
Abstract
MDM2, a ubiquitin E3-ligase of the RING family, has a key role in regulating p53 abundance. During normal non-stress conditions p53 is targeted for degradation by MDM2. MDM2 can also target itself and MDMX for degradation. MDMX is closely related to MDM2 but the RING domain of MDMX does not possess intrinsic E3-ligase activity. Instead, MDMX regulates p53 abundance by modulating the levels and activity of MDM2. Dimerization, mediated by the conserved C-terminal RING domains of both MDM2 and MDMX, is critical to this activity. Here we report the crystal structure of the MDM2/MDMX RING domain heterodimer and map residues required for functional interaction with the E2 (UbcH5b). In both MDM2 and MDMX residues C-terminal to the RING domain have a key role in dimer formation. In addition we show that these residues are part of an extended surface that is essential for ubiquitylation in trans. This study provides a molecular basis for understanding how heterodimer formation leads to stabilization of MDM2, yet degradation of p53, and suggests novel targets for therapeutic intervention.
Similar content being viewed by others


Main
The tumor suppressor protein p53 plays a central role in maintaining the integrity of the genome by inducing cell cycle arrest and apoptosis in response to stress.1 Loss of the anti-proliferative activities of p53 is central to the development of most malignancies in humans, therefore control of p53 abundance is critical. In normal unstressed cells, p53 protein is unstable and present at very low levels due to its ubiquitylation by mouse double minute (MDM2).2 A hallmark of stress pathways is the rapid increase in p53 abundance due to a block in its degradation. The exact mechanism by which p53 is stabilized is unclear, although a series of post-translational modifications to itself, MDM2 and the closely related protein MDMX (also known as MDM4), are thought to dissociate the p53-MDM2 complex leading to increased levels of p53.3 Consistent with this, disruption of either the MDM2 or MDMX genes in mice causes activation of p53 and results in death early in embryogenesis. However, the lethal effect of mutating either gene can be rescued if p53 is also disrupted.4
MDM2 is a member of the really interesting new gene 1 (RING) domain family of E3 ubiquitin ligases (Figure 1a). Like other RING domain proteins, MDM2 functions as an adaptor protein, simultaneously binding to a cognate E2 ubiquitin-conjugating enzyme and a substrate protein, resulting in transfer of ubiquitin to the substrate and subsequent degradation by the proteasome. In this manner p53 is constantly targeted for degradation by MDM2 during normal non-stress conditions, as are other proteins, notably MDM2 itself and MDMX.5, 6 Like MDM2, MDMX directly inhibits p53 due to its ability to bind to the transactivation domain of p53 and block transcription, but in contrast to MDM2, MDMX is not under the transcriptional control of p53. Phosphorylation plays a key role in regulating MDMX and promotes its degradation in response to DNA damage, leading to stablization of p53.7, 8 Although MDMX contains a RING domain that is very similar to the RING domain of MDM2, it does not possess intrinsic E3 ubiquitin ligase activity. Instead, MDMX controls p53 abundance by modulating the levels and activity of MDM2.9, 10, 11 Dimerization, mediated by the conserved C-terminal RING domains of both MDM2 and MDMX, is critical to this activity.12 While the MDM RING domains can form homodimers, heterodimers form preferentially resulting in reduced auto-ubiquitylation of MDM2 and increased p53 ubiquitylation.13 MDMX therefore serves to stabilize MDM2, and keep p53 levels and activity low in healthy cells.10, 11, 14

The MDM2/MDMX RING domain heterodimer mediates ubiquitylation. (a) Schematic showing the domain structure of MDM2. The line indicates the region included in expression constructs. (b) Sequence alignment of the MDM RING domains with the secondary structure and zinc coordinating residues indicated. The arrow indicates the N terminus of MDM2s. The position of ubiquitin attachment in MDMX is also indicated. (c) Ubiquitylation assays containing GST-tagged MDM RING homo- and heterocomplexes as indicated. E1 and E2 were omitted in lanes 2 and 3, respectively. (d) Analysis of the products of ubiquitylation assays that contained purified soluble RING domain dimers in the presence or absence of the E2 as indicated. (e) Comparison of the activities of heterodimers containing MDM2 and either MDMX–wt or MDMX–K442A. Soluble MDM2 and ubiquitin migrate at the same position. Ubiquitylated products were detected using α-ubiquitin antibodies and lower panels were stained with Coomassie Blue
RING domains are about 60 amino acids in length and can be recognized by the conserved residues that coordinate the two zinc ions (Figure 1b). RING domain-dependent dimerization has been reported previously. For example, BRCA1 becomes activated upon heterodimer formation with its RING protein partner, BARD1, as does Ring1b with its partner Bmi1.15, 16 While disruption of the structurally important zinc centers in MDM2 causes loss of E3 ligase activity,17 the C-terminal five residues are also essential for both dimerization and E3 activity.18 In addition, C-terminal mutant forms of MDM2 that lack E3 ligase activity as MDM2 homodimers, are active when bound to wild-type MDMX. This suggests that MDMX can contribute directly to the activity of MDM2, and that the C-terminal region of MDMX is critical for this.19
MDM2 and MDMX clearly function together, as well as independently, to regulate p53 levels. Here we report the crystal structure of the MDM2/MDMX RING domain heterodimer and provide a molecular basis for understanding how heterodimer formation leads to stabilization of MDM2, and degradation of p53. These studies suggest novel targets for therapeutic intervention.
Results
Minimal MDM RING domain constructs retain E3-ligase activity
Domain boundaries of the MDM2 and MDMX RING domains were determined by comparison of the sequence to other RING domains (Figure 1a). Minimal MDM2 (residues 432–491, referred to as MDM2s) and MDMX (residues 421–490) RING domains were expressed as glutathione-S-transferase (GST) fusion proteins (Figure 1b). The E3 ligase activity of the MDM2s RING domain was confirmed by measuring the E2-dependent addition of ubiquitin to GST-MDM2s RING (Figure 1c; Supplementary Figure 1). No E3-ligase activity was observed for GST-MDMX RING. However, the GST-fused heterodimer retained significant activity (Figure 1c).
MDM2s RING, MDMX RING, and the MDM2s/MDMX RING dimers were then cleaved from GST and purified (Supplementary Figure 2). While MDMX and MDM2s/MDMX eluted at a position expected for a dimer, indicating formation of a stable complex, the MDM2s RING homodimer had poor solution properties and could not be purified. A second, slightly longer, MDM2 RING (residues 417–491, referred to as MDM2l) construct was expressed and purified. It also aggregated when cleaved from GST, although the GST-fused protein formed stable homo- and heterodimers that had significant E3-ligase activity (Figure 1c), and the soluble MDM2l/MDMX heterodimer could be purified (Supplementary Figure 2).
The RING domain alone of MDM2 has previously been observed to mediate ubiquitylation of itself when present as a homodimer.13 To determine if MDM2/MDMX RING domain heterodimers were also targeted for auto-ubiquitylation we used soluble RING domain dimers in ubiquitylation reactions. Purified MDMX RING was not active, but both heterodimers retained E2-dependent E3-ligase activity (Figure 1d). The presence of the ubiquitylated protein ladder with the GST fused proteins, compared to the single ubiquitylated band for the isolated RING domains, indicates that GST is also a substrate for ubiquitylation. These results show that the isolated RING domain of MDM2 is an active E3 ligase and that in vitro the RING domains themselves are substrates for auto-ubiquitylation.
Ubiquitylation of MDMX RING
To determine which of the RINGs was ubiquitylated in the isolated MDM2/MDMX heterodimer, we analyzed the results of the ubiquitylation assays with both RING domain heterodimers (Figure 1d). Because the same sized ubiquitylated band was seen whether the heterodimers contained MDM2s or MDM2l RING domains, MDMX RING, but not MDM2 RING, must have been ubiquitylated (Figure 1d). Mass-spectrometry after in-gel tryptic digestion of the isolated bands identified lysine 442 of MDMX RING as the site of ubiquitylation, no ubiquitylation of MDM2 was apparent. To confirm this, a K442A mutant of MDMX was prepared, and a heterodimer with MDM2 RING was formed. When purified MDM2/MDMX-K442A heterodimer was used in assays, ubiquitylation of mutant MDMX by MDM2 was abolished (Figure 1e).
These studies demonstrate that the MDMX subunit of the MDM2/MDMX RING domain heterodimer is targeted for ubiquitylation. The absence of MDM2 ubiquitylation in the heterodimer implies that ubiquitin is not attached to the RING domain that acts as the E3 ligase, but rather to its dimeric partner in trans. Although not ubiquitylated in heterodimers, homodimeric MDM2 RING is ubiquitylated.13 We propose that in MDM2 homodimers, a similar mechanism occurs with one subunit acting as an E3 ligase while the other subunit is the substrate.
Structure of the MDM2/MDMX RING domain heterodimer
To investigate the molecular details of the MDM2/MDMX RING domain dimer, and develop a model for its function as a ubiquitin E3-ligase, we determined the crystal structure of the MDM2s/MDMX RING domain heterodimer to 2.2 Å (Figure 2a, Table 1). Electron density for the first nine residues of MDMX was missing, suggesting that these residues were disordered. Therefore, we also solved the structure of the MDM2l/MDMX RING domain heterodimer. Electron density for four additional residues of MDM2, and two additional residues of MDMX, was apparent but the remaining residues (11 and 7 residues respectively) could not be placed suggesting that the deletion used to generate the shorter MDM2 construct had not significantly destabilized the N terminus of MDMX. Because the structure of MDM2l/MDMX was more complete it was used in all further comparisons.

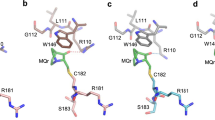
Structure of the MDM2/MDMX RING domain heterodimer. (a) Cartoon diagram of the MDM2l/MDMX RING domain heterodimer structure. MDM2 RING is shown in orange and MDMX RING in yellow, with the zinc ions and coordinating residues shown as spheres and sticks, respectively. (b) Details of the heterodimer interface between the C terminus of MDMX RING (yellow sticks), with β2 and the N terminus (top loop) of MDM2 RING (orange surface). Hydrogen bonds are shown as lines and the key residues that make cross dimer contacts are indicated
The RING domains of both MDM2 and MDMX are nearly indistinguishable (r.m.s.d of 0.56 Å over the 60 core Cα atoms) and have a compact structure with two zinc ions at the core that is common to all RING domain structures (Figure 2a; Supplementary Figure 3a). In contrast, greater differences are seen between the MDM2 RING domain from the heterodimer and the monomers of the MDM2 homodimeric RING structure (r.m.s.d of 1.8 Å over the Cα atoms of residues 436–491) (Supplementary Figure 3b). An absence of constraints in the NMR structure,13 reflected by regions of disorder, probably accounts for most of the differences.
Comparison of the individual MDM RING domains to the pdb gives the highest similarity to the BRCA1 RING domain (r.m.s.d. of 2.39 Å over 50 residues) and the RING domain of Ring1b (2.84 Å over 50 residues),16 the U-box proteins, PRP19 (3.20 Å over 44 residues)20 and CHIP (3.28 Å over 47 residues)21 also overlay well. The main difference between the MDM RING domains and other RING structures is in the position of the ligands that coordinate the second Zn2+ion. Unlike all other RING domains, four residues separate the third and fourth ligands in MDM RING domains, and both ligands are histidines, yet the coordinating sidechains and Zn2+ ion overlay closely (data not shown). In the MDM RING domains β3 is also extended relative to other RING domains, although the contacts with β1 and its position are conserved.
The MDM2/MDMX RING domain heterodimer forms a highly symmetrical structure with equivalent residues from the N- and C-terminal flanking sequences, together with the three β-strands from each monomer, involved in dimer formation. In total a surface area of 1375 Å2 (1054 Å2) of MDM2l (MDM2s) is buried. The main contacts involve interaction of β3 and the C-terminal residues from one subunit, with β2 from the other subunit, such that the core of the dimer is effectively formed by a six-stranded β-barrel (Figure 2a). The inside of the barrel is filled by the sidechains of hydrophobic residues. The C-terminal five residues of either MDM2 or MDMX are involved in 13 of 15 interface hydrogen bonds. Notably, six residues (L483, I485, V487, F488, I489 and A490 in MDMX) from the C terminus of each monomer are buried as a consequence of dimer formation (Figure 2b). Additional contacts that appear to stabilize the dimer involve residues (L430, A434 and I435 from MDM2) at the N terminus that are part of the irregular structure that extends across the dimer interface (Figure 2). The arrangement of the MDM2/MDMX heterodimer is similar to that of the MDM2 homodimer,13 although when superimposed, using just the MDM2 monomer of the heterodimer, the second monomer in the homodimer is offset (Supplementary Figure 3b). Very few experimental constraints defined the NMR structure of the homodimer and this probably accounts for at least some of the difference.13
Thus while residues flanking the RING domain are required for dimerization, the MDM dimer structures contrast with the structures of the CHIP,21 BRCA122 and Ring1b dimers,16, 23 which are predominantly stabilized by interaction of N- and/or C- terminal helices. The critical role for the flanking C-terminal residues agrees with studies that showed that mutation or deletion of the C-terminal residues disrupts dimer formation.18, 19
E2 recruitment by MDM2
Since specific ubiquitylation of K442 in MDMX of the heterocomplex was observed (Figure 1e), although a number of lysine residues are present in the MDM RING domain heterodimer (Supplementary Figure 3c), we sought to investigate the mechanism by which auto-ubiquitylation occurs. Because MDM2 RING alone can mediate ubiquitylation,13 but MDMX RING cannot (Figure 1c and d), we reasoned that the MDM2 RING domain contained a critical binding site for the E2 that was also present within the heterodimer. The putative E2-binding site on the MDM2 RING domain was predicted by comparison with the E2/E3 complex of UbcH7 and the c-Cbl RING domain.24 When MDM2 RING is superimposed on the c-Cbl RING domain, there are no clashes with UbcH5b25 that has been positioned by superposition on UbcH7 (Figure 3a). To assess the role of the predicted interface residues we systematically mutated these to alanine. The interaction between UbcH5b and the RING domain complexes could not be detected by pull-down experiments because the affinity of the E2 for the MDM2 RING domain is low. Instead, the ability of the E2 to functionally interact with the RING domain mutants was determined by measuring the ability of the mutant GST-MDM2 RING homodimers to mediate auto-ubiquitylation (Figure 3b). As observed by others, hydrophobic contacts appear critical to recruitment of the E2 since mutation of I440, L468 and P476 abolished activity. Residues adjacent to the hydrophobic patch formed by I440, L468 and P476 are also important since mutation of R479 disrupted activity, while mutation of V439 and R471 reduced activity (Figure 3c). Together the residues that disrupt activity define a surface on the face of MDM2 that is consistent with, but larger than the E2-binding site observed in other RING domains.16, 24

Interaction of MDM2 RING with E2 UbcH5b. (a) A putative MDM2-E2 ligase complex was constructed by superimposing the MDM2 RING domain (orange) onto the RING domain of Cbl (green) from the crystal structure (PDB 1fbv) of a complex between Cbl and UbcH7 (pink). UbcH5b (tan) was then superimposed onto UbcH7 to predict an UbcH5b-MDM2 complex. (b) Indicated residues in the predicted UbcH5b-MDM2 interaction site were mutated to alanine. The activity of each GST-tagged MDM2 homodimer mutant was analyzed using in vitro auto-ubiquitylation assays. (c) The putative primary E2-binding interface of MDM2, residues within four Å of UbcH5b are represented in ball and stick format. The sidechains of mutated residues are shown as spheres colored according to their effect on ubiquitylation activity as determined in (b) (wild type activity, green; partial activity, cyan; inactive mutants, red). (d) Mutation of Y489A renders the MDM2 homodimer (lane 3) inactive while the heterodimer (lane 4) regains activity. (e) MDM2 homodimers with the indicated mutations were inactive. In b, d and e the lower panel shows the MDM2 input (Coomassie Blue stained)
Many of the surface exposed residues within MDM2 that are required for recruitment of the E2 are conserved in MDMX, although some of the surrounding residues differ (Figure 1b). To determine if these differences render MDMX inactive we mutated residues in MDMX to the MDM2 equivalents (AGA470RNK, EK441QG, K486L or K435E), but none of these changes were able to engender E3 ligase activity to the MDMX RING domain (Supplementary Figure 4). This suggests that no single change is pivotal, instead a number of small differences may contribute to the inactivity of MDMX, and changes distant from the site of E2 recruitment may be important. In addition the oligomeric status of MDMX is uncertain and if predominantly monomeric as suggested by Tanimura et al.,12 this may contribute to its inactivity.
All studies of ubiquitylation processes are confounded by our limited appreciation of the mechanistic details. However, in MDM RING domains the C-terminal residues appear to directly contribute to E3 ligase activity because single point mutations of solvent exposed residues (e.g., Y489A), that do not disrupt dimer formation, nevertheless significantly diminished activity (Figure 3d, lane 3).19 In addition, the C-terminal residues of MDMX are functional because the E3 ligase activity of an inactive homodimeric MDM2 C-terminal mutant could be restored when mixed with wild-type MDMX (Figure 3d, lane 4). However, when MDMX that contained mutation of an equivalent C-terminal residue (F488) was added to wt MDM2 RING, activity was abolished.19 When mapped onto the structure of the heterodimer, F488 is positioned between the putative E2-binding site on MDM2 and K442 (MDMX) that we identified as the site of ubiquitylation on MDMX RING. To more carefully evaluate the role of V477 and V439, which lie at the edge of the E2 binding site in MDM2, we mutated both residues to glutamic acid. When assayed as homodimers both proteins were now inactive (Figure 3e), emphasizing the extended nature of the surface required for functional interaction with the E2.
Together these studies suggest that an enlarged secondary surface that extends across the dimer interface, and depends on the C terminus of the subunit that does not provide the primary binding surface, is required for E3 ligase activity (Figure 4a). While only MDM2 has a functional primary E2-binding site, the C-terminal residues (β3 and the residues that follow) of either MDM2 or MDMX can provide the secondary interface. Thus the symmetrical MDM2 homodimers have two primary and secondary interfaces and the E2 could be recruited by either monomer, leading to ubiquitylation of the other subunit. However, in the MDMX/MDM2 heterodimer the primary E2 interaction site is provided by MDM2 while the secondary interface depends on the C-terminal residues of MDMX, thus MDM2 is not ubiquitylated.

Model to account for the activity of the MDM2/MDMX heterodimer. (a) Putative complex of the MDM2 RINGl/MDMX RING heterodimer (orange/yellow) with a model of the ubiquitin∼UbcH5b conjugate (blue/tan) contacting the primary binding site. Sidechains of residues that disrupt activity are shown as red spheres. (b) Model to account for the stabilization of MDM2 by MDMX. At low MDMX levels, MDM2 exists as homodimers and mediates ubiquitylation of itself (left), while when MDMX levels equal those of MDM2, heterodimers preferentially form, and MDM2 is spared from ubiquitylation (middle). When MDMX levels are high, excess MDMX (either monomeric or dimeric) binds to p53, which is now protected from degradation (right)
Discussion
Numerous studies have shown that loss of p53 function is tightly linked to cancer development and activation of p53 is associated with death of tumor cells.1 While inactivation of p53, due to deletion or mutation of the p53 gene, accounts for ∼50% of tumors in humans, the rest express wild-type p53 and abnormalities in p53 regulation account for defective signaling. MDM2 is a key negative regulator of p53, and in human sarcomas accumulation of MDM2 leads to increased degradation of p53, preventing the activation of apoptotic pathways. Overexpression of MDMX has also been reported to have a similar effect and contribute to tumor formation.26 Therefore, in tumors that retain wild-type p53, modulation of MDM activity is a potential target for therapeutic intervention.
MDM proteins are modular in nature and the RING domain of MDM2 is essential, as well as sufficient, for E3 ligase activity and dimerization. The structure of the MDM2/MDMX RING domain heterodimer reveals a key role for the C-terminal residues from both RING domains in dimer formation (Figure 2) and is in agreement with previous reports where deletion or mutation of a number of C-terminal residues (e.g., I485E in MDM2) was shown to disrupt both homo- and heterodimer formation.19, 27 The structure of the MDM2/MDMX RING domain heterodimer is similar to the MDM2 RING domain homodimer structure13 indicating that a major rearrangement is not associated with heterodimer formation (Supplementary Figure 3). The conserved conformation of the C-terminal residues in MDM2 and MDMX suggests that other C-terminal RING domains such as IAPs,28 which have a similar C-terminal sequence, will have a similar structure.
Previous studies have shown that residues in the α-helix and the zinc chelating loops of RING domains are required for their interaction with E2's.16, 24 Consistent with these studies, residues predicted to mediate E2 recruitment disrupt the activity of MDM2 (Figure 3). In addition to this primary interface, our mutagenesis experiments and those of others suggest that a larger surface,19, 27 which extends across the dimer interface, and therefore depends on dimer formation, is also required for E3 activity (Figure 4a). The role of the extended surface is uncertain but it may aid E2 recruitment or E2∼ubiquitin movement.
In addition to mediating transfer of ubiquitin to substrate proteins, such as p53, MDM2 also ubiquitylates itself and MDMX. While the sites of ubiquitylation within full-length MDM proteins have not been reported, and there may be several, our studies show that K442 in the RING domain of MDMX is specifically ubiquitylated in the heterodimer (Figure 1e). The MDM2 subunit of the heterodimer was not ubiquitylated, yet the MDM2 RING domain homodimer mediates auto-ubiquitylation of itself.13 These results are consistent with a model that favors transfer of ubiquitin to the MDM subunit that does not interact with the E2 (Figure 4). Thus, in the heterodimer, MDM2 is not ubiquitylated because MDMX does not have a primary E2-binding site. To enable ubiquitin transfer to K442 of MDMX, which is distant from the primary E2-binding site on MDM2, we propose that the E2∼ubiquitin complex interacts with the extended surface that includes the heterodimer interface, restricting transfer of ubiquitin to specific nearby target residues (Figure 4a). In the case of MDM2 RING homodimers it is expected that one of the lysines at positions 466, 467, 469 or 470, along the α-helix in MDM2, would be targeted for ubiquitylation. In support of this, acetylation of MDM2 by p300 regulates its E3 ligase activity and requires lysines 466, 467, 469 and 470.29 However, Itahana et al.30 recently reported that the E3 ligase activity of MDM2 was not required for degradation of MDM2 as disruption of the RING domain did not result in stabilization of MDM2. Since this study used a RING domain mutant that would be misfolded and preclude dimer formation, it is also possible that the mutant MDM2 is destabilized in other ways.
The proposed model, whereby the MDM subunit that does not initially interact with the E2 is preferentially ubiquitylated, predicts that when MDM2 exists as homodimers it will target itself for degradation (Figure 4b). However, when it forms heterodimers, ubiquitylation of MDM2 would be diminished because MDMX does not recruit the E2, and MDMX itself would be preferentially ubiquitylated. Consistent with this prediction, MDM2 and MDMX preferentially form heterodimers,12, 13, 14, 27 and when MDMX is present MDM2 is spared from degradation, whereas at low MDMX levels MDM2 ubiquitylates itself and is destabilized.9, 31 Other modifications, such as phosphorylation of MDMX, modulate the extent to which ubiquitylation occurs.8
Degradation of p53 by MDM2 requires the RING domain and C-terminal residues of MDM2.19 Because C-terminal point mutants of MDM2 as well as C-terminally truncated forms cannot degrade p53, yet p53 degradation can occur if MDMX is added, a common mechanism is probably responsible for transfer of the E2∼ubiquitin conjugate from MDM2 to either p53, or MDMX and MDM2. Interaction between p53, and MDM2 or MDMX, primarily depends on binding of the N-terminal transactivation domain of p53 with a deep cleft in the p53-binding domain at the N-terminus of the MDMs.32 Thus not only would MDM2/MDMX heterodimer formation result in the stabilization of MDM2, but it is likely to increase ubiquitylation and degradation of p53. Some studies support this conclusion,8, 14 while others present conflicting data and report stabilization of p53 by MDMX.33 However, Gu et al.9 showed that the effect of MDMX on p53 levels was dependent on the ratio of MDMX to MDM2, with ratios of MDMX/MDM2 less than 2 : 1 resulting in p53 degradation, while ratios greater than 2 : 1 resulted in stabilization of p53. This is consistent with our model since at high levels of MDMX, monomers or homodimers will predominate (Figure 4b). The oligomeric status of MDMX is uncertain but both forms of MDMX would be expected to bind p53 and sequester it away from the active MDM2 homo- or heterodimers.
While a number of structures of fragments of MDM2 and p53 have been reported, the structure of neither protein in their entirety has been determined, and sequence analysis suggests that some regions are disordered.34 However, in p53, only lysine residues at the C terminus of the protein are specifically targeted for ubiquitylation.35 The E3 ligase activity of other RING domains (c-Cbl and Rbx1), and the specificity of target protein ubiquitylation, has been shown to depend on a structural scaffold that precisely positions the substrate.36 It is therefore likely that when p53 is bound to MDM2, it is oriented so that the C terminus is precisely positioned relative to the MDM RING domains, so that transfer of ubiquitin to specific lysine residues can occur.
Disruption of MDM2 function is an attractive therapeutic target. Indeed, nutlin-3, a peptidomimetic that disrupts the p53-MDM2 interaction, activates p53 pathways both in vitro and in vivo in human cell lines that possess wild-type p53 and overexpress MDM2.32 In a second approach, Vousden and colleagues established that it is possible to stabilize p53 by directly inhibiting the E3-ligase activity of MDM2.37 While the mechanism of action of the compounds was not determined, and some cross reactivity with other E3's was observed, these studies indicate that if molecules can be developed that are specific for MDMs, they should be effective. Our studies suggest that it might be possible to obtain MDM-specific E3 ligase inhibitors by targeting the MDM2/MDMX RING domain dimer interface rather than the primary E2 binding site that is common to many RING domain E3-ubiquitin ligases.
Materials and Methods
Plasmids and mutagenesis
Human MDM2 (Accession number: Q00987) residues 432–491 (MDM2s) and residues 417–491 (MDM2l), and MDMX (Accession number: O15151) residues 421–490 were cloned into pGEX-6p3 and expressed as GST fusion proteins in Escherichia coli. Purified proteins have seven additional N-terminal residues, GPLGSGT, as a result of cloning. The QuikChange site-directed mutagenesis kit (Stratagene) was used to generate mutants. UbcH5b and ubiquitin were expressed as a His tag fusion protein from pET21d and pQE80L, respectively.
Expression and purification of proteins
All RING domain constructs were expressed in E. coli BL21(DE3) at 18°C overnight using LB media. To prepare heterodimers a GST-MDM2 RING cell pellet was sonicated in lysis buffer (50 mM Tris, 500 mM NaCl, 2 mM DTT, pH 8.5) and the soluble protein was bound to glutathione sepharose. Resin bound GST-MDM2 was then mixed with the GST-MDMX soluble fraction that had been lysed in the same buffer. Samples were then washed and cleaved from GST using GST fused 3C protease. The soluble fraction was purified using a Sephadex 75 column (Amersham) in 50 mM Tris, 500 mM NaCl, pH 8.5 (Supplementary Figure S1). MDMX RING was prepared in a similar manner. Ubc5Hb was expressed in E. coli at 37°C and purified from clarified lysate by IMAC using Ni-NTA.
Crystallization
Crystals of the MDM2s/MDMX RING domain heterodimer were grown at 18°C by the vapor diffusion method. Sitting drops (200 nl) were set up using the Mosquito robot (TTP LabTech Ltd.) and contained a 3 : 1 ratio of protein (3–5 mg/ml in 50 mM Tris, 500 mM NaCl, pH 8.5) and crystallization buffer (1.8 M NH4(SO4)2, 0.5 M NaCl, 0.1 M Na Citrate, pH 6.5). Crystals formed in 1–2 days in the space group P21 with cell dimensions of a=54.15 Å, b=41.70 Å, c=77.04 Å, α=γ=90.00, β=109.14.
Structure solution and refinement
Diffraction data from crystals of MDM2s/MDMX were collected at beamline BL9-2 at the SSRL and processed with MOSFLM and SCALA from the CCP4 package.38 Processing statistics are shown in Table 1. The structure was solved using the MAD method around the K-edge of zinc anomalous absorption, using the autoSHARP software interface.39 The positions of eight zinc atoms were clearly located using SHELXD, corresponding to two zinc atoms per monomer and two heterodimers in the asymmetric unit. Electron density maps following density modification in SOLOMON were clearly interpretable and an initial model was built by Arp/Warp.40 This model was used as a starting point for refinement in REFMAC5, followed by iterative cycles of manual rebuilding in COOT and further refinement. Final refinement statistics are displayed in Table 1.41
Diffraction data from the MDM2l/MDMX RING domain heterodimer crystals were collected using Cu-Kα radiation, and the structure was solved by molecular replacement using Phaser and the MDM2s/MDMX RING domain complex as a search model.
E3 ubiquitin ligase assays
Ubiquitylation reactions contained 1.8 μM UbcH5b and either 127 nM (soluble MDM) or 42 nM (resin-bound MDM) of E1 (Sigma) for assays. Reactions containing 12 μM soluble protein or 5 μM GST-fusion proteins were incubated for 90 min at 37°C in 20 μL of 20 mM Tris, pH 7.5, 50 mM NaCl, 60 μM ubiquitin (Sigma) or His-tagged ubiquitin, 5 mM ATP, 2 mM MgCl2 and 2 mM DTT. Except where indicated, 2 × SDS-PAGE sample buffer was then added to the reactions and samples were resolved by 16% or 12% SDS-PAGE and transferred onto nitrocellulose membrane (BioRad) blots for analysis using α-GST, α-ubiquitin (Santa Cruz) or α-His antibodies (Amersham).
To identify the site of ubiquitylation in the MDMX RING domain, reactions contained 70 μM of soluble heterodimer. Proteins were resolved by 16% SDS-PAGE and bands were cut out for in-gel tryptic digest. Mass spectrometry analyses were used to identify fragments with a ubiquitin specific weight adduct that was only present in ubiquitylated samples. Ubiquitylation of lysine 442 in MDMX was verified by MS/MS analysis.
Abbreviations
- GST:
-
glutathione-S-transferase
- MDM:
-
mouse double minute
- RING:
-
really interesting new gene 1
References
Vousden KH, Lane DP . p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8: 275–283.
Yang Y, Li CC, Weissman AM . Regulating the p53 system through ubiquitination. Oncogene 2004; 23: 2096–2106.
Shmueli A, Oren M . Regulation of p53 by MDM2: fate is in the numbers. Mol Cell 2004; 13: 4–5.
Brooks CL, Gu W . p53 ubiquitination: MDM2 and beyond. Mol Cell 2006; 21: 307–315.
Stommel JM, Wahl GM . Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J 2004; 23: 1547–1556.
de Graaf P, Little NA, Ramos YF, Meulmeester E, Letteboer SJ, Jochemsen AG . Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J Biol Chem 2003; 278: 38315–38324.
LeBron C, Chen L, Gilkes DM, Chen J . Regulation of Mdmx nuclear import and degradation by Chk2 and 14-3-3. EMBO J 2006; 25: 1196–1206.
Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A et al. DNA damage-induced phosphorylation of MDMX at serine 367 activates p53 by targeting MDMX for MDM2-dependent degradation. Mol Cell Biol 2005; 25: 9608–9620.
Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG et al. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem 2002; 277: 19251–19254.
Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M . HDMX stimulates HDM2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA 2003; 100: 12009–12014.
Sharp DA, Kratowicz SA, Sank MJ, George DL . Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 1999; 274: 38189–38196.
Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M . MDM2 interacts with MDMX through their RING finger domains. FEBS Lett 1999; 447: 5–9.
Kostic M, Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE . Solution structure of the HDM2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol 2006; 363: 433–450.
Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM . RING domain-mediated interaction is a requirement for MDM2's E3 ligase activity. Cancer Res 2007; 67: 6026–6030.
Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem 2001; 276: 14537–14540.
Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van Lohuizen M, Sixma TK . Structure and E3-ligase activity of the RING-RING complex of polycomb proteins Bmi1 and RING1b. EMBO J 2006; 25: 2465–2474.
Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM . MDM2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 2000; 275: 8945–8951.
Poyurovsky MV, Priest C, Kentsis A, Borden KL, Pan ZQ, Pavletich N et al. The MDM2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J 2007; 26: 90–101.
Uldrijan S, Pannekoek WJ, Vousden KH . An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J 2007; 26: 102–112.
Vander Kooi CW, Ohi MD, Rosenberg JA, Oldham ML, Newcomer ME, Gould KL et al. The Prp19 U-box crystal structure suggests a common dimeric architecture for a class of oligomeric E3 ubiquitin ligases. Biochemistry 2006; 45: 121–130.
Zhang M, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou C et al. Chaperoned ubiquitylation – crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell 2005; 20: 525–538.
Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE . Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol 2001; 8: 833–837.
Li Z, Cao R, Wang M, Myers MP, Zhang Y, Xu RM . Structure of a Bmi-1-RING1b polycomb group ubiquitin ligase complex. J Biol Chem 2006; 281: 20643–20649.
Zheng N, Wang P, Jeffrey PD, Pavletich NP . Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 2000; 102: 533–539.
Ozkan E, Yu H, Deisenhofer J . Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc Natl Acad Sci USA 2005; 102: 18890–18895.
Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R et al. Amplification of MDMX (or MDM4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol 2004; 24: 5835–5843.
Singh RK, Iyappan S, Scheffner M . Hetero-oligomerization with MDMX rescues the ubiquitin/NEDD8 ligase activity of RING finger mutants of MDM2. J Biol Chem 2007; 282: 10901–10907.
Silke J, Kratina T, Chu D, Ekert PG, Day CL, Pakusch M et al. Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc Natl Acad Sci USA 2005; 102: 16182–16187.
Wang X, Taplick J, Geva N, Oren M . Inhibition of p53 degradation by MDM2 acetylation. FEBS Lett 2004; 561: 195–201.
Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindström MS et al. Targeted Inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 2007; 12: 355–366.
Stad R, Little NA, Xirodimas DP, Frenk R, van der Eb AJ, Lane DP et al. MDMX stabilizes p53 and MDM2 via two distinct mechanisms. EMBO Rep 2001; 2: 1029–1034.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848.
Marine JC, Dyer MA, Jochemsen AG . MDMX: from bench to bedside. J Cell Sci 2007; 120: 371–378.
Bell S, Klein C, Muller L, Hansen S, Buchner J . p53 contains large unstructured regions in its native state. J Mol Biol 2002; 322: 917–927.
Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT . Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol 2000; 20: 8458–8467.
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002; 416: 703–709.
Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV et al. Small molecule inhibitors of Hdm2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005; 7: 547–559.
CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994; 50: 760–763.
Vonrhein C, Blanc E, Roversi P, Bricogne G . Automated structure solution with autosharp. Methods Mol Biol 2006; 364: 215–230.
Perrakis A, Morris R, Lamzin VS . Automated protein model building combined with iterative structure refinement. Nat Struct Biol 1999; 6: 458–463.
Emsley P, Cowtan K . Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60: 2126–2132.
Acknowledgements
We thank Rayleen Fredericks-Short and Nelly Olova for excellent technical assistance, Torsten Kleffmann and the Centre for Protein Research (University of Otago) for mass spectrometry analysis, Mark Hinds for helpful discussions, and Andrew Mercer and Anthony Braithwaite for reagents. The SSRL Structural Molecular Biology program acknowledges the NCRR (Grant No P41 RR001209), a component of the NIH for funding. This work was supported by the Marsden Fund (NZ) (CLD), and PDM is a recipient of a Health Sciences Career Development Award (University of Otago).
Author information
Authors and Affiliations
Corresponding author
Additional information
Data deposition: The atomic co-ordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID 2vje and PDB ID 2vjf).
Edited by: B Zhivotovsky
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Linke, K., Mace, P., Smith, C. et al. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ 15, 841–848 (2008). https://doi.org/10.1038/sj.cdd.4402309
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402309
Keywords
This article is cited by
-
1H, 15N and 13C backbone resonance assignments of the acidic domain of the human MDM2 protein
Biomolecular NMR Assignments (2023)
-
Structure of the nutrient-sensing hub GATOR2
Nature (2022)
-
1H, 15N and 13C backbone resonance assignments of the acidic domain of the human MDMX protein
Biomolecular NMR Assignments (2022)
-
MDMX is essential for the regulation of p53 protein levels in the absence of a functional MDM2 C-terminal tail
BMC Molecular and Cell Biology (2021)
-
Critical role of cysteine-266 of SIE3 in regulating the ubiquitination and degradation of SIP1 transcription factor in Lotus japonicus
Planta (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}