
Abstract
Familial Adenomatous Polyposis (FAP) results from a germline mutation in the APC gene. A new mutation rate of 4–9×10−6 mutations/gametes/generation has been reported. In other familial cancer syndromes a bias for paternal origin of new mutations has been described. This bias is probably due to a larger number of cell divisions during spermatogenesis compared to oogenesis; giving a larger susceptibility for mutagenesis. We report here a molecular genetic analysis of 26 FAP patients with putative de novo APC mutations. In 15 families the novel origin of the mutations was confirmed by haplotyping and sequencing. Analysis of 10 of these mutations, in which the parental origin could be established, gave a 6 : 4 distribution in favour of maternal origin. This is in agreement with a 1 : 1 distribution and does not indicate an expected paternal bias. Moreover, no parental age effect was identified. We propose that APC germline mutations are not premeiotic events but more likely arise during the meiosis. This would give an equal susceptibility for mutagenesis during spermatogenesis and oogenesis, respectively. The model is in concordance with the previously established difference between APC somatic mutations, as being a mitotic event and APC germline mutations, as being a meiotic event. The confirmation of 15 de novo mutations by a molecular genetic approach is in fine agreement with previous results based on clinical records.
Similar content being viewed by others
Introduction
Familial Adenomatous Polyposis (FAP) is a predisposition to colorectal cancer and other malignancies inherited in an autosomal dominant mode. The estimated birth frequency in Scandinavia is approximately 1 : 13 500–1 : 21 500.1,2,3
The main clinical feature of the disease is the development of numerous polyps (adenomas) in the colon and rectum and the penetrance is close to 100% at age 40 years. Without surgical intervention most patients would die from colorectal adenocarcinoma before the age of 40–50 years. Extracolonic manifestations are frequently observed and include upper gastrointestinal tumours, osteomas, epidermoid cysts, desmoid tumours and congenital hypertrophy of the retinal epithelium. FAP exhibits variable expression with marked inter- and intrafamilial variation.
FAP is caused by germline mutations in the tumour suppressor gene Adenomatous Polyposis Coli (APC, OMIM 175100), which maps to chromosome 5q21 and was identified by a positional cloning approach in 1991.4,5,6,7 The APC gene product is a 2843-amino acid polypeptide, which is suggested to repress expression of the c-MYC oncogene by binding and degrading β-catenin.8 The mutation spectrum is very heterogeneous, with hundreds of different mutations scattered primarily throughout the 5′ region of the 8.5-kb large coding region. The majority of these mutations are chain-terminating mutations resulting in truncated APC proteins (review in 9). Germline mutations causing FAP may be inherited or occur de novo. In two recent publications the rate of new mutations was reported to be between 4 and 9×10−6 mutations/gametes/generation, respectively1,2 and the proportion of FAP cases due to new mutations was between 11 and 25%.
In an increasing number of monogenetic disorders patterns of new mutations have been elucidated.10,11,12 The main part of disorders have shown a marked bias towards paternal origin of the new mutation (eg Apert syndrome12, multiple endocrine neoplasia type 2B10, retinoblastoma11, and achondroplasia13) often combined with an age effect since de novo mutations often appear with older fathers. Only a few disorders (for example von Hippel-Lindau disease) have failed to demonstrate a paternal bias.14
The present study presents the results of molecular analysis of families in which FAP apparently is caused by a new mutation. To our knowledge this is the first molecular genetic analysis of a larger group of APC de novo mutations. In 88% of the informative families presence of a new mutation was confirmed. The parental origin of the new mutation was established in 10 of the families and showed no evidence of an expected paternal bias. This could indicate that the event leading to the APC germline mutations happens during meiosis and not during mitotic replications prior to the meiosis.
Materials and Methods
Patients and families
The Danish Polyposis Register was established in 1971. The register included 508 patients with histologically verified polyposis in 165 families diagnosed during the period 1.1.1910 to 1.8.2001. We included all families (n=26) from the register with (a) an identified APC germline mutation and (b) a family history compatible with a new mutation. In seven families the affected individual had not reproduced, and in five families both parents died. From the identified 26 families blood samples from all available first-degree relatives of the affected individual(s) (n=102) were selected for direct sequencing and haplotyping. Information on the parental age at birth was extracted from the Danish Polyposis Register.
Mutation analysis
DNA was extracted from venous blood using QIAmp DNA Blood Mini Kit according to the method described by the manufacturer (supplied by Kebo Lab, Albertslund, Denmark).
Mutations in the APC gene was detected by direct sequencing of the relevant genomic DNA fragment amplified by the polymerase chain reaction (PCR). For the sequence analysis, the amplification primers designed by Groden et al4 were used. Direct sequencing of genomic PCR product was performed in an ABI 377 DNA sequencer (Applied Biosystems, Naerum, Denmark) by use of dye-terminator or dye-primer cycle sequencing, according to the manufacturer's manual (Applied Biosystems). Sequence reactions were carried out either on a Vistra DNA labstation 635 or by hand. All results were confirmed by analysis of two independently collected blood samples and were sequenced in both directions.
Haptotyping
Haplotyping was performed using six PCR-based microsatellite markers. The order of loci on chromosome 5q21-22 is: CEN - D5S2084 - D5S659 - D5S82 - APC - D5S346 - D5S2070 - D5S1975 - TEL.15,16 The marker sets were spanning 16 cM and D5S346 mapped within 70 kb of the APC gene.17 Amplification of the CAYN5.64c repeat (D5S82) was performed with CA primer as described by Breukel et al18 and TG primer 5′-TGAGGGCCAACGGATTATGA-3′.
PCR was performed in a total reaction volume of 22 μl, using 100 ng of genomic DNA, 0.2 μM of each primer, 50 μM of each dNTPs, 1.0 mM MgCl, 0.625 U Promega Taq polymerase (supplied by Bie & Berntsen A-S, Rodovre, Denmark), and 1× Promega Reaction Buffer. The solution was run on a Techne Genius thermal cycler with one cycle of 95°C, 4 min followed by 26 cycles of 95°C, 30 s; 55°C, 30 s; 72°C, 30 s; and a final extension at 72°C for 4 min. One primer of each pair was 5′ABI dye labelled. The products were separated on an ABI 377 sequencer and were analysed with GeneScan Software 2.02 (Applied Biosystems).
To identify the mutated allele, the mutation status of at least one child of the proband was determined and by a subsequent haplotyping of these two individuals the mutated and the normal allele of the affected individual were identified. The parental origin was determined by a subsequent haplotyping of one or more parent/sibling.
To verify a new mutation, all available siblings and parents of the proband were haplotyped to find an individual with the mutated allele but without the mutation. If the proband had not reproduced, the new mutation was verified by the finding of both alleles of the proband in siblings or parents without the mutation.
Statistical analysis
The parental ages at the birth of new mutation cases were compared with that expected from the general population in 1950 (the median birth year of the patients). The expected maternal age at birth were calculated using 5 year fertility rates.19,20 The expected paternal age is assessed by adding 3.5 years to the maternal age at birth, since Danish women in average were 3.8 years younger than their spouses.21 The 95% confidence interval is calculated using two sided t distribution.
The parental origin of de novo mutations was analysed calculating one-tailed 95% significance test, using exact binomial probability. The expected parental biases were set at 10 : 1, because the secondary spermatocyte at reproductive age has undergone at least 10 times the number of mitotic divisions of a woman (see Discussion).
Results
Parental origin
To confirm that apparent new mutations in fact were new mutations, DNA samples were collected from all accessible members from 26 families in whom a new mutation seemed likely, evaluated from the pedigree. In nine of these families DNA was not available from siblings or parents of the affected individual and the new mutation status could not be verified. Direct sequencing and haplotyping of the remaining 17 families confirmed the new mutation in 15 families. Two families exhibited a mutual founder mutation (Figures 1 and 2). In six families both parents were available and the de novo mutation was determined by direct sequencing alone. Haplotyping was performed to exclude non-paternity. In the remaining nine families only one parent was available for testing, and the new mutation was confirmed by subsequent sequencing and haplotyping of one or several siblings and children.

Distribution of the included families. The bottom two bars show the distribution of the 26 included families between new mutations, familial cases and undetermined ones. The top bar distributes the de novo mutations according to the parental origin.

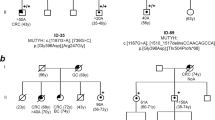
Haplotypes of the families. Pedigrees of FAP families with confirmed APC de novo mutation. Family number and mutation description are above the pedigree. (A) The six families with a maternal origin of the APC new mutation. (B) The four families with a paternal origin of the APC new mutation. (C) The five families where the parental origin of the APC new mutation could not be determined. General symbols: squares, males; circles, females; all symbols with a diagonal, deceased. Open symbols, clinically unaffected; half filled, clinically and histologically verified FAP; solid, colorectal cancer. Probands are indicated with an arrow. The parental age at birth of the proband are given below the symbols. Alleles of the microsatellites are determined in length (bp) and inferred results are in brackets. Haplotypes are displayed as vertical bars, and only the supposed disease haplotype is shown. Mut neg/mut pos/NT above the haplotype indicate if the family mutation has been identified by direct sequencing in the person or has not been tested (NT).
By haplotyping we were able to determine the parental origin of the de novo APC germline mutation in 10 families. Six families were found with a maternal origin and four families with paternal origin (Figure 2). This distribution differs significantly from a paternal bias of 10 : 1 (P<0.001), and is in agreement with a 1 : 1 distribution.
Parental age
Parents to the 10 new mutations with identified parental origin had a mean age at the child's birth of 27.1 years (ci ±3.6); father's age: 27.5 (ci ±5.9) and mother's age: 26.8 (ci ±6.3). If the five families with de novo mutations but without an identified parent of origin were included the mean paternal and maternal ages at child's birth were 28.4 (ci ±2.7) and 27.0 (ci ±3.4) years, respectively. These ages are not significantly different from the expected of 31.0 and 27.5 years.19,20,21
Mutations
The 15 proven new mutations were mainly four or five base pair deletions, or non-sense mutations and do not differ from what would be expected from the literature when considering all germline APC mutations (Table 1Table 1).
We searched the APC database (http://perso.curie.fr/thierry.soussi/APC.html) and the Human Gene Mutation Database22 and found that four of the mutations had not previously been published.
These novel APC variants consisted of two one base deletions in codon 771 and 1219 respectively, and in one family we found an insertion of CC in codon 881. As shown in Table 1, the mutation is a duplication of the wildtype CC. The last novel mutation is a 4 bp deletion in the context of a 3 bp repeat (codon 1160–1161). All four mutations resulted in a frameshift and introduced downstream stopcodons (Table 1).
Discussion
Several studies of other dominantly inherited disorders have shown a paternal bias for point mutations10,11,12,13 and a maternal bias for larger structural changes.23,24 Since families in this study have only small mutations (similar to point mutations) a marked paternal bias was expected, but there was no evidence of a bias for a paternal origin of de novo APC gene mutations and no parental age effect was identified. The sample size of 10 gave a 78% probability of detecting a maternal/paternal ratio of 1 : 10. Paternal bias has been described as caused by the differences between spermatogenesis and oogenesis. A secondary oocyte will have undergone 24 mitotic divisions regardless of the woman's age, whereas a secondary spermatocyte in a man at age 28 years will have undergone approximately 335 mitotic divisions with the number rising every year.25 If the germline mutations arose during these premeiotic mitotic divisions one would expect a marked bias for a paternal origin and a striking parental age effect as seen in disorders like Apert syndrome12 and multiple endocrine neoplasia type 2B.10 Paternal bias is frequently observed but the parental origin of different genetic disorders is complex and the present results resembles those found in von Hippel-Lindau disease.14
The failure to observe an underlying paternal bias among FAP patients could be explained by a selective disadvantage of the mutated male germ cell or by maternal imprinting. RNA assays have, however, shown both APC alleles to be transcribed,26 which makes imprinting unlikely and selective disadvantage of germ cells with a point mutation, has to our knowledge never been described. We therefore find it most likely that the observed proportion reflects the true proportion in formation of de novo APC germline mutations.
Another remarkable feature of APC germline mutations is the clear distinction (especially in location) from somatic APC mutations as observed in the APC mutation database (http://perso.curie.fr/thierry.soussi/APC.html) and from a review of the literature,27 for instance >85% of somatic mutations are clustered between codons 1280 and 1500 compared to only 29% of germline mutations in this region. Up till now this difference has been assigned a difference in selection of germline vs somatic APC mutations. A difference could also arise due to a pronounced founding effect. However, about 80% of FAP families have their own unique germline mutation – and in the remaining cases of hot spot mutations a founder effect has not been described. It is therefore to be assumed that the difference between germline mutations and somatic mutations is not due to any founding bias.
Taken together the present results and the difference between germline and somatic mutations indicate that the APC germline mutations arise during the meiotic division since both the male gamete and the female gamete undergoes this division only once giving unity in parental origin. Moreover the meiotic division is identical in male and female giving no sex difference in type of mutation and finally the meiotic division differs from the mitotic division where somatic mutations arise. There is no regular DNA replication during meiosis, but by the use of antibodies Plug et al28 has localised DNA polymerase β (pol β) along homologous chromosomes as they synapse and progress through prophase of meiosis I in spermatocytes as well as in oocytes. The data were consistent with a role for pol β in both semiconservative DNA synthesis and in repair synthesis during early stages of meiosis.28
Whereas the polymerase α (among others) is believed to participate in replication of genomic DNA at mitosis – pol β does not seem to play a direct role in this process (review in 29). Several studies have shown that the error rate during DNA synthesis is non-random. The error rate is sequence dependent and dependent of the DNA polymerase, that is pol α and pol β has different fidelity and they have different mutation hot spots.30,31 This could explain the different locations of somatic mutations and germline mutations – somatic mutations arising during the mitotic replication by pol α and the germline mutations arising during meiosis by pol β.
An APC mutational mosaic resembles a de novo APC mutation by mutation screening and pedigree analysis. However, in mosaic individuals only a fraction of the blood leukocytes contain the APC mutation, implying that the new mutation has emerged postmeiotic after at least some mitotic divisions in the embryo. According to the proposed model, APC mutational mosaic cannot arise by the same mechanism as the true de novo mutations, but rather by the same mitotic mechanism as somatic APC mutations. Farrington and Dunlop32 have identified two individuals being APC mutational mosaics, one being a non-sense mutation (APC 4192G→T). A literature search revealed at least four colorectal tumours harbouring the same somatic APC mutation33,34 and only one family with this mutation as a germline mutation.35 No somatic nor germline reports of the other mutation published by Farrington and Dunlop32 was found. As only two mosaic mutations has been described it is difficult to draw extended conclusions, but it is noticeable that the result is in concordance with our proposed model, with the APC germline mutations being of meiotic origin and differing from somatic mutations as found in mosaic individuals (mitotic mutation very early in embryo genesis) and colorectal cancers.
The APC mutation rate has been calculated to be 9×10−6 and 4×10−6 mutations per gamete per generation.1,2 We determined the de novo mutation status in nine cases previously examined by Bisgaard et al.1 Six cases were then grouped as unknown and three as new mutations. By direct sequencing and haplotyping the three new mutations were verified, four of the unknown cases were found to be new mutations, and two to be inherited cases. These few cases (n=9) which are selected for an identified new mutation and a family history compatible with a new mutation confirm the former assumed distribution between new mutations and unknown cases.
The mutation rate calculated by clinical findings by Bisgaard et al1 is supported by the present molecular genetic approach. The Swedish Polyposis register2 had identified 145 families in a population of approximately nine million inhabitants, while the Danish Polyposis register comprised almost the same number of families from a population of five million. The most likely explanation of the difference between the Danish and Swedish mutation rates calculated by direct method may be related to a lower number of included de novo mutations in the Swedish material.
The results raise the questions why the APC gene is susceptible to genetic changes during the meiosis when most other examined genes are apparently not. And moreover, why the APC gene may not be susceptible to mitotic changes prior to meiosis. Additional knowledge of the meiotic process and the impact of gene structure may clear this out.
To our knowledge this is the first analysis of de novo APC germline mutations. The parental origin of the mutations showed a surprising lack of bias towards paternal origin. We have presented a model of mutation formation explaining this difference. The model is in concordance with the known difference between germline and somatic APC mutations. In addition the results supports a former reported APC new mutation rate.
References
Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J . Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate Hum Mutat 1994 3: 121–125
Björk J, Akerbrant H, Iselius L, Alm T, Hultcrantz R . Epidemiology of familial adenomatous polyposis in Sweden: changes over time and differences in phenotype between males and females Scand J Gastroenterol 1999 34: 1230–1235
Järvinen HJ . Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival Gut 1992 33: 357–360
Groden J, Thliveris A, Samowitz W et al. Identification and characterization of the familial adenomatous polyposis coli gene Cell 1991 66: 589–600
Joslyn G, Carlson M, Thliveris A et al. Identification of deletion mutations and three new genes at the familial polyposis locus Cell 1991 66: 601–613
Kinzler KW, Nilbert MC, Su LK et al. Identification of FAP locus genes from chromosome 5q21 Science 1991 253: 661–665
Nishisho I, Nakamura Y, Miyoshi Y et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients Science 1991 253: 665–669
He TC, Sparks AB, Rago C et al. Identification of c-MYC as a target of the APC pathway Science 1998 281: 1509–1512
Polakis P . Mutations in the APC gene and their implications for protein structure and function Curr Opin Genet Dev 1995 5: 66–71
Carlson KM, Bracamontes J, Jackson CE et al. Parent-of-origin effects in multiple endocrine neoplasia type 2B Am J Hum Genet 1994 55: 1076–1082
Kato MV, Ishizaki K, Shimizu T et al. Parental origin of germ-line and somatic mutations in the retinoblastoma gene Hum Genet 1994 94: 31–38
Moloney DM, Slaney SF, Oldridge M et al. Exclusive paternal origin of new mutations in Apert syndrome Nat Genet 1996 13: 48–53
Wilkin DJ, Szabo JK, Cameron R et al. Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome Am J Hum Genet 1998 63: 711–716
Richards FM, Payne SJ, Zbar B, Affara NA, Ferguson-Smith MA, Maher ER . Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene Hum Mol Genet 1995 4: 2139–2143
Dib C, Faure S, Fizames C et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites Nature 1996 380: 152–154
Olschwang S, Laurent-Puig P, Melot T, Thuille B, Thomas G . High resolution genetic map of the adenomatous polyposis coli gene (APC) region Am J Med Genet 1995 56: 413–419
Spirio L, Joslyn G, Nelson L, Leppert M, White R . A CA repeat 30-70 KB downstream from the adenomatous polyposis coli (APC) gene Nucleic Acids Res 1991 19: 6348
Breukel C, Tops C, van Leeuwen C et al. CA repeat polymorphism at the D5S82 locus, proximal to adenomatous polyposis coli (APC) Nucleic Acids Res 1991 19: 5804
Vital Statistics. Denmark Statistics; Copenhagen, Denmark 1951
Ægteskaber, fødte og døde 1956–1969. Denmark Statistics; Copenhagen, Denmark 1973
Ægteskaber, fødte og døde 1941–1955. Denmark Statistics; Copenhagen, Denmark 1962
Krawczak M, Cooper DN . The human gene mutation database Trends Genet 1997 13: 121–122
Grimm T, Meng G, Liechti-Gallati S, Bettecken T, Muller CR, Muller B . On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis J Med Genet 1994 31: 183–186
Lazaro C, Gaona A, Ainsworth P et al. Sex differences in mutational rate and mutational mechanism in the NF1 gene in neurofibromatosis type 1 patients Hum Genet 1996 98: 696–699
Vogel F, Motulsky AG, Vogel F, Motulsky AG (eds). Human Genetics: problems and approaches Berlin: Springer Verlag 1997 pp 385–430
Powell SM, Petersen GM, Krush AJ et al. Molecular diagnosis of familial adenomatous polyposis N Engl J Med 1993 329: 1982–1987
Miyaki M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Konishi M . Familial polyposis: recent advances Crit Rev Oncol Hematol 1995 19: 1–31
Plug AW, Clairmont CA, Sapi E, Ashley T, Sweasy JB . Evidence for a role for DNA polymerasebeta inmammalian meiosis Proc Natl Acad Sci USA 1997 94: 1327–1331
Hubscher U, Nasheuer HP, Syvaoja JE . Eukaryotic DNA polymerases, a growing family Trends Biochem Sci 2000 25: 143–147
Kunkel TA . Misalignment-mediated DNA synthesis errors Biochemistry 1990 29: 8003–8011
Osheroff WP, Jung HK, Beard WA, Wilson SH, Kunkel TA . The fidelity of DNA polymerase beta during distributive and processive DNA synthesis J Biol Chem 1999 274: 3642–3650
Farrington SM, Dunlop MG . Mosaicism and sporadic familial adenomatous polyposis Am J Hum Genet 1999 64: 653–658
Spirio LN, Samowitz W, Robertson J et al. Alleles of APC modulate the frequency and classes of mutations that lead to colon polyps Nat Genet 1998 20: 385–388
Miyaki M, Konishi M, Kikuchi-Yanoshita R et al. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors Cancer Res 1994 54: 3011–3020
Lamlum H, Ilyas M, Rowan A et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson's ‘two-hit’ hypothesis Nat Med 1999 5: 1071–1075
Antonarakis SE . Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group Hum Mutat 1998 11: 1–3
Acknowledgements
Vibeke Reichardt and Vibeke Trandbohus are thanked for technical assistance in the mutation identification process. We are also grateful to Marianne Schwartz for providing lab-space. The work was funded with a scholarship from H:S Sundhedsfaglige Forskningsråd
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ripa, R., Bisgaard, M., Bülow, S. et al. De novo mutations in familial adenomatous polyposis (FAP). Eur J Hum Genet 10, 631–637 (2002). https://doi.org/10.1038/sj.ejhg.5200853
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200853
Keywords
This article is cited by
-
Novel read through agent: ZKN-0013 demonstrates efficacy in APCmin model of familial adenomatous polyposis
Journal of Molecular Medicine (2023)
-
A de novo pathogenic variant in the MSH6 gene in a 52 years-old woman
Familial Cancer (2022)
-
Trabecular and cortical mandibular bone investigation in familial adenomatous polyposis patients
Scientific Reports (2021)
-
Somatic APC mosaicism and oligogenic inheritance in genetically unsolved colorectal adenomatous polyposis patients
European Journal of Human Genetics (2018)
-
Breast and ovarian cancer predisposition due to de novo BRCA1 and BRCA2 mutations
Oncogene (2016)
