483) and the loci associated with HDL-C (LRIG3, GFRA1, IGF1R, and DCTPP1) and all-cause mortality (TNFRSF1A, TNFRSF1B, B2M, and CST3). As a representative example of the Hi-C assays, results from an African-American female MESA participant homozygous for the SCARB1 rs10846744 effect C allele showed significant cis interactions between LAG3 and LRIG3 (chr12: 58872149; interaction score [IS] = 17.4; n = 22 read pairs; P < 0.0001 based on two-sided Student t-test) (Fig. 4). No significant trans interactions were observed between LAG3 and GFRA1 (chr10:116056925), IGF1R (chr15:98648539) or DCTPP1 (chr16: 30423615).

We performed in situ Hi-C analysis in EBV-transformed B lymphoblasts from two female (one African-American and one Hispanic) MESA carriers homozygous for the SCARB1 rs10846744 reference G and two female (one African-American and one Hispanic) MESA carriers homozygous for the effect C alleles. The in situ Hi-C analysis was performed as recommended by the 4D Nucleome Consortium using the four base pair cutter DpnII restriction enzyme and high read depth next gen sequencing (NGS) to maximize resolution of the high frequency interactions between chromatin contacts (https://www.4dnucleome.org/protocols.html). Each cell library underwent deep NGS at read depths between 1.4–3.3 billion and this was done independently twice as technical replicates for each cell library. Bioinformatic analysis was conducted using Hi-C Pro software with binning of the matrix at different resolutions and iterative correction and eigenvector decompensation normalization of the matrix for each of the four libraries62,63. Readouts were all valid paired-end reads and corresponding high frequency contact interaction scores. We then used data generated from the million binning resolution and filtered it based on the LAG3 chromosomal coordinates (chr12:677250-6778455) using human assembly GRCh38/hg38 (https://genome.ucsc.edu/cgi-bin/hgGateway), which yielded both cis (chr12) and trans interactions. We set interaction scores at the LAG3 locus arbitrarily at 1 and then compared interaction scores from direct and indirect cis and trans interactions. Student t-test was performed with P ≤ 0.05 considered statistically significant. The results shown are representative from one of the MESA cell lines, with the schematic representing the near cis (panel a) and cis interactions (panel b) from the LAG3 locus on chromosome 12.
For loci associated with all-cause mortality, significant cis interactions were observed between LAG3 and TNFRSF1A (chr12: 6328757, IS = 3308; n = 13932 read pairs; P < 0.0001 based on two-sided Student t-test). Significant trans interactions were observed between LAG3 and B2M (chr15: 44711487, IS = 11.95; n = 23 read pairs; P < 0.0001 based on two-sided Student t-test), but not with TNFRSF1B (chr1: 12166948) or CST3 (chr20: 23626706).
Discussion
LAG3 is an important immune checkpoint molecule with continued interest in its role as an immuno-oncology therapeutic agent34. We were the first to report the association of LAG3 protein deficiency with HDL-C and increased risk of MI, and had identified independent predictors for plasma LAG3 protein15. In the present study, we examined the association of variants within ±500 kb of the LAG3 locus in TOPMed MESA and FHS to identify those significantly associated with plasma LAG3 protein concentrations and clinical outcomes. We identified the LAG3 rs3782735 variant as significantly associated with plasma LAG3 protein levels, and neighboring C1S rs7970720 variant as significantly associated with peripheral vascular disease (ABI) in the MESA population (Supplementary Fig. S1). The LAG3 rs3782735 is an intronic variant that appears to reside within an enhancer region, has been associated with multiple myeloma risk in women, but has not yet been associated with lipids, CHD or mortality risk35.
We next examined the association of LAG3 with plasma proteins in the TOPMed SOMAscan proteomics datasets available in MESA and FHS. We identified 183 LAG3-associated proteins between MESA and FHS, with pathway analysis revealing sixty-four high disease matches in malignancy, autoimmune diseases, neurological disorders, vascular diseases including MI (AKT1, CCL5, CD163, CDH5, CST3, CXCL12, ENG, IL6R, LTA, PF4, PPBP, PSMA6, SELL, TEK, THBS2, TLR4, TNFRSF1A, VCAM1) and heart disease (ANGPT2, CMA1, CST3, CXCL12, DSCAM, ENG, JAG1, NOTCH1, PF4, PPBP, TLR4, TNFRSF1A, TNFRSF1B, VCAM1). Under pathway analysis, we identified high score matches for fluid shear stress and atherosclerosis (AKT3, AKT1, PDGFB, AKT2, BCL2, CDH5, VCAM1, PDGFA, TNFRSF1A). Additionally, when we performed pathway analysis for proteins positively associated with LAG3 we found high score matches for pigment epithelium-derived factor (PEDF) induced signaling while negatively associated proteins had high score matches for innate immune system pathways. These significant LAG3-associated proteins identified in both MESA and FHS provide a framework to examine this network in mediating various inflammatory and non-inflammatory diseases.
Given that plasma LAG3 was shown to be inversely associated with HDL-C, we examined the association of the 183 LAG3-associated overlapping proteins in MESA and FHS with HDL-C using adjusted models as shown in Fig. 3. In MESA and FHS, GFRA1 showed an inverse association with HDL-C, while LRIG3, IGF1R, and DCTPP1 were all positively associated with HDL-C. A PubMed literature search did not reveal a known connection between these four proteins and LAG3 or connections with HDL-C except for LRIG3, suggesting novel findings of these proteins with lipid metabolism. LRIG3 has been shown to be associated with HDL-C in both humans and animals36,37,38. In particular, FHS investigators examined single-locus and epistasis variants on HDL-C and identified a region near LRIG3 as being significant36. In another study in African-American subjects, the region near LRIG3 was found to be significantly associated with incident congestive heart failure (CHF)37. In Lrig3−/− mice, investigators showed that aging mice demonstrated cardiac hypertrophy and low plasma HDL-C levels38.
In MESA plasma LAG3 was shown to be inversely associated with increased MI risk15. Therefore, we examined the association of the overlapping 183 proteins with CHD and all-cause mortality in MESA and FHS as shown in Fig. 3. In MESA, there were significant associations of EFNA5, ENG, IL1R1, and NRCAM with CHD risk but the number of events were extremely small and thus caution was taken in interpreting these results. In MESA, we did not observe significant association with all-cause mortality. In FHS, there were 18 proteins significantly associated with all-cause mortality, with 4 proteins nominally associated with mortality in MESA. Abers et al39. recently reported the significant independent association of soluble TNFRSF1A with mortality in patients hospitalized with COVID-19. Schnabel et al40. reported that TNFRSF1B (also known as TNFR2) was significantly associated with cardiovascular disease and mortality in FHS. Zaghlool et al41. performed a step-wise epigenome-wide association analysis with plasma proteomics measured by SOMAscan in two cohorts (Cooperative Health Research in the Region of Augsburg and Qatar Metabolomics Study on Diabetes), and identified nine association pathways. One of these was identified as a chronic inflammatory pathway represented by NLRC5 and included LAG3, B2M, CD48, CXCL10, FCGR3B, CD163, and CXCL11. We have now identified a LAG3-association network with all-cause mortality that includes B2M (a component of the class I major histocompatibility complex molecule involved in antigen presentation42) and CD48 (Supplementary Data 12). We also identified that LAG3 and B2M have significant trans chromatin interactions, which is consistent with the trans contacts between NLRC5 and LAG3 and B2M reported by Zaghlool et al41. Also of importance, these authors identified an inverse association of LAG3 with HDL-C, confirming our initial observation.
Pioneers in the field of 3D chromatin architecture have developed and continue to refine methodologies that evaluate the effect of cis and trans gene-gene interactions on downstream gene regulation43,44,45,46. We used Hi-C assays to evaluate unbiased global chromatin interactions (many-to-many) without immunoprecipitation followed by high depth NGS. We then used a number of bioinformatic programs to assess the quality of the technical and biological replicates in order to evaluate the chromatin interactions from the LAG3 locus47,48,49. In particular, HiCcompare allows statistical analysis of technical and biological replicates for cis interactions, while trans interactions can be challenging50. We observed that LAG3 has strong chromatin interactions with C1S, LRIG3, and TNFSRSF1A, all in close proximity to the LAG3 locus, which is consistent with observations that chromatin contact interactions are strongest with small differences in genomic distance. While we did observe significant LAG3 chromatin interactions with trans contacts, we want to proceed with caution as the absolute value of the interaction scores were low as well as the number of read pairs. It could be possible there are biological effects with the LAG3 trans chromatin contacts but we acknowledge there are post-translational processes that more likely influence LAG3 protein interactions. Nonetheless, the significant trans interactions lends support to the hypothesis that trans chromatin contacts could explain some of the variance in the LAG3-associated protein network.
In conclusion, we have identified a common LAG3 variant that is associated with plasma LAG3 protein levels, while adjacent genes in significant chromatin contact with the LAG3 locus were associated with HDL-C (LRIG3) and clinical outcomes such as all-cause mortality (TNFRSFA1 and B2M). This LAG3-associated network analysis identifies novel targets for further study in HDL metabolism, cardiovascular diseases, and all-cause mortality.
Methods
Trans-omics for precision medicine (TOPMed)
This study was approved by the TOPMed Publications & Presentations Steering Committees with data access provided by an approved project (#21279) through a database of Genotypes and Phenotypes (dbGaP) application. The study cohorts that comprise this analysis include MESA and FHS.
Study participants
MESA is a longitudinal study of subclinical CVD and risk factors that predict progression to clinically overt CVD or progression of the subclinical disease. The first clinic visits (Exam 1) occurred from 2000 to 2002 in 6,814 participants recruited from 6 field centers across the United States, and all participants were free of clinical CVD at Exam 1. The self-reported ancestry distribution is approximately 38% White, 28% African-American, 22% Hispanic, and 12% Asian (predominantly of Chinese descent)51. MESA has been enhanced by many ancillary studies focused on specific phenotypic and exposure domains. One ancillary study (MESA Family Study) exclusively recruited African-American and Hispanic family members specifically for genetic studies. In contrast, the FHS is a single community-based cohort initiated in 1948. Three generations of participants have been recruited, and the majority of them were white individuals of European ancestry. Participants were invited to attend physical examinations every 2–8 years. The current study was restricted to the Offspring cohort participants who attended their fifth clinical examination cycle during 1991–199552.
All MESA participants provided written informed consent for participation at the six field sites, and FHS participants provided written informed consent for participation at the single site in Framingham, MA. MESA and FHS study protocols were both reviewed and approved by the Institutional Review Boards (IRBs) at each of the participating study sites, as well as at the University of Virginia (AM), Boston University Medical Campus (HL) and the University of Connecticut Health (AR).
Genotype data
We used the TOPMed Freeze 8 whole genome sequencing (WGS) data, focused on the region within ±500 kb of the LAG3 gene for examination of our genotype data. TOPMed WGS was conducted at a mean depth of >30X using Illumina HiSeq X Ten instruments. Variant discovery and genotype calling was performed jointly across all studies by the TOPMed Informatics Research Center (IRC) using the GotCloud pipeline. Variant quality control (QC) was performed by the IRC using support vector machine filtering to identify high quality variants. WGS sample QC including sample identity and consent checks was performed by the TOPMed Data Coordinating Center.
Proteomic data
For MESA Exam 2 (n = 5623), LAG3 protein ELISA kits were purchased from RayBiotech, Inc. (Norcross, GA) and LAG3 was measured per the manufacturer’s instructions15. Briefly, aliquots of fasting plasma samples stored at −80 °C were thawed, diluted 3-fold, and then 100 µl were used for duplicates per sample for plasma LAG3 measurement. The intra-assay coefficient of variance was <10% and inter-assay CV < 12%. The R2 for the standard curve, which was run for each plate, had mean values >0.98. This ELISA has not been validated for clinical use. The single-stranded DNA aptamer-based SOMAscan™ proteomics platform was used to assay plasma samples from MESA Exams 1 (years 2000–2002) and 5 (years 2010–2012), and from FHS Exam 5 (years 1991–1995). All MESA samples were profiled with Version 1.3k. FHS samples were either profiled with Version 1.1k with 1124 aptamers or with Version 1.3k with 1305 aptamers. All assays were performed using SOMAscan reagents according to the manufacturer’s detailed protocol53. The strength of this platform is its ability to measure multiple proteins in a single small aliquot. As reported by SOMALogic, the assay measures >1300 proteins from a small biological sample (150 µl) with low limits of detection and 5% median coefficient of variation53,54. This technology uses DNA aptamers (short single-stranded oligonucleotides) that bind to proteins and is quantified by hybridization to custom DNA microarrays. The units are reported as Relative Fluorescent Units (RFU) that are directly proportional to the amount of target protein.
Phenotype data
Phenotypes of this study were obtained from MESA Exam 1, Exam 2 (years 2002–2004), and Exam 5. The MESA Family Study (years 2004–2006) examination data and event data were combined with MESA Exam 1 data. Fasting blood samples were drawn and processed using a standardized protocol and sent for measurement of lipid levels55. LDL-C was calculated using the Friedewald formula. Triglycerides were measured using a glycerol-blanked enzymatic method with the Triglyceride GB reagent on the Roche COBAS FARA centrifugal analyzer. Measures of subclinical atherosclerosis examined included the presence (defined by a cutoff of coronary artery calcium [CAC] > 0) or absence of CAC, calcium Agatston score (phantom-adjusted) and ultrasound measurements of intima-media thickness (IMT) in millimeters (mm) for common and internal carotid IMT, and the ankle brachial index (ABI)56. Systolic blood pressure (SBP) measurements in the bilateral brachial, dorsalis pedis, and posterior tibial arteries were obtained in the supine position using a hand-held Doppler instrument with a 5-mHz probe. Cardiovascular events including stroke, MI, CHD and all-cause mortality were adjudicated by a MESA committee of neurologists, cardiologists, and physician epidemiologists, who provided a detailed description of the cardiovascular event adjudication process57. In FHS, the Friedewald formula was used to calculate LDL-C, and a modification of the Kessler-Lederer method was used to measure triglyceride levels58,59.
Phenotypes for genetic association
In MESA, eleven phenotypes were used for genetic association study analyses: plasma LAG3 protein levels as measured by ELISA (Exam 2) and SOMAscan proteomic assay (Exams 1 and 5), HDL-C, LDL-C, triglycerides, CAC presence/absence, Agatston calcium score (among those with presence of CAC), common carotid IMT, internal carotid IMT, ABI, stroke, and MI. All of the phenotypes were examined as quantitative traits, except for CAC (presence/absence), stroke and MI which were examined as dichotomous traits. We performed inverse normal transformations for LAG3 protein levels, and performed natural-log transformation for Agatston calcium score, common carotid IMT, and internal carotid IMT due to non-normal distribution of these outcome traits. Genetic analysis of clinical events in MESA was carried out based on MESA adjudication through the year 2017.
Genetic association analysis in MESA
We approached discovery for genetic association analyses in MESA in two ways (Fig. 1). First, we stratified data by race/ethnic groups, and performed genetic association analyses in each race/ethnic group using EPACTS software for quantitative phenotypes (https://github.com/statgen/EPACTS) and SNPTEST software for dichotomous outcome traits60. Quantitative trait analysis was conducted including all participants with available genotype and phenotype data, and family structure was accounted using linear mixed models in EPACTS, while the dichotomous trait analysis was conducted for a subset of participants with first-degree relatives removed. We then meta-analyzed the genetic association results across all four race/ethnic groups by fixed effects model using METAL61. Second, we combined all four MESA race/ethnic groups into one simple pooling group without weighting and performed genetic association analyses using EPACTS and SNPTEST. Analyses were conducted across MESA Exams 1, 2 and 5 due to the availability of LAG3 protein measurements at these examination times (Table 1).
The association of genetic variants with eleven phenotypes was conducted with adjustment for some of the following covariates: age, sex, study site, principal components (PCs) of ancestry (2 PCs for White, 1 PC for Chinese, 1 for African-American, and 3 for Hispanic, and 5 PCs for race/ethnic pooled analyses), self-reported race/ethnicity (pooled-group analysis only), HDL-C, LDL-C, triglycerides, body mass index (BMI), fasting glucose, SBP, diastolic blood pressure (DBP), current smoking, former smoking, and lipid medication use. Covariate adjustment for all lipid variables (HDL-C, LDL-C, and triglycerides) was omitted in the analysis of these three traits as phenotypic outcomes.
In genetic association analysis, we excluded variants with heterozygosity count (HC) less than 30 overall (for quantitative traits) or HC less than 30 among cases (for dichotomous traits). We then applied the false discovery rate (FDR) method to control for inflation of the type I errors due to multiple comparisons. Variants with FDR < 0.05 were considered statistically significant in this study.
Validation study
We performed validation analyses for genetic association analysis of LAG3 protein levels in two ways (Fig. 1). First, we selected significant variants from genetic association analysis/meta-analysis results of MESA (discovery) and investigated the association of these variants in FHS (validation) that mainly focuses on white individuals of European ancestry. Second, we selected significant variants from FHS (discovery) and verified if they were also significant in MESA (validation). Genetic association analysis in FHS followed the same approach as that used for pooled analysis in MESA, with the exception that analysis in FHS did not require covariate adjustment for race/ethnicity and study site; as noted for MESA, the linear mixed models implemented in EPACTS do account for family relationships that are present in the FHS data.
LAG3 protein association analysis
To identify proteins associated with LAG3, we performed linear regression analysis using inverse normal transformed protein levels (in MESA) and log-transformed protein levels (in FHS) for LAG3 levels as an independent variable and the individual proteins as dependent variables. These regression analyses were carried out separately in MESA Exam 1, MESA Exam 5, and FHS Offspring Exam 5 using SOMAscan proteomic measurements. In MESA, regression analyses were carried out for an unrelated subset of participants using linear regression, while in FHS related individuals were included and familial relationship adjusted using linear mixed models. LAG3 protein association analysis included covariate adjustment for age, sex, study site (in MESA), race/ethnicity (in MESA), PCs of ancestry, pack-years of smoking (in MESA), current smoking (in FHS), lipid medication, plate ID and batch (in FHS). In FHS, regression analyses did not require adjustment for race/ethnicity and study site as all of the FHS participants were white and from a single study site. In MESA, all proteomics tests were carried out in a single batch, so analyses did not require adjustment for batch. Independent discovery and validation of LAG3-associated proteins in MESA and FHS proceeded following the approach described for genetic association analysis of LAG3 protein levels (Fig. 1).
For a set of 183 LAG3-associated proteins identified by discovery and supported by validation in both MESA and FHS, we further carried out regression analyses to examine their association with HDL-C, CHD and all-cause mortality. Protein association was conducted using adjudicated events in MESA through the year 2017. In MESA, we examined associations with inverse normal transformed protein levels, while in FHS, we used log-transformed protein levels. Association of HDL-C with protein levels was examined by linear regression with covariate adjustment for age, sex, study site (in MESA), race/ethnicity (in MESA), PCs of ancestry, BMI, triglycerides, pack-years of smoking (in MESA), current smoking (in FHS), current alcohol use, LAG3 protein level, plate ID and batch (in FHS). Association of baseline protein levels with CHD and all-cause mortality was examined under a Cox proportional hazards model with covariate adjustment for age, sex, race/ethnicity (in MESA), PCs of ancestry, BMI, total cholesterol, HDL-C, SBP, DBP, pack-years of smoking (in MESA) and current smoking status (in FHS) and batch (in FHS).
Chromatin capture analysis
We performed in situ Hi-C analysis in EBV-transformed B lymphoblasts from two female (one African-American and one Hispanic) MESA carriers homozygous for the SCARB1 rs10846744 reference G and two female (one African-American and one Hispanic) MESA carriers homozygous for the effect C alleles. The in situ Hi-C analysis was performed as recommended by the 4D Nucleome Consortium using the four base pair cutter DpnII restriction enzyme and high read depth next gen sequencing (NGS) to maximize resolution of the high frequency interactions between chromatin contacts (https://www.4dnucleome.org/protocols.html). Each cell library underwent deep NGS at read depths between 1.4–3.3 billion and this was done independently twice as technical replicates for each cell library. Bioinformatic analysis was conducted using Hi-C Pro software with binning of the matrix at different resolutions and iterative correction and eigenvector decompensation normalization of the matrix for each of the four libraries62,63. Readouts were all valid paired-end reads and corresponding high frequency contact interaction scores. We first used HiCcompare to evaluate the quality of the technical replicates of each library50. We then used data generated from the million binning resolution and filtered it based on the LAG3 chromosomal coordinates (chr12:677250–6778455) using human assembly GRCh38/hg38 (https://genome.ucsc.edu/cgi-bin/hgGateway), which yielded both cis (chr12) and trans interactions. We set interaction scores at the LAG3 locus arbitrarily at 1 and then compared interaction scores from direct and indirect cis and trans interactions. Student t-test was performed with P ≤ 0.05 considered statistically significant.
GeneAnalytics LAG3 protein network analysis
We inputted the protein list generated from the LAG3 protein association analyses into GeneAnalytics (GA) (https://ga.genecards.org/#input). Based on GA proprietary algorithms, scores that were identified as high (adjusted p ≤ 0.0001) were analyzed for the LAG3 protein network analysis.
Statistics and reproducibility
The sample sizes for the MESA population was 3867 and for FHS study it was 1913. Multivariate linear regression analyses were performed using fully adjusted models and FDR ≤ 0.05 was considered significant. For the HiC chromatin capture assays, two-sided Student’s t-test was performed and P ≤ 0.05 was considered significant. Statistical software programs used were JMP v15 or R.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data access for MESA (phs001416) and FHS (phs000974) was approved by the TOPMed Publications & Presentations Steering Committees with data access provided by an approved project (#21279) through a database of Genotypes and Phenotypes (dbGaP) application. Hi-C chromatin capture data are available from dbGAP by using MESA phs000209 as the identifier. Interested researchers would have to apply to TOPMed and dbGaP in order to obtain access to these datasets. Source data underlying Fig. 3a, b are presented in Supplementary Data 14–15, respectively.
References
Sidney, S. et al. Comparative trends in heart disease, stroke, and all-cause mortality in the United States and a large integrated healthcare delivery system. Am. J. Med 131, 829–836 e821 (2018).
Casper, M. et al. Changes in the geographic patterns of heart disease mortality in the United States: 1973 to 2010. Circulation 133, 1171–1180 (2016).
Sidney, S. et al. Heterogeneity in national U.S. mortality trends within heart disease subgroups, 2000-2015. BMC Cardiovasc Disord. 17, 192 (2017).
Ridker, P. M. et al. HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: an analysis from the JUPITER trial. Lancet 376, 333–339 (2010).
Handelsman, Y. & Shapiro, M. D. Triglycerides, atherosclerosis, and cardiovascular outcome studies: focus on omega-3 fatty acids. Endocr. Pr. 23, 100–112 (2017).
Lin, F. J. et al. Residual risk factors to predict major adverse cardiovascular events in atherosclerotic cardiovascular disease patients with and without diabetes mellitus. Sci. Rep. 7, 9179 (2017).
Barter, P. J. et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med 357, 2109–2122 (2007).
Ko, D. T. et al. High-density lipoprotein cholesterol and cause-specific mortality in individuals without previous cardiovascular conditions: the CANHEART study. J. Am. Coll. Cardiol. 68, 2073–2083 (2016).
Group, H. T. C. et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med 371, 203–212 (2014).
Group, H. T. R. C. et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N. Engl. J. Med 377, 1217–1227 (2017).
Madsen, C. M., Varbo, A. & Nordestgaard, B. G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur. Heart J. 38, 2478–2486 (2017).
Voight, B. F. et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 380, 572–580 (2012).
Rodriguez, A. High HDL-cholesterol paradox: SCARB1-LAG3-HDL axis. Curr. Atheroscler. Rep. 23, 5 (2021).
Schwartz, G. G. et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med 367, 2089–2099 (2012).
Golden, D. et al. Lymphocyte activation gene 3 and coronary artery disease. JCI Insight 1, e88628 (2016).
Manichaikul, A. et al. Association of SCARB1 variants with subclinical atherosclerosis and incident cardiovascular disease: the multi-ethnic study of atherosclerosis. Arterioscler Thromb. Vasc. Biol. 32, 1991–1999 (2012).
Manichaikul, A. et al. Association of the lipoprotein receptor SCARB1 common missense variant rs4238001 with incident coronary heart disease. PLoS ONE 10, e0125497 (2015).
Naj, A. C. et al. Association of scavenger receptor class B type I polymorphisms with subclinical atherosclerosis: the Multi-Ethnic Study of Atherosclerosis. Circ. Cardiovasc Genet 3, 47–52 (2010).
Zanoni, P. et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351, 1166–1171 (2016).
Grallert, H. et al. Eight genetic loci associated with variation in lipoprotein-associated phospholipase A2 mass and activity and coronary heart disease: meta-analysis of genome-wide association studies from five community-based studies. Eur. Heart J. 33, 238–251 (2012).
Huard, B., Tournier, M., Hercend, T., Triebel, F. & Faure, F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur. J. Immunol. 24, 3216–3221 (1994).
Triebel, F. LAG-3: a regulator of T-cell and DC responses and its use in therapeutic vaccination. Trends Immunol. 24, 619–622 (2003).
Bruniquel, D., Borie, N., Hannier, S. & Triebel, F. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics 48, 116–124 (1998).
Workman, C. J. et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J. Immunol. 182, 1885–1891 (2009).
Workman, C. J. et al. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J. Immunol. 172, 5450–5455 (2004).
Woo, S. R. et al. Differential subcellular localization of the regulatory T-cell protein LAG-3 and the coreceptor CD4. Eur. J. Immunol. 40, 1768–1777 (2010).
Li, N. et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 26, 494–504 (2007).
Subramanyam, M., W., G., Nabioullin, R. & Tepper, M. A. Soluble human lymphocyte activation gene-3 modulates allospecific T cell responses. Int. Immunol. 10, 679–689 (1998).
Ng, T. H. et al. Regulation of adaptive immunity; the role of interleukin-10. Front Immunol. 4, 129 (2013).
Battaglia, M., Gregori, S., Bacchetta, R. & Roncarolo, M. G. Tr1 cells: from discovery to their clinical application. Semin Immunol. 18, 120–127 (2006).
Gagliani, N. et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med 19, 739–746 (2013).
Asseman, C., Leach, M. S., Coffman, M. W., Powrie, R. L. & An, F. essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med 190, 995–1003 (1999).
Zhu, Z. et al. Function of T regulatory type 1 cells is down-regulated and is associated with the clinical presentation of coronary artery disease. Hum. Immunol. 79, 564–570 (2018).
Smylie, M. G. Use of immuno-oncology in melanoma. Curr. Oncol. 27, S51–S58 (2020).
Lee, K. M. et al. Common single nucleotide polymorphisms in immunoregulatory genes and multiple myeloma risk among women in Connecticut. Am. J. Hematol. 85, 560–563 (2010).
Li, M. et al. Genome-wide association analysis of total cholesterol and high-density lipoprotein cholesterol levels using the Framingham Heart Study data. BMC Med. Genet. 11, 55 (2010).
Smith, N. L. et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ. Cardiovasc. Genet. 3, 256–266 (2010).
Hellstrom, M. et al. Cardiac hypertrophy and decreased high-density lipoprotein cholesterol in Lrig3-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 310, R1045–R1052 (2016).
Abers, M. S. et al. An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI Insight 6, https://doi.org/10.1172/jci.insight.144455 (2021).
Schnabel, R. B. et al. Multiple inflammatory biomarkers in relation to cardiovascular events and mortality in the community. Arterioscler. Thromb. Vasc. Biol. 33, 1728–1733 (2013).
Zaghlool, S. B. et al. Epigenetics meets proteomics in an epigenome-wide association study with circulating blood plasma protein traits. Nat. Commun. 11, 15 (2020).
Ploegh, H. L., O., H. & Strominger, J. L. Major histocompatibility antigens: the human (HLA-A, -B, -C) and murine (H-2K, H-2D) class I molecules. Cell 24, 287–299 (1981).
Cope, N. F. & Fraser, P. Chromosome conformation capture. Cold Spring Harb. Protoc. 2009, pdb prot5137 (2009).
Nagano, T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013).
Nagano, T. et al. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat. Protoc. 10, 1986–2003 (2015).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Li, D., Hsu, S., Purushotham, D., Sears, R. L. & Wang, T. WashU epigenome browser update 2019. Nucleic Acids Res 47, W158–W165 (2019).
Stansfield, J. C., Cresswell, K. G., Vladimirov, V. I. & Dozmorov, M. G. HiCcompare: an R-package for joint normalization and comparison of HI-C datasets. BMC Bioinformatics 19, 279 (2018).
Bild, D. E. et al. Multi-ethnic study of atherosclerosis: objectives and design. Am. J. Epidemiol. 156, 871–881 (2002).
Feinleib, M., Kannel, W. B., Garrison, R. J., McNamara, P. M. & Castelli, W. P. The Framingham offspring study. Design and preliminary data. Prev. Med 4, 518–525 (1975).
Gold, L., Walker, J. J., Wilcox, S. K. & Williams, S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. N. Biotechnol. 29, 543–549 (2012).
Gold, L. et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 5, e15004 (2010).
Tsai, M. Y. et al. Cholesteryl ester transfer protein genetic polymorphisms, HDL cholesterol, and subclinical cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 200, 359–367 (2008).
Criqui, M. H. et al. The ankle-brachial index and incident cardiovascular events in the MESA (Multi-Ethnic Study of Atherosclerosis). J. Am. Coll. Cardiol. 56, 1506–1512 (2010).
Yeboah, J. et al. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation 120, 502–509 (2009).
Wilson, P. W., Abbott, R. D., Garrison, R. J. & Castelli, W. P. Estimation of very-low-density lipoprotein cholesterol from data on triglyceride concentration in plasma. Clin. Chem. 27, 2008–2010 (1981).
Nemeth, P. M., Hitchins, O. E., Solanki, L. & Cole, T. G. Fluorometric procedures for measuring triglyceride concentrations in small amounts of tissue and plasma. J. Lipid Res. 27, 447–452 (1988).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet 39, 906–913 (2007).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Pal, K., Forcato, M. & Ferrari, F. Hi-C analysis: from data generation to integration. Biophys. Rev. 11, 67–78 (2019).
Acknowledgements
A. R. was supported by NIH R01HL131862 and an endowment from the Linda and David Roth Chair of Cardiovascular Research. Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for “NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study” (phs000974) was performed at the Broad Institute of MIT and Harvard (HHSN268201500014C and 3R01HL092577-06S1). WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-13S1 and HHSN268201500014C). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I). TOPMed MESA Multi-Omics (HHSN2682015000031/HSN26800004). The MESA and MESA SHARe projects are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for the Multi-Ethnic Study of Atherosclerosis (MESA) projects are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420, UL1TR001881, DK063491, and R01HL105756. MESA Family is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support is provided by grants and contracts R01HL071051, R01HL071205, R01HL071250, R01HL071251, R01HL071258, R01HL071259, UL1TR001881, DK063491, and by the National Center for Research Resources, Grant UL1RR033176. The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutes can be found at http://www.mesa-nhlbi.org. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The Framingham Heart Study (FHS) acknowledges the support of Contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the National Heart, Lung and Blood Institute and grant supplement R01 HL092577-06S1 for this research. We also acknowledge the dedication of the FHS study participants without whom this research would not be possible. R. S. V. is supported in part by the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine.
Author information
Authors and Affiliations
Contributions
Original concept and approval of the paper proposal from TOPMed: A.R. and A.M. Data analysis from MESA and FHS: A.M., H.L., C.K., C.Y. and A.R. First draft of the paper: A.R., A.M. and H.L. Critical review of the data analysis and draft manuscripts: S.S.R., K.D.T., X.G., J.I.R., W.C.J., E.C., R.P.T., J.P.D., Y.L., R.S.V., L.A.C., R.E.G., C.B.C., D.J., M.P.C., T.B., G.J.P., A.R., A.M. and H.L. All authors reviewed all revisions to the manuscript and consented to its submission.
Corresponding author
Ethics declarations
Competing interests
A.R. reports inventorship rights to patents held by University of Connecticut Health and Lipid Genomics. A.R. is the founder of Lipid Genomics. The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Peter Penson, Chew-Kiat Heng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Chiea Chuen Khor, Karli Montague-Cardoso, and George Inglis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Manichaikul, A., Lin, H., Kang, C. et al. Lymphocyte activation gene-3-associated protein networks are associated with HDL-cholesterol and mortality in the Trans-omics for Precision Medicine program. Commun Biol 5, 362 (2022). https://doi.org/10.1038/s42003-022-03304-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03304-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
