
Abstract
Chemokines such as C-C motif ligand 5 (CCL5) regulate immune cell trafficking in the tumor microenvironment (TME) and govern tumor development, making them promising targets for cancer therapy. However, short half-lives and toxic off-target effects limit their application. Oncolytic viruses (OVs) have become attractive therapeutic agents. Here, we generate an oncolytic herpes simplex virus type 1 (oHSV) expressing a secretable single-chain variable fragment of the epidermal growth factor receptor (EGFR) antibody cetuximab linked to CCL5 by an Fc knob-into-hole strategy that produces heterodimers (OV-Cmab-CCL5). OV-Cmab-CCL5 permits continuous production of CCL5 in the TME, as it is redirected to EGFR+ glioblastoma (GBM) tumor cells. OV-Cmab-CCL5 infection of GBM significantly enhances the migration and activation of natural killer cells, macrophages and T cells; inhibits tumor EGFR signaling; reduces tumor size; and prolongs survival of GBM-bearing mice. Collectively, our data demonstrate that OV-Cmab-CCL5 offers a promising approach to improve OV therapy for solid tumors.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others


Data availability
Source data for Figs. 1–8 and Extended Data Figs. 1–10 are provided as Source Data files. All other data supporting the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018).
Ladomersky, E. et al. The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Front. Pharmacol. 10, 200 (2019).
Nduom, E. K., Weller, M. & Heimberger, A. B. Immunosuppressive mechanisms in glioblastoma. Neuro. Oncol. 17, vii9–vii14 (2015).
Ooi, Y. C. et al. The role of regulatory T-cells in glioma immunology. Clin. Neurol. Neurosurg. 119, 125–132 (2014).
Calinescu, A. A. et al. Overview of current immunotherapeutic strategies for glioma. Immunotherapy 7, 1073–1104 (2015).
Perng, P. & Lim, M. Immunosuppressive mechanisms of malignant gliomas: parallels at Non-CNS Sites. Front. Oncol. 5, 153 (2015).
Keophiphath, M., Rouault, C., Divoux, A., Clement, K. & Lacasa, D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler. Thromb. Vasc. Biol. 30, 39–45 (2010).
Khalid, A. et al. Recent advances in discovering the role of CCL5 in metastatic breast cancer. Mini Rev. Med. Chem. 15, 1063–1072 (2015).
Aldinucci, D. & Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014, 292376 (2014).
Dangaj, D. et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 35, 885–900 e810 (2019).
Ma, W., He, H. & Wang, H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 19, 40 (2018).
Friedman, G. K. et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas.N. Engl. J. Med. 384, 1613–1622 (2021).
Xu, B. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. 37, 45–54 (2019).
Han, J. et al. TGFbeta treatment enhances glioblastoma virotherapyby inhibiting the innate immune response. Cancer Res. 75, 5273–5282 (2015).
Chen, X. et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 7, 27764–27777 (2016).
Renner, C. et al. Cure of xenografted human tumors by bispecific monoclonal antibodies and human T cells. Science 264, 833–835 (1994).
Krishnamurthy, A. & Jimeno, A. Bispecific antibodies for cancer therapy: a review. Pharmacol. Ther. 185, 122–134 (2018).
Kontermann, R. E. & Brinkmann, U. Bispecific antibodies. Drug Discov. Today 20, 838–847 (2015).
Wei, H. et al. Structural basis of a novel heterodimeric Fc for bispecific antibody production. Oncotarget 8, 51037–51049 (2017).
Liu, B. L. et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 10, 292–303 (2003).
Kitange, G. J. et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro. Oncol. 11, 281–291 (2009).
Rehman, H., Silk, A. W., Kane, M. P. & Kaufman, H. L. Into the clinic: talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 4, 53 (2016).
Conry, R. M., Westbrook, B., McKee, S. & Norwood, T. G. Talimogene laherparepvec: first in class oncolytic virotherapy. Hum. Vaccin. Immunother. 14, 839–846 (2018).
Raman, S. S., Hecht, J. R. & Chan, E. Talimogene laherparepvec: review of its mechanism of action and clinical efficacy and safety. Immunotherapy 11, 705–723 (2019).
Tian, L. et al. Targeting Fc receptor-mediated effects and the ‘Don’t Eat Me’ signal with an oncolytic virus expressing an Anti-CD47 antibody to treat metastatic ovarian cancer. Clin. Cancer Res. 28, 201–214 (2022).
Ma, R. et al. An oncolytic virus expressing IL15/IL15Ralpha combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 81, 3635–3648 (2021).
Mollica Poeta, V., Massara, M., Capucetti, A. & Bonecchi, R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front. Immunol. 10, 379 (2019).
Ran, G. H. et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct. Target Ther. 7, 205 (2022).
Soria, G. & Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 267, 271–285 (2008).
Chada, S., Ramesh, R. & Mhashilkar, A. M. Cytokine- and chemokine-based gene therapy for cancer. Curr. Opin. Mol. Ther. 5, 463–474 (2003).
Whiteside, T. L. Cytokines and cytokine measurements in a clinical laboratory. Clin. Diagn. Lab. Immunol. 1, 257–260 (1994).
Elgundi, Z., Reslan, M., Cruz, E., Sifniotis, V. & Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 122, 2–19 (2017).
Scott, A. M., Wolchok, J. D. & Old, L. J. Antibody therapy of cancer. Nat. Rev. Cancer 12, 278–287 (2012).
Adams, G. P. & Weiner, L. M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 23, 1147–1157 (2005).
Reichert, J. M., Rosensweig, C. J., Faden, L. B. & Dewitz, M. C. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 23, 1073–1078 (2005).
Melero, I., Hervas-Stubbs, S., Glennie, M., Pardoll, D. M. & Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 7, 95–106 (2007).
Jonker, D. J. et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 357, 2040–2048 (2007).
Graham, J., Muhsin, M. & Kirkpatrick, P. Cetuximab. Nat. Rev. Drug Discov. 3, 549–550 (2004).
Lustig, R. Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol. Ther. 5, 1242–1243 (2006).
Cho, J. et al. Glioblastoma-derived epidermal growth factor receptor carboxyl-terminal deletion mutants are transforming and are sensitive to EGFR-directed therapies. Cancer Res. 71, 7587–7596 (2011).
Chen, Z. & Hambardzumyan, D. Immune microenvironment in glioblastoma subtypes. Front. Immunol. 9, 1004 (2018).
Wang, Q. et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56 e46 (2017).
Martinez-Lage, M. et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 7, 203 (2019).
Zhang, B., Shen, R., Cheng, S. & Feng, L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 8, 2897–2907 (2019).
Harris, J., Sengar, D., Stewart, T. & Hyslop, D. The effect of immunosuppressive chemotherapy on immune function in patients with malignant disease. Cancer 37, 1058–1069 (1976).
Iorgulescu, J. B., Reardon, D. A., Chiocca, E. A. & Wu, C. J. Immunotherapy for glioblastoma: going viral. Nat. Med. 24, 1094–1096 (2018).
Woroniecka, K. I., Rhodin, K. E., Chongsathidkiet, P., Keith, K. A. & Fecci, P. E. T-cell dysfunction in glioblastoma: applying a new framework. Clin. Cancer Res. 24, 3792–3802 (2018).
Woroniecka, K. et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 24, 4175–4186 (2018).
Mirzaei, R., Sarkar, S. & Yong, V. W. T cell exhaustion in glioblastoma: Intricacies of immune checkpoints. Trends Immunol. 38, 104–115 (2017).
Schuessler, A. et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 74, 3466–3476 (2014).
Brown, C. E. et al. Regression of Glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016).
Shah, W. et al. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4(+)FOXP3(+) regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell Mol. Immunol. 8, 59–66 (2011).
Huang, Y. et al. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget 6, 17462–17478 (2015).
Yang, I. et al. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J. Clin. Neurosci. 17, 1381–1385 (2010).
Ayoub, D. et al. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. MAbs 5, 699–710 (2013).
Terada, K., Wakimoto, H., Tyminski, E., Chiocca, E. A. & Saeki, Y. Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 13, 705–714 (2006).
Gismondi, A. et al. Proline-rich tyrosine kinase 2 and Rac activation by chemokine and integrin receptors controls NK cell transendothelial migration. J. Immunol. 170, 3065–3073 (2003).
Huang, Y. et al. CRK proteins selectively regulate T cell migration into inflamed tissues. J. Clin. Invest. 125, 1019–1032 (2015).
Han, Z. et al. 15-deoxy-Delta12,14 -prostaglandin J2 reduces recruitment of bone marrow-derived monocyte/macrophages in chronic liver injury in mice. Hepatology 56, 350–360 (2012).
Wang, Y. et al. The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat. Immunol. 20, 10–17 (2019).
Dong, W. et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. 9, 1422–1437 (2019).
Acknowledgements
This work was supported by grants from the National Institutes of Health (NIH) (NS106170, AI129582, CA247550, CA264512, CA266457 and CA223400 to J.Y.; CA210087, CA265095 and CA163205 to M.A.C.), the Leukemia and Lymphoma Society (1364-19 to J.Y.) and a 2021 Exceptional Project Award from Breast Cancer Alliance (to J.Y.). The authors appreciate the Pathology Core of Shared Resources at City of Hope Beckman Research Institute and National Medical Center for performing H&E and immunohistochemical staining experiments.
Author information
Authors and Affiliations
Contributions
J.Y., M.A.C. and L.T. conceived and designed the project. L.T., B.X., Y.C., Z.L., J.W., R.M., S.C. and W.H. conducted experiments. L.T. and J.Z. performed data analyses. L.T. and J.Y. wrote the paper. L.T., J.Y., M.A.C., E.A.C. and B.K. reviewed and/or revised the paper. J.Y. and M.A.C. acquired funding. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.A.C., J.Y., L.T., B.K. and E.A.C. have relevant or nonrelevant oncolytic virus patents awarded or pending. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks John Bell, Michael Platten and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Concentration of CCL5 secreted by GBM cells, and expression of CCL5 receptors on NK cells, macrophages, and T cells.
(a) Concentration of CCL5 secreted by human GBM cell lines (U251T2, Gli36ΔEGFR, and GBM30), measured by ELISA (n = 3 independent experiments). (b-e) CCR1 and CCR5 expression on human NK cells (b, n = 6 independent donors), macrophages (c, n = 5 independent donors), CD4+ T cells (d, n = 3 independent donors), and CD8+ T cells (e, n = 3 independent donors), measured by flow cytometry. Error bars indicate the standard deviations (s.d.) and data are presented as mean ± s.d..
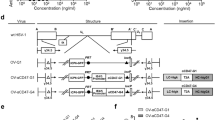
Extended Data Fig. 2 Comparison between Cmab-hCCL5 and hCCL5-Cmab and characterization of OV-Cmab-hCCL5.
(a, b) Detection of purified Cmab-hCCL5 or hCCL5-Cmab bound to U251T2 cells, measured by flow cytometry after staining Cmab-hCCL5- or hCCL5-Cmab-incubated tumor cells with anti-Fc-APC (a) or anti-hCCL5-APC (b). Cmab-hCCL5 and hCCL5-Cmab were purified from lentivirus-infected CHO cells. IgG1 isotype served as control. (c, d) Detection of Cmab-hCCL5 in the supernatant, collected from OV-Cmab-hCCL5-infected U251T2 GBM cell culture, that was associated with U251T2 cells, measured by flow cytometry after staining supernatant-incubated U251T2 cells with anti-Fc-APC (c) or anti-hCCL5-APC (d). (e) Cmab-hCCL5 produced in the supernatant of OV-Cmab-hCCL5-infected U251T2 cells at different MOIs (n = 3 independent experiments). (f) Cell viabilities of OV-Cmab-hCCL5 infection at three indicated MOIs were measured at 24 hours post infection (hpi), 48 hpi, and 72 hpi (n = 3 independent experiments). (g) The ability of OV-Q1 and OV-Cmab-hCCL5 to induce oncolysis of GBM cells, measured by real-time cell analysis (RTCA) The experiment was performed twice with similar data. (h) U251T2 cells were infected with OV-Q1 or OV-Cmab-hCCL5 at an MOI of 2. The supernatant was harvested at the indicated time points to assess viral production using a plaque assay with Vero cells (n = 3 independent experiments). Experiments in a-d were representative of three independent experiments with similar results. Error bars indicate the standard deviations (s.d.), and data are presented as mean ± s.d. (e, f, h). Statistical analyses were performed by one-way ANOVA with P values corrected for multiple comparisons by Bonferroni method (e).
Extended Data Fig. 3 Migration of immune cells induced by OV-Cmab-hCCL5.
NK cells, macrophages, CD4+ T cells, and CD8+ T cells in the upper chamber induced by OV-Cmab-hCCL5- or OV-Q1-infected U251T2 GBM cells at an MOI of 2 in the lower chamber, measured by transwell assay. 72 hours after the transwell assay was set up, immune cells in the lower chamber were quantified by flow cytometry. Error bars indicate the standard deviations (s.d.) and data are presented as mean ± s.d.. Statistical analyses were performed by one-way ANOVA with P values corrected for multiple comparisons by Bonferroni method (n = 3 independent donors).
Extended Data Fig. 4 OV-Cmab-hCCL5 improves oncolytic virotherapy in nude mice bearing GBM30 cells.
(a) Survival of GBM30 tumor-bearing nude mice treated with OV-Q1, OV-Cmab-hCCL5, or vehicle control. Survival was estimated by the Kaplan–Meier method and compared by log-rank test (n = 7 mice). (b) Luciferase imaging of GBM30-FFL GBM mice with the indicated treatments 15 and 20 days post tumor implantation.
Extended Data Fig. 5 Construction and characterization of OV-Cmab-mCCL5.
(a, b) Detection of purified Cmab-mCCL5 bound to CT2A-hEGFR cells, measured by flow cytometry after staining Cmab-mCCL5-incubated tumor cells with anti-Fc-APC (a) or anti-mCCL5-PE (b). Cmab-mCCL5 was purified from lentivirus-infected CHO cells. IgG1 isotype served as control. (c) mCCL5 and human Fc levels in Cmab-mCCL5 in the concentrated supernatant from engineered CHO cells or OV-Q1- or OV-Cmab-mCCL5-infected CT2A-hEGFR cells, detected by immunoblotting. (d, e) mCCL5 (d) and Cmab (e) of the Cmab-mCCL5 fusion protein in the supernatant from OV-Cmab-mCCL5-infected CT2A-hEGFR cells, quantified by ELISA. Cmab-mCCL5 purified from engineered CHO cells with known concentrations served as standards (n = 3 independent experiments). (f) Migration of murine NK cells, macrophages, CD4+ T cells, and CD8+ T cells induced by Cmab-mCCL5 purified from engineered CHO cells, measured using a transwell assay. Recombinant mCCL5 (rmCCL5, 100 ng/ml) served as positive control. Experiments in a-c were repeated with three independent experiments with similar results. Error bars indicate the standard deviations (s.d.), and data are presented as mean ± s.d. (d-f). Statistical analyses were performed by 2-sided Student’s t test (f, n = 3 mice/ biologically independent samples; rmCCL5 versus control and Cmab-mCCL5 versus IgG1 isotype are only compared).
Extended Data Fig. 6 Cmab-mCCL5 promotes innate and adaptive immunocyte activation in vitro.
(a) Cytotoxicity of mouse primary NK cells against Cmab-mCCL5 pre-treated-CT2A-hEGFR cells, measured by 51Cr release. Control versus Cmab-mCCL5, P = 0.0009. A linear mixed model was used to account for the underlying variance and covariance structure (n = 3 mice). (b) CD69 expression, measured by flow cytometry, on mouse primary NK cells co-cultured with CT2A-hEGFR cells in the presence of Cmab-mCCL5 purified from engineered CHO cells or IgG1 isotype control (n = 3 independent mice). (c) ADCP of mouse macrophages induced by Cmab-mCCL5 purified from engineered CHO cells, targeting CT2A-hEGFR cells. CT2A-hEGFR target cells were prelabeled with CFSE and then were co-cultured with mouse macrophages in the presence of Cmab-mCCL5 purified from engineered CHO cells or IgG1 isotype control. The percentage of mouse macrophages that had phagocytosed labeled tumor cells was measured by flow cytometry, determined by CFSE+ macrophages (n = 3 independent mice). (d) Cytokine RNA expression levels, measured by real-time RT-PCR, of mouse macrophages co-cultured with CT2A-hEGFR cells at a ratio of 1:1 with or without Cmab-mCCL5 for 6 hours. Macrophages were purified by cell sorting to extract total RNA for generating cDNA for real-time RT-PCR (n = 3 independent mice). Error bars indicate the standard deviations (s.d.) and data are presented as mean ± s.d. (a-d). Statistical analyses were performed by one-way ANOVA with P values corrected for multiple comparisons by Bonferroni method (n = 3 mice).
Extended Data Fig. 7 OV-Cmab-mCCL5 prolongs the survival of glioblastoma-bearing mice in an immunocompetent mouse model.
(a) Survival of CT2A-hEGFR tumor-bearing mice treated with OV-Q1, OV-Cmab-mCCL5, or vehicle control. Survival was estimated by the Kaplan–Meier method and compared by log-rank test (n = 7 or 8 mice). Of note, tumors remained in the OV-Cmab-mCCL5-treated group on the day of mouse sacrifice (day 50). (b) C57BL/6J mice bearing CT2A-hEGFR cells were intracranially treated with 2 × 105 PFU of indicated oncolytic virus or vehicle control (saline) 3 days post tumor implantation. Eleven days post tumor implantation, brains were collected from mice treated with saline, OV-Q1, or OV-Cmab-mCCL5 for standard H&E staining. Data of three mice in each group are shown. Scale bar, 2 mm. (c) Scheme for main Fig. 6f. Mice bearing CT2A-hEGFR cells were treated with saline, OV-Cmab-mCCL5, OV-Q1 + mCCL5 delivered by an osmotic pump, or OV-Q1+ Cmab-mCCL5 delivered by an osmotic pump.
Extended Data Fig. 8 Production of mCCL5 and oHSV in the GBM TME.
(a) Cmab-mCCL5 production in the brains collected from GBM mice treated with the indicated different doses of OV-Cmab-mCCL5. Mice were sacrificed on day 3 post treatment. Error bars indicate s.d., and statistical analyses were performed by one-way ANOVA with P values corrected for multiple comparisons by Bonferroni method (n = 5 mice in the saline and 2 × 105 PFU groups and n = 4 mice in the 1 × 105 PFU and 4 × 105 PFU groups). (b, c) Immunohistochemical (IHC) analysis of mCCL5, oHSV, and H&E staining of the brains collected from mice treated with saline, OV-Q1, or OV-Cmab-mCCL5. Slides with brain tissue isolated from the experimental mice were subjected to H&E and IHC staining, and anti-HSV antibody and anti-mCCL5 antibody are used for detecting mCLL5 production and oHSV (OV-Q or OV-Cmab-mCCL5) existence, respectively. Images with high and low magnification are shown in (b, scale bars, 100 μm) and (c, scale bar, 2 mm), respectively. The boxed images in (c) are shown at higher power in (b).
Extended Data Fig. 9 OV-Cmab-mCCL5 inhibits tumor EGFR signaling and promoted the infiltration of innate and adaptive murine immune cells into the GBM TME in a dose-dependent manner.
(a) Immunoblotting of p-AKT and p-EGFR from protein harvested from the brains of mice treated with OV-Q1, OV-Cmab-mCCL5, or saline (n = 3 independent mice). (b) Total number of immune cells in main Fig. 7b-d, measured by flow cytometry (n = 4 independent mice in the OV-Q1 group and n = 5 independent mice in the OV-Cmab-mCCL5 group and the saline group). (c, d) Representative flow cytometry data of immune cells infiltrated into the brains of mice treated with saline, OV-Q1, or OV-Cmab-mCCL5. Summary data are shown in main Fig. 7c. (e) Immune cell infiltration into the brains of mice treated with indicated different doses of OV-Cmab-mCCL5 for two days (n = 5 independent mice in each group). (f) Survival of CT2A-hEGFR GBM-bearing mice treated with indicated different doses of OV-Cmab-mCCL5. Saline served as control. Survival was estimated by the Kaplan–Meier method and compared by log-rank test (n = 7 independent mice in the 2 × 105 PFU group and n = 6 in all other groups). For panels b and e, error bars indicate s.d. and data are presented as mean ± s.d., and statistical analyses were performed by one-way ANOVA with P values corrected for multiple comparisons by Bonferroni method. Experiments in c and d were representative results of one of four to five mice in each group with similar data.
Extended Data Fig. 10 OV-Cmab-mCCL5 improves abscopal control of GBM in brain.
(a) Representative MRI images of a bilateral CT2A-hEGFR immunocompetent GBM model treated with saline, OV-Q1, or OV-Cmab-mCCL5 only on the right side of the brain. Data of two out of three to four mice in each group with similar data are shown. (b) The total number of immune cells, NK cells, macrophages, T cells, CD4+ T cells, and CD8+ T cells as well as the ratio of CD4+ T cells to CD8+ T cells in the untreated left side of the brain of the mice with the right side of the brain treated as indicated, measured by flow cytometry. Error bars indicate the standard deviations (s.d.) and data are presented as mean ± s.d.. Statistical analyses were performed by one-way ANOVA with P values corrected for multiple comparisons by the Bonferroni method (n = 5 independent mice in each group). (c) Therapeutic effects of OV-Cmab-mCCL5 when treating the two-side model with 1-day or 3-day intervals. Survival was estimated by the Kaplan–Meier method and compared by log-rank test (n = 6 mice).
Supplementary information
Supplementary Information
Supplementary Figures 1-3 and Supplementary Table 1
Source data
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 1
Statistical Source Data.
Source Data Fig. 2
Statistical Source Data.
Source Data Fig. 3
Statistical Source Data.
Source Data Fig. 4
Statistical Source Data.
Source Data Fig. 5
Statistical Source Data.
Source Data Fig. 6
Statistical Source Data.
Source Data Fig. 7
Statistical Source Data.
Source Data Fig. 8
Statistical Source Data.
Source Data Extended Data Fig. 1
Statistical Source Data.
Source Data Extended Data Fig. 2
Statistical Source Data.
Source Data Extended Data Fig. 3
Statistical Source Data.
Source Data Extended Data Fig. 4
Statistical Source Data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 5
Statistical Source Data
Source Data Extended Data Fig. 6
Statistical Source Data
Source Data Extended Data Fig. 7
Statistical Source Data.
Source Data Extended Data Fig. 8
Statistical Source Data.
Source Data Extended Data Fig. 9
Unprocessed western blots.
Source Data Extended Data Fig. 9
Statistical Source Data.
Source Data Extended Data Fig. 10
Statistical Source Data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tian, L., Xu, B., Chen, Y. et al. Specific targeting of glioblastoma with an oncolytic virus expressing a cetuximab-CCL5 fusion protein via innate and adaptive immunity. Nat Cancer 3, 1318–1335 (2022). https://doi.org/10.1038/s43018-022-00448-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-022-00448-0
This article is cited by
-
Understanding the immunosuppressive microenvironment of glioma: mechanistic insights and clinical perspectives
Journal of Hematology & Oncology (2024)
-
Mechanistic insights and the clinical prospects of targeted therapies for glioblastoma: a comprehensive review
Experimental Hematology & Oncology (2024)
-
Improving the therapeutic efficacy of oncolytic viruses for cancer: targeting macrophages
Journal of Translational Medicine (2023)
-
Neoantigen-targeted TCR-engineered T cell immunotherapy: current advances and challenges
Biomarker Research (2023)
-
Oncolytic virotherapy: basic principles, recent advances and future directions
Signal Transduction and Targeted Therapy (2023)
