
Abstract
Aims
Age-related macular degeneration (AMD) is considered a complex genetic disease, although the genetic influences are not yet fully understood. Genetic analysis is hampered by the late onset of disease and the difficulty in obtaining multigenerational families. To investigate this problem further we studied our population of early onset drusen cases. The Arg345Trp mutation on exon 10 of the EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1) gene causes two clinical phenotypes of early onset drusen (Doyne honeycomb retinal dystrophy and Malattia Leventinese), yet does not appear to be involved in other early onset drusen phenotypes or typical AMD. We wished to ascertain the involvement of the EFEMP1 gene in our population of sporadic and familial subjects presenting with early onset drusen and their affected relatives.
Methods
Individuals presenting with drusen/end-stage maculopathy at 60 years or under were identified from retinal clinics in Melbourne. All available first- and second-degree relatives were also examined. In all, 116 ethnically matched controls were collected from the same community for comparison.
Results
Single stranded conformational polymorphism (SSCP) analysis and subsequent sequencing revealed four previously described and three novel sequence variations. Most occurred at similar frequencies in the case and control populations and were not thought to be disease associated.
Conclusion
The term early onset drusen encompasses a wide range of phenotypes and our findings indicate that it is likely that more than one gene is involved in its causation. It is essential that these clinical phenotypes are well described and categorised to allow greater possibility of success in the search for other disease genes.
Similar content being viewed by others

Introduction
Age-related macular degeneration (AMD) is an increasingly common cause of blind registration in the elderly population in Western societies. Although it is clear from family1 and twin studies2 that there is a genetic component to the disease, the actual genes involved remain an enigma. Genetic analysis has in part been hampered by difficulty in collecting multigenerational families, due to the late onset of disease and the possible confounding influence of other environmental factors.
Early stage disease, referred to as age-related maculopathy (ARM), typically presents with soft drusen (>63 μm), which are often asymptomatic. Progression can occur to geographic atrophy or choroidal neovascularisation with potentially severe disability, and at this stage is referred to as AMD. The prevalence of AMD is very much age dependent: 0.21% between 55 and 64 years, 0.85% between 65 and 74 years, 4.59% between 75 and 84 years, and 13.05% over 85 years (pooled figures from three studies).3
Typical ARM and AMD changes can be found in people younger than 60 years although this is rare. Data from the Melbourne Visual Impairment Project indicated that only 0.8% of the general population under 60 years have 10 or more medium-sized drusen (125–250 μm). Thus this rare early onset group is a very valuable resource for genetic studies as they would be more likely to have a greater genetic contribution to their maculopathy compared to those who develop the disease at a later age.
Two most well-recognised forms of early onset drusen are Doyne honeycomb retinal dystrophy (DHRD) and Malattia Leventinese (ML), both of which exhibit a dominant pattern of genetic inheritance. DHRD is diagnosed if drusen are found around the optic nerve head, while in ML the drusen run in radial streaks from the centre of the macula. Both diseases typically present between the second and fourth decade. Of all the inherited maculopathies, these are thought to have the most in common with the ARM/AMD phenotype.
The mutational event responsible for DHRD and ML is a C>T base pair transition at codon 345 of the EFEMP1 gene (Arg345Trp mutation) that causes an arginine-to-tryptophan change.4 The exact function of the EFEMP1 gene is not known, but has been found to accumulate within and beneath RPE cells in areas of drusen in ML and AMD. It does not accumulate in normal eyes with drusen or in retina without pathology.5
The Arg345Trp mutation has been investigated in other early onset drusen phenotypes6 and in typical sporadic and familial AMD.4, 6 However, this mutation does not appear to be implicated in these diseases.
Since the EFEMP1 gene is known to be responsible for certain drusen phenotypes (DHRD/ML), and can be implicated pathophysiologically in drusen formation, it is important that the potential for its involvement in other drusen phenotypes is fully investigated.
Two studies have investigated the role of the other exons of the EFEMP1 gene in a small number of sporadic (n=147, n=168) and familial (n=38) cases of early onset drusen. Both excluded the involvement of this gene in their populations: however, only one of these studies examined the untranslated exons 1 and 2.
The purpose of this study was to extend the previous studies and examine the entire EFEMP1 gene (untranslated exons and 10 coding exons) in a larger than previously described group of sporadic and familial cases of early onset drusen.
Materials and methods
Patient selection
All subjects were initially identified as part of our AMD inheritance study, through outpatient clinics at the Royal Victorian Eye and Ear Hospital or through referral from private clinics. Medical histories were taken together with information on ethnicity and family history. Eye examinations were undertaken by an ophthalmologist on all the subjects enrolled in the study, and fundus photographs were taken. Written informed consent that had institutional ethical approval was obtained from all the subjects recruited. The study was conducted in accordance with the declaration of Helsinki as revised in 1989 and the National Health and Medical Research Council of Australia's statement on ethical conduct in research on humans, revised in 1999.
For the purposes of this study, index cases were individuals who were 60 years and under who presented with soft drusen (>63 μm) typical of ARM in one or both eyes, or who had late stage changes (neovascular or atrophic lesions) by the time of presentation. We examined available relatives of all ages with or without a history of eye disease.
In all, 116 control subjects were collected from the same community as part of the large population-based epidemiological study, the Melbourne Visual Impairment Project. Our control population was older than the index cases (ie 60 years and over) in order to exclude an early onset drusen phenotype. All these individuals were examined by an ophthalmologist and were deemed to be controls if they had no retinal pathology, apart from a few hard drusen (less than 10 hard drusen of less than 63 μm).
Genetic techniques
A sample of 10 ml of whole blood was collected from each patient and genomic DNA isolated using a standard phenol/chloroform extraction procedure.9 The EFEMP1 gene consists of 12 exons, with exons 1 and 2 being untranslated8 and exons 3–10 defined as the coding exons.8 However, exons 1 and 2 do contain a number of control elements involved in transcription and alternative splicing.10 All 12 exons were therefore screened using a polymerase chain reaction (PCR)-based approach. The sequence of primers used in this analysis (available on request) were derived from the EFEMP1 gene sequence published by the National Center for Biotechnology Information (NCBI). All primers were designed to have a length of 18–30 nucleotides and a G/C content of 40–60%. PCR amplification of each exon was performed using these primer pairs with Taq DNA polymerase in a 25 μl reaction. Amplification parameters consisted of a denaturation step of 95°C for 5 min, followed by 40 cycles of 94°C for 30 s, annealing temperature for 30 s, and 72°C for 30 s, followed by a final extension step of 72°C for 10 min.
All the 12 exons and their adjacent intronic boundaries were screened by single stranded conformational polymorphism (SSCP) for the presence of mutations or single nucleotide polymorphisms (SNP) using previously described conditions.11 In brief, 3 μl of a formamide-based denaturing mixture was added to the PCR mixture. The mixture was then denatured at 95°C for 5 min, and 4–5 μl of the mix was loaded onto a 6% polyacrylamide gel. Gels were run under constant temperature conditions of 19°C for 4–6 h at 7 w or overnight for 15 h at 3 w.
Exon 10 was screened first to identify any individual who may be carrying the DHRD/ML Arg345Trp mutation, so that they could be excluded from further analysis.
Individuals who displayed a mobility shift during screening of the 12 exons were bidirectionally dideoxy-sequenced and analysed using an ABI 377 automated sequencer.
Results
A total of 54 index patients presenting with early onset drusen were examined with an age range of 24–60 years (mean=50. 3 years). Of the 21 cases with a family history of maculopathy the mean age was 51.7 years and in 33 single or sporadic cases the mean age was 49.4 years. In all, 35 affected relatives were also examined: these included parents, siblings, children (first degree) or aunts and nieces/nephews (second degree). Their age range was 36–88 years.
There was considerable intrafamilial variability of macular features (Figure 1), ranging from early onset drusen, similar to that seen in ARM, to typical late onset AMD. There was also a great variety in the macular features seen between index cases (Figure 2). Even in early stage disease, the distribution, number, and size of drusen differed between individuals, but this variability was in keeping with that seen in typical older onset ARM. Also, many young onset patients presented with late stage disease typical of AMD (35.2%).

Colour fundus photographs showing daughter (a) and father (b) aged 24 and 57 years, respectively, presenting with bilateral asymptomatic early onset drusen. Note the difference in drusen appearance between father and daughter.

Colour fundus photographs showing sporadic female aged 46 years presenting with bilateral drusen. Note again the different size and location of the drusen compared to the index case in Figure 1a.
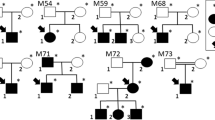
One of the largest families seen consisted of six affected individuals (Figure 3). The index case (II:5) was diagnosed with maculopathy at the age of 59 years and had three sisters (II:2, II:3, II:4) affected with late stage AMD at ages 70, 69, and 70 years, respectively. Her younger brother (II:7) was found to have asymptomatic bilateral drusen at the age of 67 years during our examination of the family. One female sibling (II:3) had a daughter with asymptomatic soft drusen diagnosed at the age of 45 of years (III:1).

Pedigree of large family with six members affected with early onset drusen and ARM/AMD.
During SSCP analysis of exon 10, we detected one family (father and daughter) who were found to have a mobility shift that was identical to the DHRD positive control with the Arg345Trp mutation. Direct sequencing confirmed the presence of this mutation. The daughter had been diagnosed with early onset drusen at the age of 46 years (Figure 4a). Her father was diagnosed with typical AMD at the age of 75 years (Figure 4b) and had a disciform scar in the left eye and geographic atrophy in the right eye at examination. Neither was thought to show obvious features of DHRD or ML on examination by their treating ophthalmologist or by the authors of this paper. Following the sequence results and with the benefit of retrospective examination some features could be observed that would be consistent with a DHRD/ML phenotype (few drusen around the optic nerve head and subtle radial macular drusen). They were therefore excluded from further analysis in this early onset drusen study.

Colour fundus photographs showing daughter (a) and father (b) who were found to possess the Arg345Trp mutation on the SSCP analysis.
SSCP analysis of the EFEMP1 gene revealed band shifts in six of the 12 exons (exons 1, 5, 6, 10, 11, and 12). Duplication of the gel revealed identical band shifts in the same samples as the original gels. All six exons had banding profiles, where nucleotide changes were identified following sequence analysis (Table 1 ). The four previously described nucleotide variations were found and three novel sequence variations were discovered. Most occurred at similar frequencies in the case and control populations, and were thus not thought to be disease associated. The nucleotide change that was seen in exon 12 occurred in the control population at a frequency of 0.87% and was not detected at all in the cases. This difference in frequency was not significant (P=1.00). At such a low frequency, a significant difference may not have been seen due to the small number of cases in the study. Thus, its appearance in only the controls is not unexpected and its likely involvement in the disease is therefore limited.
Discussion
In this study, we have been able to exclude the involvement of sequence changes within the coding and untranslated regions of the EFEMP1 gene in the causation of our early onset drusen phenotypes.
A previous study excluded the involvement of the coding region (exons 3–12) of the EFEMP1 gene in 14 sporadic cases of early onset drusen.7 This ‘early onset’ group included a wide range of phenotypes and an individual who developed drusen at the age of 63 years. Tarttelin et al8 found that the untranslated region (exons 1 and 2) and coding exons of the EFEMP1 gene were not involved in disease causation in three families and 16 sporadic cases of early onset drusen. In our study, we were able to exclude the involvement of both the untranslated region and coding region in 20 familial and 33 sporadic cases (once the father/daughter family with the DHRD mutation were removed from the study). The sequence variations that we did find within this gene were present in similar frequencies in both cases and controls, implying that they are unlikely to be associated with disease.
The mutation technique of SSCP has been in use since 1989 and a review of the technique reported the mutation detection rate to be 90% in fragments up to 200 bp12 and 80% in fragments between 200 bp and 350 bp in size13. The conditions described for the SSCP analysis in this study allowed reproducible detection of nucleotide alterations. Di-deoxy nucleotide sequencing did not reveal the presence of false negatives through SSCP and the 80–90% detection rate for this technique is comparable to a number of other currently used mutation detection techniques.
Thus far, the Arg345Trp mutation is the only mutation found within either the coding or adjacent intronic regions of the EFEMP1 gene in familial or sporadic early onset drusen. This may suggest that the Arg345Trp mutation is responsible for producing a specific drusen phenotype (drusen around the optic nerve head in DHRD and radial drusen in ML). However, other mutational events may also be capable of causing this phenotype. Toto et al (2002)14 described a family with ML that did not have this mutation and we have also described an individual with classic Doyne phenotype from a family of early onset drusen who did not possess this mutation (ARVO Abstract 2003). Although the absence of mutations within the coding region of EFEMP1 suggests a lack of involvement for this gene in the disease, it does not totally rule out its involvement. Recent reports have indicated that mutations within intronic regions of genes can also alter gene expression. These can arise through pseudoexon activation, where a splice site is created,15 or where a silencer/enhancer sequence is disrupted.16 We have not screened the entire intronic region (approximately 54 kilobase pairs) of the EFEMP1 gene and we are not aware of other studies that have undertaken this analysis. Therefore, it still remains possible that further analysis will reveal an intronic change within this gene that may play a role in the disease.
Together, these findings indicate that it is likely that more than one gene is involved in the causation of early onset drusen. The term early onset drusen encompasses a wide range of phenotypes, and subgrouping these further will greatly assist the investigation of the genetic influences on these diseases.
References
Heiba IM, Elston RC, Klein BE, Klein R . Sibling correlations and segregation analysis of age-related maculopathy: the Beaver Dam Eye Study. Genet Epidemiol 1994; 11: 51–67.
Klein ML, Mauldin WM, Stoumbos VD . Heredity and age-related macular degeneration. Observations in monozygotic twins. Arch Ophthalmol 1994; 112(7): 932–937.
Smith W, Assink J, Klein R, Mitchell P, Klaver CCW, Klein BEK et al. Risk factors for age-related macular degeneration. Ophthalmology 2001; 108: 697–704.
Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet 1999; 22: 199–202.
Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D et al. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci USA 2002; 99: 13067–13072.
Guymer RH, McNeil R, Cain M, Tomlin B, Allen PJ, Dip CL et al. Analysis of the Arg345Trp disease-associated allele of the EFEMP1 gene in individuals with early onset drusen or familial age-related macular degeneration. Clin Exp Ophthalmol 2002; 30: 419–423.
Sauer CG, White K, Kellner U, Rudolph G, Jurklies B, Pauleikhoff et al. EFEMP1 is not associated with sporadic early onset drusen. Ophthal Genet 2001; 22: 27–34.
Tarttelin EE, Gregory-Evans CY, Bird AC, Weleber RG, Klein ML, Blackburn J et al. Molecular genetic heterogeneity in autosomal dominant drusen. J Med Genet 2001; 38: 381–384.
Sambrook J, Fritsch EF, Mariatas T . Molecular Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbour Press: 1989 Cold Spring Harbour, NY.
Lecka-Czernik B, Lumpkin Jr CK, Goldstein S . An overexpressed gene transcript in senescent and quiescent human fibroblasts encoding a novel protein in the epidermal growth factor-like repeat family stimulates DNA synthesis. Mol Cell Biol 1995; 15(1): 120–128.
Orita M, Iwanhana H, Kanazawa H, Hayashi K, Sekiya T . Detection of polymorphisms of human DNA by gel electrophoresis as single stranded conformation polymorphisms. Proc Natl Acad Sci USA 1989; 86: 2766–2770.
Nataraj AJ, Olivos-Glander I, Kusukawa N, Highsmith Jr WE . Single-strand conformation polymorphism and heteroduplex analysis for gel-based mutation detection. Electrophoresis 1999; 20(6): 1177–1185.
Hayashi K, Yandell DW . How sensitive is PCR-SSCP? Hum Mutat 1993; 2(5): 338–346.
Toto L, Parodi MB, Baralle F, Casari G, Ravalico G, Romano M . Genetic heterogeneity in Malattia Leventinese. Clin Genet 2002; 62: 399–403.
Tuffery-Giraud S, Saquet C, Chambert S, Claustres M . Pseudoexon activation in the DMD gene as a novel mechanism for Becker muscular dystrophy. Hum Mutat 2003; 21(6): 608–614.
Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM, Arlt W et al. Mutations in the genes encoding 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet. 2003; 34(4): 434–439.
Acknowledgements
This work was funded by Perpetual Trustees, The Royal Victorian Institute for the Blind, The Helen Macpherson Smith Trust, The LEW Carty Charitable Fund and the Eye, Ear, Nose and Throat Research Institute and was presented at the Royal College of Ophthalmologists (UK) Annual Congress 2003. We thank Dr Hien Vu for her statistical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors have no proprietary interests in the work presented
Rights and permissions
About this article
Cite this article
Narendran, N., Guymer, R., Cain, M. et al. Analysis of the EFEMP1 gene in individuals and families with early onset drusen. Eye 19, 11–15 (2005). https://doi.org/10.1038/sj.eye.6701435
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6701435
Keywords
This article is cited by
-
Doyne honeycomb retinal dystrophy – functional improvement following subthreshold nanopulse laser treatment: a case report
Journal of Medical Case Reports (2019)
-
Doyne honeycomb retinal dystrophy/malattia leventinese induced by EFEMP1 mutation in a Chinese family
BMC Ophthalmology (2018)
-
Molecular genetics of AMD and current animal models
Angiogenesis (2007)
