
Abstract
Bcr-Abl is the cause of Philadelphia-positive (Ph+) leukemias and also constitutes their principal therapeutic target, as exemplified by dramatic effects of imatinib mesylate. However, mono-targeting of Bcr-Abl does not always achieve complete leukemia eradication, and additional strategies those enable complete elimination of leukemic cells are desired to develop. Here we demonstrate that INNO-406, a much more active Bcr-Abl tyrosine kinase inhibitor than imatinib, augments the activities of several proapoptotic Bcl-2 homology (BH)3-only proteins (Bim, Bad, Bmf and Bik) and induces apoptosis in Ph+ leukemia cells via Bcl-2 family-regulated intrinsic apoptosis pathway. ABT-737, an inhibitor of antiapoptotic Bcl-2 and Bcl-XL, greatly enhanced the apoptosis by INNO-406, even in INNO-406-less sensitive cells with Bcr-Abl point mutations except T315I mutation. In contrast, co-treatment with INNO-406 and other pharmacologic inducers of those BH3-only proteins, such as 17-allylaminogeldanamycin, an heat shock protein-90 inhibitor, or PS-341, a proteasome inhibitor, did not further increase the BH3-only protein levels or sensitize leukemic cells to INNO-406-induced apoptosis, suggesting a limit to how much expression levels of BH3-only proteins can be increased by anticancer agents. Thus, double-barrelled molecular targeting for Bcr-Abl-driven oncogenic signaling and the cell protection by antiapoptotic Bcl-2 family proteins may be the rational therapeutic approach for eradicating Ph+ leukemic cells.
Similar content being viewed by others


Main
The treatment of Philadelphia chromosome-positive (Ph+) leukemias has improved markedly owing to the development of inhibitors of Bcr-Abl tyrosine kinase (TK). The first member of such inhibitors is imatinib mesylate (imatinib). However, resistance to imatinib can develop by various molecular mechanisms, such as Bcr-Abl overexpression, mutations within the imatinib-binding site of Bcr-Abl, expression of a drug efflux pump or compensatory overexpression of related Src family kinases. In particular, point mutations in bcr-abl constitute the most commonly occurring and intractable.1, 2, 3, 4 It is anticipated that these resistance mechanisms can be overcome by developing more potent Bcr-Abl inhibitors that possess higher affinity for Bcr-Abl and are active against imatinib-insensitive mutated Bcr-Abl proteins. As a result, several ‘second-generation’ Bcr-Abl inhibitors, including dasatinib and nilotinib, have been developed.5, 6 We have developed INNO-406 (formerly NS-187), which blocks Bcr-Abl autophosphorylation in vitro 25- to 55-fold more efficiently than imatinib and is at least 10 times more effective than imatinib in suppressing the growth of Bcr-Abl-positive (Bcr-Abl+) leukemic tumors in vivo. Moreover, INNO-406 inhibits the TK activities of most of the imatinib-resistant-mutated Bcr-Abl proteins except T315I mutant.7, 8
However, it appears that a few leukemic cells often survive exposure to imatinib or second-generation Bcr-Abl inhibitors.9, 10 Indeed, mathematical modeling studies suggest that complete eradication of Bcr-Abl+ leukemic cells may require simultaneous targeting of other vital molecules.11, 12 Here, we have sought to identify additional therapeutic targets by characterizing the molecular mechanism by which the blockade of Bcr-Abl signaling kills Ph+ leukemia cells. Our study reveals that INNO-406-induced cell death is regulated by the interplay of pro- and antiapoptotic Bcl-2 family proteins that share one to four ‘Bcl-2 homology domains (BH1, 2, 3 and BH4)’. Of particular importance are the BH3-only proteins, which are a subgroup of proapoptotic Bcl-2 proteins that share only BH3 region and are essential for initiating apoptosis.13, 14 As we found that several BH3-only proteins, Bim, Bad, Bmf and Bik, play essential roles in INNO-406-induced apoptosis, we assessed whether co-treatment with a drug that activates these BH3-only proteins would enhance the killing of Bcr-Abl+ leukemia cells by INNO-406. The drugs tested included 17-allylaminogeldanamycin (17-AAG), a heat shock protein (HSP)-90 inhibitor and PS-341, a proteasome inhibitor; both have previously been shown to elevate Bim, Bad, Bmf or Bik expression.15, 16, 17, 18 We also examined whether the killing of INNO-406 could be enhanced by co-treatment with ABT-737, a drug that decreases the antiapoptotic barrier imposed by the antiapoptotic Bcl-2 family members Bcl-2, Bcl-XL and Bcl-w.19, 20 Our study shows that simultaneously targeting both pro- and antiapoptotic molecules augments the killing achieved with INNO-406, even in Bcr-Abl+ leukemias bearing imatinib-resistant Bcr-Abl point mutations. Notably, like Ph+ leukemias, most cancers maintain and propagate themselves by resisting apoptosis and enhancing cell proliferation. Inhibition of signaling for cell proliferation causes the cell cycle arrest as well as the initiation of apoptotic cell death. Thus, a double-barrelled therapeutic strategy like the one described here that targets both apoptosis resistance and enhanced proliferation of tumor cells may also be suitable for other cancers.
Results
INNO-406 induces apoptosis in Bcr-Abl+ leukemias
We first confirmed that INNO-406 kills Bcr-Abl+ cells by treating the chronic myeloid leukemia (CML)-derived cell lines K562, BV173 and MYL and bcr-abl-transformed wild-type (wt) murine fetal liver-derived myeloid progenitor cells (wt bcr-abl+ FLCs) with this drug (Figure 1a, d and data not shown). As INNO-406 caused DNA fragmentation in all four lines, it appears to induce apoptosis (Figure 1b and data not shown). To identify the apoptotic pathway that is stimulated by INNO-406, we examined the effect of INNO-406 on genetically engineered K562 subclones that overexpressed human Bcl-2 (K562/Bcl-2) or a dominant-interfering mutant of Fas-associated death domain (FADD)/MORT1 (K562/FADD-DN), which inhibits death receptor-induced apoptosis; both molecules were labeled with an N-terminal Flag tag (Figure 1c).21, 22, 23 We found that Bcl-2 overexpression blocked INNO-406-induced K562 cell death, whereas FADD-DN had no effect. Moreover, BV173R, a subclone of BV173, which expresses higher levels of Bcl-2 than parental BV173 cells, was less sensitive to INNO-406-induced cell death than BV173 (Figure 1c, d). These results demonstrate that INNO-406 induces apoptosis of Bcr-Abl+ cells via the Bcl-2 family-regulated intrinsic apoptotic pathway.

INNO-406 kills bcr-abl transformed cells via the intrinsic apoptotic pathway. (a) Survival of K562 and bcr-abl transformed wt FLC after 48 or 24 h treatment with 50 nM INNO-406, respectively. Viable cell ratio was measured by flow cytometric analysis for the uptake of PI. (b) DNA fragmentation in K562 or bcr-abl transformed wt FLC treated with vehicle (−) or 50 nM INNO-406 for 48 or 24 h (+), respectively. M; molecular marker. (c) Western blot analysis of the expression of Bcl-2, BimEL, Mcl-1 or transgenic Flag-tagged Bcl-2 or FADD-DN proteins in K562 or BV173 cells and their subclones. (d) Survival of (i) K562/Bcl-2 (open square□), K562/shBim (triangle▴), K562/FADD.DN (cross ×) and parental K562 cells (closed square▪), as well as (ii) BV173R/shBim (closed square▪), BV173R (closed diamond♦) and parental BV173 cells (triangle▴) after 48 h treatment with INNO-406
Recently, Bim, a member of the proapoptotic BH3-only subgroup of the Bcl-2 family, was found to participate in imatinib-induced apoptosis of Ph+ leukemia cells.15, 22, 24 Importantly, we found that the K562/short hairpin (sh) Bim subline, in which Bim expression is repressed by RNA interference (RNAi), was more resistant to INNO-406 than parental K562. However, K562/shBim cells were not as resistant to INNO-406 as K562 cells overexpressing Bcl-2, indicating that other proapoptotic proteins cooperate with Bim. Notably, BV173R/shBim cells were more resistant to INNO-406-induced cell death than BV173R cells (Figure 1c, d), demonstrating that Bim knockdown and Bcl-2 overexpression can cooperate to render Bcr-Abl+ cells resistant to INNO-406. This suggests that simultaneously targeting the Bim-mediated pathway via INNO-406 and inhibiting pro-survival Bcl-2 and some of its homologues may improve the efficacy of therapies against Ph+ leukemia.
Effect of INNO-406 on the expression of BH3-only proteins
To characterize further the pathway by which INNO-406 induces apoptosis, we examined the effects of this drug on mechanisms that regulate the activity of Bim and the other BH3-only family proteins, including Bad, Bik, Bmf, Hrk, Noxa and Puma. BH3-only proteins are regulated by a range of transcriptional and post-translational mechanisms.25 We therefore first examined the effect of INNO-406 on the transcription of BH3-only genes in K562, K562/Bcl-2 and K562/shBim cells. INNO-406 elevated bim mRNA levels in K562 and K562/Bcl-2 cells. In the K562/shBim cells, the RNAi reduced basal bim mRNA levels to about half of those seen in parental K562 and K562/Bcl-2 cells, but treatment with INNO-406 still slightly increased bim mRNA levels, albeit to a lesser extent compared with parental K562 cells. bad mRNA was readily detectable in all K562 subclones and treatment with INNO-406 did not affect this (Figure 2a). In contrast, the basal levels of bmf and bik mRNA were very low in all K562 subclones, but treatment with INNO-406 significantly increased their levels (Figure 2b). INNO-406 did not cause any changes in the levels of hrk, noxa or puma expression (data not shown). Thus, INNO-406 upregulates the transcription of bim, bmf and bik, but not bad, hrk, noxa or puma.

Treatment with INNO-406 elevates levels of bim, bmf and bik mRNA and the corresponding proteins, and causes dephosphorylation of Bim and Bad. (a, b) Effect of 6 or 18 h treatment with 50 nM INNO-406 on the mRNA levels of bim and bad (a) and bmf and bik (b) in K562, K562/Bcl-2 and K562/shBim cells. #P<.05 and *P<.005 for untreated control. (c, d) Effect of 1.5 μM imatinib (IM) or 50 nM INNO-406 treatment on the BH3-only protein levels in K562 (c) and wt.bcr-abl+. FLCs (d), as measured by Western blotting analysis
Next, we examined the effects of INNO-406 on the expression of Bim, Bad, Bmf, Bik, Puma and Noxa protein. Treatment of K562 cells with INNO-406 elevated BimEL levels within 6 h and also increased its mobility on sodium dodecyl sulfate-polyaclylamide gel electophoresis (SDS-PAGE), which is a hallmark of the dephosphorylation that is known to activate Bim.22, 26 Treatment with INNO-406 also increased the levels of Bmf and Bik. Although INNO-406 did not change the overall levels of Bad, it induced Bad dephosphorylation (Figure 2c). These findings were also the cases with bcr-abl-transformed FLCs. Within 6 h, the levels and migration speed of BimEL increased, Bad became dephosphorylated, and the levels of Bmf and to low extent also those of Bik increased (Figure 2d). In contrast, Puma or Noxa protein was not increased, but decreased, after INNO-406 treatment (Supplementary Figure 1).
Loss of Bim, Bad or Bmf partially inhibits INNO-406-induced apoptosis of Bcr-Abl-transformed murine FLC
The mechanisms of INNO-406-induced apoptosis were further examined by comparing bcr-abl transformed FLCs from wt, bcl-2 transgenic, bim−/−, bad−/−, bmf−/− or bim−/−bad−/− embryos. As shown in Figure 3, most bcr-abl transformed wt FLCs had died within 48 h exposure to >50 nM INNO-406. In contrast, the bcl-2 transgenic FLCs (vav.bcl-2 transgenic+ bcr-abl+.FLCs) were highly resistant, whereas the bim−/− and to a lesser extent also the bad−/− and bmf−/−.bcr-abl+.FLCs showed partial resistance. The bim−/−bad−/−.bcr-abl+.FLCs were more resistant to INNO-406-induced cell death than the bim−/− or the bad−/−. bcr-abl+.FLCs, but not as much as vav.bcl-2 transgenic+ bcr-abl+.FLCs. These results suggest that these three BH3-only proteins, Bim, Bad and Bmf, have overlapping functions in INNO-406-induced apoptosis.

Cell killing effects of INNO-406 are represented by viable cell ratio after 48-h treatment of wt.bcr-abl+.FLCs (open diamond⋄), bim−/−.bcr-abl+.FLCs (closed triangle▴), bad−/−.bcr-abl+.FLCs (square▪), bmf−/−.bcr-abl+.FLCs (diamond♦), bim−/−bad−/−.bcr-abl+.FLCs (open triangle▵) and vav.bcl-2 transgenic+bcr-abl+.FLCs (cross ×)
17-AAG does not enhance INNO-406-induced killing of Bcr-Abl+ leukemia cells
17-AAG inhibits Bcr-Abl signaling by a different mechanism to that used by INNO-406, namely, 17-AAG inhibits the HSP-90 chaperone protein and thereby promotes the degradation of its client Bcr-Abl protein (Figure 4a).16 Notably, Akt, a major downstream signaling molecule of Bcr-Abl that regulates the expression of Bim and Bad, is also known to be a client of HSP-90.27 Thus, 17-AAG may restore the Bim and Bad levels and their proapoptotic activity in bcr-abl-expressing cells. Indeed, treatment of K562 and BV173 cells with 17-AAG caused an increase in the levels of Bim, Bmf and Bik, Bad dephosphorylation and a faint reduction in the levels of antiapoptotic Mcl-1 (Figure 4b, c). 17-AAG caused a dose-dependent apoptosis over 48 h that plateaued around 0.5 μM and the apoptosis by 17-AAG was partially inhibited by Bim knockdown and almost entirely blocked by Bcl-2 overexpression. Contrary to our expectation, co-treatment with 17-AAG did not augment INNO-406-induced killing of K562 or BV173 cells (Figure 5a, Supplementary Figure 2a), and the two agents together cannot enhance the activation of these BH3-only proteins further (Figure 4b).

Effect of PS-341 or 17-AAG co-treatment with INNO-406 on caspase-8 activity and p210 Bcr-Abl, caspase-8 and BH3-only protein levels. K562 cells were treated for 24 h. (a, b) p210 Bcr-Abl, caspase-8 (a) and BH3-only protein (b) levels were determined by Western blot analysis. (c) Effect of 6 or 24 h treatment with INNO-406 (25 nM), 17-AAG (1 μM) or PS-341 (50 nM) on the expression of Bcl-2, Bcl-XL and Mcl-1 in K562 cells. (d) Caspase-8 activity by INNO-406, PS-341, 17-AAG or their combinations

ABT-737 effectively augments INNO-406 in killing bcr-abl transformed cells. K562, K562/shBim and K562/Bcl-2 cells were treated with (a) 17-AAG, (b) PS-341 or (c) ABT-737 alone (vehicle (diamond♦) or together with 5 nM (square ▪), 10 nM (triangle▴) or 25 nM (cross ×) INNO-406. The percentages of viable cells after 48 h of treatment are shown
Effect of combined treatment or Bcr-Abl+ cells with INNO-406 and PS-341
Even though PS-341 treatment did not affect Bcl-2 or Bcl-XL level, but accumulated Mcl-1 in K562 cells, it elevated caspase-8 activity (Figure 4a,c,d). This indicates that it operates by a pathway that differs from intrinsic apoptosis. PS-341 also increased Bim levels but did not affect Bmf or Bik levels or promote Bad dephosphorylation, and its combination with INNO-406 did not further elevate the Bim levels in K562 cells (Figure 4b). PS-341 killed K562 and BV173 in a dose-dependent manner and was unaffected by Bcl-2 overexpression or Bim knockdown. When combined, PS-341 and INNO-406 killed parental and Bim-knockdown K562 cells and BV173 cells more effectively than either drug alone. Moreover, when INNO-406-induced apoptosis was blocked by Bcl-2 overexpression, PS-341 retained its killing effect (Figure 5b, Supplementary Figure 2b). Thus, PS-341 and INNO-406 kill Bcr-Abl+ cells via different apoptotic mechanisms and can act cooperatively.
ABT-737 dramatically augments INNO-406-induced cell death and overcomes the INNO-406 resistance imparted by Bcl-2 overexpression or Bim knockdown
ABT-737 is a BH3-mimic that inhibits the activity of the antiapoptotic Bcl-2, Bcl-XL and Bcl-w proteins, but not Mcl-1, and has been reported to augment the cell killing effects of genotoxic treatments in hematologic malignancies and lung cancers.19, 20 As INNO-406 activates several BH3-only proteins and slightly reduces Mcl-1 (Figure 2, 4c), we expected the complementary apoptosis enhancing effect between INNO-406 and ABT-737. ABT-737 killed K562, BV173 and MYL cells in a dose-dependent manner and this was not strongly impeded by Bim knockdown or Bcl-2 overexpression. Significantly, ABT-737 strongly augmented the INNO-406-induced apoptosis of K562, BV173 and MYL cells. Notably, ABT-737 restored the sensitivity of K562/shBim or K562/Bcl-2 to INNO-406-induced cell death (Figure 5c, Supplementary Figure 2c, 3b).
Cells expressing mutant Bcr-AblE255K or Bcr-AblH396P but not Bcr-AblT315I develop sensitivity to INNO-406 upon co-treatment with ABT-737
Although INNO-406 can inhibit the TK activities of most imatinib-resistant Bcr-Abl mutants, such as Bcr-AblE255K or Bcr-AblH396P, much higher concentrations are needed to kill cells expressing these mutants than are needed for killing cells containing wt Bcr-Abl.7 Thus, we examined whether lowering of the antiapoptotic barricade by treatment with ABT-737 could restore the ability of INNO-406 to kill leukemic cells bearing imatinib-resistant Bcr-Abl. For this, we generated Ba/F3-derived murine hematopoietic cell lines transformed with wt p210-Bcr-Abl (Ba/F3/wt bcr-abl.) or Bcr-Abl bearing the E255K (Ba/F3/E255K), T315I (Ba/F3/T315I) or the H396P mutation (Ba/F3/H396P). The T315I mutation abolishes binding by both imatinib as well as INNO-406 and cells bearing this mutation are therefore completely resistant to both compounds. When the four Ba/F3 lines were treated with INNO-406, those with the E255K and H396P mutations were clearly less sensitive than Ba/F3/wt bcr-abl cells: 500 nM INNO-406 elicited >90% death of Ba/F3/wt.bcr-abl cells but only ∼20% killing of Ba/F3 lines expressing mutant Bcr-Abl. Ba/F3/T315I bcr-abl cells were completely resistant to INNO-406 (Figure 6b). These observations were paralleled by the effect of treatment on Bcr-Abl phosphorylation, as the wt Bcr-Abl expressing cells showed marked downregulation of Bcr-Abl phosphorylation, whereas this effect was less pronounced in cells expressing the E255K mutant and absent in those expressing the T315I mutant. As expected, INNO-406 induced much less Bim upregulation in the E255K and H396P mutant Bcr-Abl cells than in Ba/F3/wt bcr-abl cells, and had no effect on the levels or phosphorylation of Bim in Ba/F3/T315I bcr-abl cells (Figure 6a and data not shown). When the cells were co-treated with ABT-737 and INNO-406, Ba/F3/T315I remained resistant to INNO-406 and only the killing effects of ABT-737 were observed. Significantly, however, the cells expressing the E225K and H369P Bcr-Abl mutants were killed almost as efficiently as the Ba/F3/wt bcr-abl cells (Figure 6b). Thus, ABT-737 is an ideal partner for INNO-406 in eradicating leukemic cells with Bcr-Abl mutations except T315I.

Co-treatment with ABT-737 and INNO-406 effectively kills cells with Bcr-Abl bearing the E225K or H369P mutation. (a) Ba/F3/wt, Ba/F3/E255K and Ba/F3/T315I were treated with 50 or 250 nM INNO-406 or with vehicle for 12 h, after which the p210 Bcr-Abl and BimEL protein levels and the phosphorylation status of p210 Bcr-Abl were determined by Western blot analysis. (b) Ba/F3/E255K, Ba/F3/H396P and Ba/F3/T315I cells were treated with various concentrations of INNO-406 alone (vehicle (closed diamond♦) or together with 2.5 μM (square▪), 5 μM (open triangle▵) or 10 μM (open diamond⋄) ABT-737. The percentages of viable cells after 48 h of treatment are shown. The effect of INNO-406 alone on Ba/F3/wt.bcr-abl cells is indicated by arrows (closed triangles▴)
17-AAG plus ABT-737 produces more cell killing effects even in cells expressing mutant Bcr-AblT315I than either drug alone
Given that 17-AAG activates Bim, Bad, Bmf and Bik and slightly reduces Mcl-1 through the blockade of Bcr-Abl (Figure 4b, c), we next examined whether cell death inducing effect by 17-AAG could be also enhanced by ABT-737. As shown in Figure 7, the combination of these two agents produced more cell death not only in INNO-406-sensitive cells (i.e. Ba/F3/wt bcr-abl, Ba/F3/E255K and Ba/F3/H396P in this study) but also, importantly, in INNO-406-insensitive leukemic cells harboring T315I mutation. These again indicate that the inhibition of antiapoptotic Bcl-2 proteins is effective approach to augment the cell killing effect by the blockade of Bcr-Abl, and this combination could be a good alternative approach for leukemic cells insensitive to Bcr-Abl TK inhibitors.

Combinatory cell killing effects between 17-AAG (0.5 μM) and ABT-737 (1 μM) for 72 h on (a) Ba/F3/wt.bcr-abl, (b) Ba/F3/E255K, (c) Ba/F3/H396P and (d) Ba/F3/T315I. Mean±S.D. of viable cell ratios are demonstrated by three independent experiments
Combination effect of INNO-406 and ABT-737 on imatinib-resistant MYL cells expressing higher level of Mcl-1
Finally, we examined the effects of INNO-406, ABT-737 and their combination on imatinib-resistant MYL-R cells. MYL-R was generated by continuous exposure up to 1.0 μM imatinib in culture, and was found to express higher level of Mcl-1, whereas the levels of Bim or other BH3-only proteins were not reduced (unpublished data) (Supplementary Figure 3a).28 When compared with MYL, MYL-R was resistant to cell death either by INNO-406 or ABT-737, and the cell death by INNO-406 was increased only when the higher concentration of ABT-737 was combined (Supplementary Figure 3b, c).
Discussion
Bcr-Abl is the principal molecular target against Ph+ leukemias; however, the mono-targeting of Bcr-Abl appears to be insufficient for the complete eradication. Clinically, a number of patients who achieve molecular remission upon imatinib treatment suffer disease relapse when the drug is discontinued.29, 30 Furthermore, Bcr-Abl inhibitors have been shown to be more cytostatic than cytotoxic against Bcr-Abl+ leukemic cells.9, 10 These suggest that the complete eradication of Bcr-Abl+ leukemic cells may require Bcr-Abl inhibitors in combination with therapies that modify the molecular pathways that control cell survival. Our study supports this premise, as we were able to markedly increase the killing of Bcr-Abl+ leukemic cells by treating cells with INNO-406 plus ABT-737, which lowers the antiapoptotic barrier. Significantly, this co-treatment regimen also overcame the partial resistance to INNO-406 of leukemic cells expressing mutant forms of Bcr-Abl, which exhibit reduced binding of TK inhibitors.
We here showed that INNO-406-induced apoptosis in Bcr-Abl+ leukemic cells is mediated by Bim and, perhaps to lesser extent, also other BH3-only proteins, such as Bad, Bmf and Bik. The blockade of Bcr-Abl by INNO-406 probably upregulates bim transcription through shutdown of the PI3 kinase pathway leading to activation of forkhead box O3A, a known transcriptional activator of bim.31 Loss of ERK1/2 or AKT kinase activity is probably responsible for de-phosphorylation and activation of Bim and Bad, respectively.32 INNO-406 induces Bmf and Bik approximately 30-fold more potently than imatinib, but how this is achieved is presently unknown.
Based on these observations, we searched for promising agents whose combination with INNO-406 would enhance its apoptotic effect. We first tested two clinically available agents, 17-AAG and PS-341, both of which are able to increase the levels of Bim, Bad, Bmf and/or Bik in cancer cells.15, 17, 18 Indeed, 17-AAG potently increased Bim, Bmf and Bik expression and caused Bad dephosphorylation in Bcr-Abl+ cells, but its combination with INNO-406 did not further increase the expression of the BH3-only proteins or enhance cell killing. A possible explanation for this could be that there is a limit to how much expression levels of BH3-only proteins can be increased. Like 17-AAG, PS-341 co-treatment with INNO-406 did not further elevate BH3-only protein levels, but it increased cell killing via a distinct pathway to that used by INNO-406. The cell death pathway activated by PS-341 is currently unclear. Although this drug triggered caspase-8 cleavage, a hallmark of the extrinsic apoptosis pathway, the K562/FADD.DN was only marginally resistant to PS-341 (data not shown). This suggests that PS-341-induced cell killing is unlikely to be mediated by the extrinsic apoptosis pathway. Instead, this drug may induce endoplasmic reticulum (ER) stress-mediated cell death via the unfolded protein response,33 although it should be noted that the involvement of caspase-8 in ER-stress-mediated cell death remains controversial. In any case, these observations indicate that seeking to elevate the effects of INNO-406 on the expression and activation of BH3-only proteins, such as Bim or Bad, by using other BH3-only protein-stimulating agents is unlikely to be an effective strategy.
Next, we examined the effect of co-treatment with ABT-737 and INNO-406. ABT-737 dramatically augmented the killing of CML-derived cell lines by INNO-406. Moreover, co-treatment with ABT-737 was able to overcome the resistance to INNO-406-induced cell death that was caused by the abrogation of the intrinsic apoptotic pathway, caused either by Bcl-2 overexpression or Bim knockdown. Although the mechanism has not been fully elucidated, Mcl-1 might have important role in the combination of INNO-406 and ABT-737.19, 34 This combination effect could be explained by the neutralization of Mcl-1 by Bim, which was not only induced by INNO-406 but also dissociated from Bim/Bcl-2 complex by ABT-737.19, 34 Indeed, the combination effect was largely abrogated in Mcl-1 highly expressing MYL-R (Supplementary Figure 3a, c). Collectively, these results suggest the involvement of Mcl-1 in determining the sensitivity for the combination therapy of INNO-406 and ABT-737.
Remarkably, ABT-737 potently sensitized cells bearing the E255K or H396P Bcr-Abl point mutations to INNO-406-induced apoptosis, such that they were killed as efficiently as INNO-406-treated cells bearing the wt Bcr-Abl protein. Imatinib in combination with ABT-737 did not achieve this effect (unpublished data). However, ABT-737 did not sensitize cells with the T315I-bearing Bcr-Abl protein to INNO-406-induced cell death, which indicates that ABT-737 can act synergistically only when its partner agent retains at least some activity against target cells. On the basis of these results, we examined the effect of treating Bcr-Abl+ leukemias with both 17-AAG and ABT-737. We were interested in particular in the effect of this treatment regimen on leukemic cells with the T315I mutation, as 17-AAG has been shown to exert anti-Bcr-Abl+ leukemia effects regardless of the Abl point mutation status.35 Although 17-AAG killed leukemic cells expressing wt Bcr-Abl less potently than INNO-406, its effects were markedly augmented by ABT-737; moreover, the co-treatment efficiently killed cells with the E255K, H369P and even the T315I point mutations (Figure 7). These observations confirm that dual targeting of Bcr-Abl and antiapoptotic Bcl-2 proteins may be an effective strategy for treating Bcr-Abl+ leukemias. In addition, ABT-737 has been already demonstrated to be active against primitive leukemic stem cells (LSCs) in acute myeloid leukemias.19 Besides sensitizing to apoptosis, ABT-737 may be also expected to eradicate Bcr-Abl+ LSCs those may escape the killing by Bcr-Abl inhibitors.9, 10
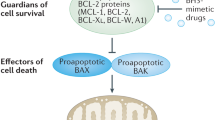
How does the dual targeting of Bcr-Abl and antiapoptotic Bcl-2 proteins synergize in eradicating Bcr-Abl+ leukemias? To maintain and expand the tumor, leukemic cells need to maintain their capacity for cell proliferation, while being resistant to apoptosis in the face of cytotoxic or environmental insults.36 In Ph+ leukemias, Bcr-Abl and its downstream signaling cascades promote both cell proliferation and resistance to apoptotic stimuli. The antiapoptotic Bcl-2 and Bcl-XL proteins can further increase apoptosis resistance of Bcr-Abl-transformed cells.37 The extent to which these two distinct, but converging, molecular pathways operate in particular leukemic cells may vary depending on the cellular context. This is shown by the differences in the responses of K562 and BV173 to INNO-406 or ABT-737. Moreover, it has been reported that coexpression of Bcl-2, but not Ras or Myc, promoted the blastic transformation of myeloproliferative disease in Bcr-Abl transgenic mice.38 This again indicates the biological significance of the acquisition of an antiapoptotic phenotype in Ph+ leukemias in addition to the oncogenic signaling by Bcr-Abl. This may help explain why blocking only the signaling cascades promoting cell proliferation does not eradicate leukemic cells, as these cells are also highly protected by an antiapoptotic molecular network (Figure 8). Consequently, the concomitant blockade of molecules involved in the latter molecular network may help to augment the therapeutic efficacies of Bcr-Abl inhibitors.

Cell death-inducing pathways utilized by INNO-406, PS-341, 17-AAG and ABT-737. ER, endoplasmic reticulum; MOMP, mitochondrial outer membrane permeabilization
In conclusion, the therapeutic effects of Bcr-Abl inhibitors, such as INNO-406, that strongly induces and activates BH3-only proteins can be significantly boosted by co-treatment with inhibitor, like ABT-737, that blocks antiapoptotic Bcl-2/Bcl-XL proteins.
Materials and Methods
Cell lines, generation of subclones and reagents
K562 is an erythroleukemia, BV173 is a pre-B leukemia and MYL is a myeloid leukemia cell line; all were derived from Ph+ CML patients in blast crisis phase. Long-term culture resulted in the isolation of the BV173R subclone, which is more resistant to imatinib-induced cell death than the parental BV173 cells. K562/Bcl-2 and K562/FADD-DN were generated as described previously; both molecules were labeled with an N-terminal Flag tag, and the former inhibits the mitochondria-mediated intrinsic apoptotic pathway, whereas the latter protein inhibits death receptor-induced apoptosis. K562/shBim and BV173R BV173R/shBim cell lines were generated by using the anti-Bim shRNA construct cloned into the pSUPER vector.21, 22, 23, 39 MYL and its imatinib-resistant subclone MYL-R were kind gifts from Dr. Hideo Tanaka (Hiroshima University, Japan).28 We generated Ba/F3-derived murine hematopoietic cell lines transformed with wt p210-Bcr-Abl (Ba/F3/wt.) or Bcr-Abl bearing the E255K (Ba/F3/E255K), T315I (Ba/F3/T315I) or the H396P mutation (Ba/F3/H396P).7, 8 Murine FLCs that were retrovirally transformed with the bcr-abl gene were previously described, according to the guidelines of the Melbourne Directorate Animal Ethics Committee.22 INNO-406 was provided by Nippon Shinyaku (Kyoto, Japan). Imatinib and PS-341 were purchased from a pharmacy. 17-AAG was purchased from Calbiochem (La Jolla, CA, USA). ABT-737 was produced by Dr. Michael Andreeff.19 Low-binding pipette tips and tubes (Sorenson BioScience Inc., Salt Lake City, UT, USA) were used for handling ABT-737 throughout this study.
Quantitative RT-PCR
Total RNA was extracted by using the Micro-to-Midi Total RNA Extraction Kit (Invitrogen, San Diego, CA, USA) and subjected to reverse transcription.40.The mRNA levels of human bad, bik, bim, bmf, hrk, noxa and puma were analyzed by using the LightCycler System (Roche Diagnostics, Sandhoferstraβe, Mannheim, Germany) with FastStart DNA Master SYBR Green I (Roche). Amplicons were validated by their melting curve and electrophoresis. The expression levels of the target mRNAs were normalized with that of the housekeeping gene β-actin. Informations on primers used are shown in Supplementary Table.
Western blotting
Protein samples were separated by SDS-PAGE and then electroblotted onto a Hybond-PDVF membrane (Amersham Biosciences, Uppsala, Sweden). The membranes were saturated with 5% (wt/vol) non-fat dry milk in PBS with 0.1% (vol/vol) Tween 20 (Sigma, Saint Luis, MO, USA). Antibodies (Abs) against β-actin (Sigma), Bcl-2 (Bcl-2–100, Upstate, Lake Placid, NY, USA), Bcl-XL (Stressgen, Victoria, Canada), Bik (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Bim (clone 3C5, produced by Dr. LA O’Reilly), Bad (Stressgen for human Bad, and Cell Signaling Technologies, Beverly, MA, USA, for murine Bad), Bmf (clone 9C10, Alexis, for human Bmf and clone 17A9 from Dr. LA O’Reilly for mouse Bmf), c-Abl (Santa Cruz), caspase-8 (BD Pharmingen, San Diego, CA, USA), FLAG (M2, Sigma), Mcl-1 (Santa Cruz), Noxa (Alexis Biochemicals, San Diego, CA, USA), phospho-Bad (Cell Signaling Technologies), phospho-tyrosine (Upstate) and Puma (ProSci Inc., Poway, CA, USA) were utilized. Ab detection was achieved by using horseradish peroxidase-conjugated secondary Abs and enhanced chemiluminescence (ECL) or ECL advance (Amersham Biosciences).
Cell death assay and DNA fragmentation assay
The incorporation of propidium iodide (PI) was used to detect dead cells in flow cytometric analysis. The DNA fragmentation assay was performed by using the Apo Ladder kit (TAKARA, Shiga, Japan).
Caspase-8 activity
Caspase-8 activity was measured by using the caspase-8 fluorometric assay kit (MBL, Nagoya, Japan) and Wallac Victor 2 Multi-label Counters (Perkin Elmer, Wellesley, MA).40
Combination cell killing effects with two agents from INNO-406, 17-AAG, PS-341 and ABT-737 on Bcr-Abl+ leukemic cells
Cells were treated with various concentrations of INNO-406, 17-AAG, PS-341 or ABT-737 alone or the combinations of two of listed agents at various concentrations as indicated in the figures. The cell death assay was performed 48 h after drug treatment.
Statistical analysis
All data were analyzed with two-sided unpaired t-tests, and a P=0.05 was considered as statistically significant. Values are expressed as mean±S.D. of triplicate independent experiments in cell death assay, quantitative RT-PCR and the measurement for caspase-8 activity.
Abbreviations
- Ab:
-
antibody
- BH:
-
Bcl-2 homology
- CML:
-
chronic myeloid leukemia
- DN:
-
dominant negative
- ECL:
-
enhanced chemiluminescence
- ER:
-
endoplasmic reticulumn
- FADD:
-
Fas-associated death domain
- FLC(s):
-
fetal liver cell(s)
- HSP-90:
-
heat shock protein-90
- imatinib:
-
imatinib mesylate
- Ph:
-
Philadelphia
- PI:
-
propidium iodide
- SDS-PAGE:
-
sodium dodecyl sulphate-polyaclylamide gel electophoresis
- TK:
-
tyrosine kinase
- wt:
-
wild type
- 17-AAG:
-
17-allylaminogeldanamycin
References
Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 2003; 101: 690–698.
Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293: 876–880.
Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003; 101: 2368–2373.
Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2: 117–125.
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL . Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004; 305: 399–401.
Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005; 7: 129–141.
Kimura S, Naito H, Segawa H, Kuroda J, Yuasa T, Sato K et al. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood 2005; 106: 3948–3954.
Yokota A, Kimura S, Masuda S, Ashihara E, Kuroda J, Sato K et al. INNO-406, a novel BCR-ABL/Lyn dual tyrosine kinase inhibitor, suppresses the growth of Ph+ leukemia cells in the central nervous system and cyclosporine A augments its in vivo activity. Blood 2007; 109: 306–314.
Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 2006; 107: 4532–4539.
Holtz MS, Slovak ML, Zhang F, Sawyers CL, Forman SJ, Bhatia R . Imatinib mesylate (STI571) inhibits growth of primitive malignant progenitors in chronic myelogenous leukemia through reversal of abnormally increased proliferation. Blood 2002; 99: 3792–3800.
Komarova NL, Wodarz D . Drug resistance in cancer: principles of emergence and prevention. Proc Natl Acad Sci USA 2005; 102: 9714–9719.
Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL et al. Dynamics of chronic myeloid leukemia. Nature 2005; 435: 1267–1270.
Huang DC, Strasser A . BH3-Only proteins-essential initiators of apoptotic cell death. Cell 2000; 103: 839–842.
Strasser A, O’Connor L, Dixit VM . Apoptosis signaling. Annu Rev Biochem 2000; 69: 217–245.
Aichberger KJ, Mayerhofer M, Krauth MT, Vales A, Kondo R, Derdak S et al. Low-level expression of proapoptotic Bcl-2-interacting mediator in leukemic cells in patients with chronic myeloid leukemia: role of BCR/ABL, characterization of underlying signaling pathways, and reexpression by novel pharmacologic compounds. Cancer Res 2005; 65: 9436–9444.
Nimmanapalli R, O’Bryan E, Kuhn D, Yamaguchi H, Wang HG, Bhalla KN . Regulation of 17-AAG-induced apoptosis: role of Bcl-2, Bcl-XL, and Bax downstream of 17-AAG-mediated down-regulation of Akt, Raf-1, and Src kinases. Blood 2003; 102: 269–275.
Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 2005; 7: 227–238.
Zhu H, Zhang L, Dong F, Guo W, Wu S, Teraishi F et al. Bik/NBK accumulation correlates with apoptosis-induction by bortezomib (PS-341, Velcade) and other proteasome inhibitors. Oncogene 2005; 24: 4993–4999.
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006; 10: 375–388.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Huang DC, Cory S, Strasser A . Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 1997; 14: 405–414.
Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DCS et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci USA 2006; 103: 14907–14912.
Strasser A, Harris AW, Huang DC, Krammer PH, Cory S . Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J 1995; 14: 6136–6147.
Kuribara R, Honda H, Matsui H, Shinjo T, Inukai T, Sugita K et al. Roles of Bim in apoptosis of normal and Bcr-Abl-expressing hematopoietic progenitors. Mol Cell Biol 2004; 24: 6172–6183.
Puthalakath H, Strasser A . Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ 2002; 9: 505–512.
Seward RJ, von Haller PD, Aebersold R, Huber BT . Phosphorylation of the pro-apoptotic protein Bim in lymphocytes is associated with protection from apoptosis. Mol Immunol 2003; 39: 983–993.
Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N . Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem 2002; 277: 39858–39866.
Ito T, Tanaka H, Kimura A . Establishment and characterization of a novel imatinib-sensitive chronic myeloid leukemia cell line MYL, and an imatinib-resistant subline MYL-R showing overexpression of Lyn. Eur H Haematol 2007; 78: 417–431.
Cortes J, Talpaz M, O’Brien S, Jones D, Luthra R, Shan J et al. Molecular responses in patients with chronic myelogenous leukemia in chronic phase treated with imatinib mesylate. Clin Cancer Res 2005; 11: 3425–3432.
Press RD, Love Z, Tronnes AA, Yang R, Tran T, Mongoue-Tchokote S et al. BCR-ABL mRNA levels at and after the time of a complete cytogenetic response (CCR) predict the duration of CCR in imatinib mesylate-treated patients with CML. Blood 2006; 107: 4250–4256.
Essafi A, Fernandez de Mattos S, Hassen YA, Soeiro I, Mufti GJ, Thomas NS et al. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene 2005; 24: 2317–2329.
Ley R, Ewings KE, Hadfield K, Cook SJ . Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ 2005; 12: 1008–1014.
Obeng EA, Carlson LM, Gutman DM, Harrington Jr WJ, Lee KP, Boise LH . Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006; 107: 4907–4916.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399.
Gorre ME, Ellwood-Yen K, Chiosis G, Rosen N, Sawyers CL . BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood 2002; 100: 3041–3044.
Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ . Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell 2004; 6: 241–249.
Cirinna M, Trotta R, Salomoni P, Kossev P, Wasik M, Perrotti D et al. Bcl-2 expression restores the leukemogenic potential of a BCR/ABL mutant defective in transformation. Blood 2000; 96: 3915–3921.
Jaiswal S, Traver D, Miyamoto T, Akashi K, Lagasse E, Weissman IL . Expression of BCR/ABL and BCL-2 in myeloid progenitors leads to myeloid leukemias. Proc Natl Acad Sci USA 2003; 100: 10002–10007.
Bouillet P, Robati M, Bath M, Strasser A . Polycystic kidney disease prevented by transgenic RNA interference. Cell Death Differ 2005; 12: 831–833.
Kuroda J, Kimura S, Segawa H, Sato K, Matsumoto S, Nogawa M et al. p53-independent anti-tumor effects of the nitrogen-containing bisphosphonate zoledronic acid. Cancer Sci 2004; 95: 186–192.
Acknowledgements
We are grateful to Professor Nakahata T, Drs. Heike T (Department of Pediatrics, Kyoto University), Huang DCS, Alexander W, Bouillet P, Puthalakath H, Cragg MS, Kaufmann T, Kelly PN (all WEHI) and Nakagawa Y (Kyoto University) for their gifts of transgenic and knockout mice, reagents, scientific advice and technical support. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Japan Leukemia Research Fund and Yasuda Medical Research Foundation (TM), the Public Trust Haraguchi Memorial Cancer Research Fund (Tokyo, Japan), Kurozumi Medical Foundation (Tokyo, Japan), Long-Term Research Grants from the TOYOBO Biotechnology Foundation (Osaka, Japan), the Uehara Memorial Biochemical Foundation (Tokyo, Japan) (JK), Grant-in-Aid of the Japan Medical Association and the Princess Takamatsu Foundation for Cancer Research (SK) and by fellowships and grants from the National Health and Medical Research Council (Australia; program no. 257502), the Leukemia and Lymphoma Society (New York; SCOR grant no. 7015) and the National Cancer Institute (NIH, US; CA 80188 and CA 43540) (AS). Supplementary information is available at Cell Death and Differentiation's website.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by C Borner
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Kuroda, J., Kimura, S., Strasser, A. et al. Apoptosis-based dual molecular targeting by INNO-406, a second-generation Bcr-Abl inhibitor, and ABT-737, an inhibitor of antiapoptotic Bcl-2 proteins, against Bcr-Abl-positive leukemia. Cell Death Differ 14, 1667–1677 (2007). https://doi.org/10.1038/sj.cdd.4402168
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402168
Keywords
This article is cited by
-
Comparative expression analysis of dasatinib and ponatinib-regulated lncRNAs in chronic myeloid leukemia and their network analysis
Medical Oncology (2022)
-
BCL2 protein signalling determines acute responses to neoadjuvant chemoradiotherapy in rectal cancer
Journal of Molecular Medicine (2015)
-
FTY720 induces apoptosis of chronic myelogenous leukemia cells via dual activation of BIM and BID and overcomes various types of resistance to tyrosine kinase inhibitors
Apoptosis (2013)
-
Association of differential gene expression with imatinib mesylate and omacetaxine mepesuccinate toxicity in lymphoblastoid cell lines
BMC Medical Genomics (2012)
-
Activation of apoptosis signaling eliminates CD34+ progenitor cells in blast crisis CML independent of response to tyrosine kinase inhibitors
Leukemia (2012)
