
Abstract
Vascular endothelial growth factor receptor 2 (VEGFR2) is a potential cell-type biomarker in clinical diagnoses. Besides, it's the target protein of many tyrosine kinase inhibitors and its expression significantly associates with clinical performance of these inhibitors. VEGFR2 detection provides an early warning for diseases and a basis for therapy and drug screening. Some methods have been developed for VEGFR2 determination. However, they are usually performed indirectly and complexly. Herein, an electrochemical biosensing platform for VEGFR2 analysis has been first proposed. It can detect the total concentrations of the VEGFR2 protein in cells lysates directly and can be used to monitor the changes of VEGFR2 expression levels induced by treatments of different inhibitors. Moreover, the inhibitor-VEGFR2 interactions are illuminated through theoretical simulation. The simulation results agree well with the experimental data, indicating the veracity of the proposed method. The electrochemical detection methodology for VEGFR2 would be promising in clinical diagnosis and drug screening.
Similar content being viewed by others

Introduction
In the progression of many pathological diseases such as chronic inflammation or cancer, vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptors (VEGFRs) play important roles as they are essential in angiogenesis and vasculogenesis1, which significantly promote tumor growth and metastatic spread2. Among these specific tyrosine kinase receptors which are regulated by VEGF3, VEGFR2 mediates most of the angiogenic functions4,5. The VEGFR2 protein is expressed at low levels in normal cells or tissues. However, in various diseases such as diabetic retinopathy, chronic lymphocytic leukemia, ovarian cancer and breast cancers, its expression is upregulated6,7,8,9,10,11. Besides, the expression of VEGFR2 is closely related to the disease stage, recurrence and outcome12,13,14. Due to its specific expression and critical role in signaling pathway of angiogenesis, it is no wonder that VEGFR2 has been considered as an appropriate target protein for the design and development of many angiogenesis inhibitors15,16. Furthermore, the expression of VEGFR2 correlates with antitumour efficacy of VEGFR2 tyrosine kinase inhibitor17,18. Thus, the analysis of VEGFR2 not only plays an important role in diagnostic analysis, but also takes a deeper look at drugs' efficacy. So simple and sensitive detection methods for VEGFR2 are significantly required in order to monitor the progress of the diseases as well as predict the curative effect of drugs.
At present, some methods including quantitative reverse-transcription polymerase chain reaction (qRT-PCR)19, western blot (WB)20 and enzyme-linked immunosorbent assay (ELISA)21 have been developed for determination of VEGFR2 expression. The qRT-PCR technique used for the analysis of VEGFR2 protein is to measure the amount of mRNA at the gene transcription level rather than protein level19. This indirect way might constrain its application scope as it is a complicated biological process from transcription to translation and there is not a necessary positive correlation between the amount of gene expression and protein expression. The WB technique can only semi-quantitatively assay protein expression level20. The ELISA is an available quantitative method to detect proteins. But it is complicated, time-consuming and needs more expensive instruments. Besides, traditional colorimetric signal readout used in ELISA also constrains its improvement in the limit of detection22. To the best of our knowledge, electrochemical technique for VEGFR2 determination has not been reported.
Recently, electrochemical determination has been applied to many fields including environmental monitoring23, food analysis24, biological analysis25 and medical detection26 due to its intrinsic advantages such as high sensitivity, portability, relatively low cost, on-line detection, rapid response and reusability27,28. A variety of functional nanomaterials has been introduced as conductive substrate or immobilization platforms for biomolecules to amplify the biosensing signals in the process of constructing electrochemical biosensors29,30. In this work, the electrodes were modified by using chitosan functionalized reduced graphene oxide (RGO) to enhance the electrical conductivity, together with a sandwich-type assay format, an electrochemical biosensing platform for the detection of VEGFR2 has been firstly developed (Figure 1). The proposed electrochemical detection method for VEGFR2 protein exhibited good applicability in real samples. To test the changes of VEGFR2 expression induced by different irritants' treatments, rhesus macaque choroid-retinal endothelial cells (RF/6A), which was close to retinal cells derived from humans and repeatedly be used to study about retinal angiogenesis and choroid angiogenesis, were selected as model cells. Three kinds of irritants (VEGF and two tyrosine kinase inhibitors) were used to regulate the expression of VEGFR2. The changes of the protein content can be monitored by our electrochemical detection system established herein. As it has been reported that molecular conformations, interactions and properties of VEGFR tyrosine kinase inhibitors associated with their drug efficiency and clinical performance31, by combining with molecular simulation of inhibitor-VEGFR2 interaction, the relationship between drug action mechanism and its efficacy was also analyzed.

Schematic representation of the electrochemical biosensing platform for VEGFR2 protein.
Results
Characterization of the chitosan-RGO composite
The main aim of the modification using graphene on the glassy carbon electrode (GCE) surface was to enhance the surface area, accelerate electron transfer and form an interface with sites for VEGFR2 capture antibody (Ab1) immobilization. As the RGO has low solubility in water, the polymer (chitosan) and reductant (L-ascorbic acid) was used to resist the formation of irreversible agglomerates32. UV-vis absorption spectroscopy and Raman spectroscopy were used to confirm the reduction (Figure S1). As shown in Figure S1A, the UV maximum absorption of the GO dispersion shifted from 230 (curve a) to 270 nm (curve b) after the reduction, at the same time the absorption intensity of RGO was higher than GO from 200 to 600 nm, indicating the restoration of the electronic conjugations33. In addition, Raman spectra of the RGO (Figure S1B, curve b) gave an increased D/G intensity ratio (1.09) compared to that of the GO (0.96) (Figure S1B, curve a), suggesting the decrease in the average size of the sp2 domains and the increase in the number of small in-plane sp2 domains34. And the Raman spectra presented us with a weak and broadened 2D peak of RGO (Figure S1B, curve b), showing that RGO possessed much more defects compared with GO35. These phenomena confirmed the formation of RGO from the reduction of GO.
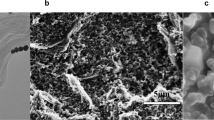
Furthermore, to confirm the effect of chitosan, the morphology of RGO with (Figure 2A) and without (Figure 2B) chitosan was analyzed by transmission electron microscope (TEM). The RGO without mediation of chitosan had obvious agglomeration, which was distinct from the chitosan-RGO with homogeneous few layers graphene nanosheets wrapped up by rough chitosan membrane. This indicated that the chitosan made contribution to the dispersion of the RGO in water through electrostatic attractions (ionic interactions)36.

TEM images of RGO with A) and without B) chitosan.
In the fabrication process of the biosensor, a comparison was made using isopyknic 0.5% chitosan in place of chitosan-RGO to fabricate the biosensor and all the other steps were the same. When they were utilized to detect the VEGFR2 protein (Figure 3), Ab2 (biotinylated VEGFR2 detection antibody)/Ag/BSA (bovine serum albumin)/Ab1/chitosan-RGO/thionine/GCE (curve b) displayed larger response of electrochemical signal than that of Ab2/Ag/BSA/Ab1/chitosan/thionine/GCE (curve a). This might attribute to that the excellent electrical conductivity of RGO accelerated electron transfer between thionine and horseradish peroxidase (HRP).

DPV responses of a) Ab2/Ag/BSA/Ab1/chitosan/thionine/GCE and b) Ab2/Ag/BSA/Ab1/chitosan-RGO/thionine/GCE.
Ag: VEGFR2 with the concentration of 43 pM. Thionine with the concentration of 30 μM. Inset: magnification of a).
Characterization of the biosensor
Cyclic voltammogram (CV) and electrochemical impedance spectroscopy (EIS) measurements were utilized to monitor the fabrication steps of the biosensor (Figure S2 and Figure 4). With the increase of scan rate ranging from 10 to 100 mV s−1, the redox peak currents of the thionine increased linearly (Figure S2), indicating the electrochemical reaction is a surface controlled process. For the biosensor fabrication, thionine was firstly attached to the GCE through subsequent coating of chitosan-RGO. So the mediator thionine could be directly and stably immobilized on the electrode surface. As shown in Figure 4A, the CVs at bare GCE (curve a) and chitosan-RGO/GCE (curve b) did not show any detectable signal, the CV at chitosan-RGO/thionine/GCE exhibited a pair of stable and well-defined redox peaks at −0.271 and −0.324 V (curve c), which corresponded to reversible oxidation (reduction) of thionine, respectively. Subsequently, covalent binding of Ab1 was achieved by using glutaraldehyde as cross-linkers for Schiff base. The loading of Ab1 led to an obvious decrease of the redox peak currents (curve d) due to the enhanced steric hindrance generated by the immobilized proteins which had inhibitory action on the electron transfer of the electrode surface. After the modified electrode was incubated in BSA, Ag and Ab2, a further decrease of the redox peak currents could be observed but not distinct (curves e, f and g). In order to further verify the successful construction of the biosensor, EIS were used to measure the changes of electrode surface. Figure 4B showed Nyquist diagrams changed gradually with the successive assembly. The equivalent circuit inserted in Figure 4B consists of the ohmic resistance of the electrolyte (Rs), the polarization resistance (Rp), Warburg impedance (W) and the constant phase angle element (CPE). The diameter of semicircle in the Nyquist plot at high frequency corresponds to the Rp, which can be calculated according to the equivalent circuit. The bare GCE exhibited a small semicircle (curve a, Rp = 129 Ω). After the electrode was modified with thionine and chitosan-RGO, the EIS was almost a straight line (curve b), implying very low electron-transfer resistance on chitosan-RGO/thionine/GCE electrode surface to the redox probe dissolved in the electrolyte solution. The stepwise assembly of Ab1, BSA, Ag and Ab2 on the chitosan-RGO/thionine/GCE electrode surface was accompanied by a gradual increase of semicircle diameters (curves c–f, Rp = 1.10 kΩ, 1.29 kΩ, 1.56 kΩ and 1.65 kΩ), indicating the increase of electron-transfer resistance. The impedance changes obtained during the modification process proved that Ab1, BSA, Ag and Ab2 had been successively assembled on the chitosan-RGO/thionine/GCE electrode surface. At last, to verify the enzymatic activity of HRP, H2O2 was added to the PBS. In Figure 4C, curve b showed the oxidation peak current of thionine decreased with its reduction peak current increased and the reduction peak potential shifted to negative, which were owing to the enzymatic catalysis of HRP to the oxidation of thionine by H2O2. The enhanced reduction peak current was served as an amplification of signal for constructing this proposed biosensor. The reaction mechanism of the enzymatic catalysis of HRP to the oxidation of thionine by H2O2 is outlined as follow37,38:



In reaction (1), H2O2 was reduced by HRP to form water and HRP(ox) denoted oxidized HRP. Then in reaction (2) HRP(ox) was reduced to HRP by thionine. Reduced and oxidized forms of thionine were represented as thionine(red) and thionine(ox), respectively. Then thionine(ox) could be reduced to thionine(red) at the electrode, in reaction (3), leading to a reduction current.

A) CVs obtained at (a) bare GCE, (b) chitosan-RGO/GCE, (c) chitosan-RGO/thionine/GCE, (d) Ab1/chitosan-RGO/thionine/GCE, (e) BSA/Ab1/chitosan-RGO/thionine/GCE, (f) Ag/BSA/Ab1/chitosan-RGO/thionine/GCE, (g) Ab2/Ag/BSA/Ab1/chitosan-RGO/thionine/GCE in 0.01 M pH 7.4 PBS. B) Nyquist diagrams of (a) bare GCE, (b) chitosan-RGO/thionine/GCE, (c) Ab1/chitosan-RGO/thionine/GCE, (d) BSA/Ab1/chitosan-RGO/thionine/GCE, (e) Ag/BSA/Ab1/chitosan-RGO/thionine/GCE and (f) Ab2/Ag/BSA/Ab1/chitosan-RGO/thionine/GCE in 10 mM [Fe(CN)6]3−/4− containing 0.1 M KCl. C) CVs of Ab2/Ag/BSA/Ab1/chitosan-RGO/thionine/GCE in 0.01 M pH 7.4 PBS (a) without and (b) with 4 mM H2O2. Ag concentration was 43 pM. The concentration of thionine was 30 μM.
VEGFR2 determination
Under the optimized conditions (chitosan-RGO composite (0.25 mg mL−1), incubation time of VEGFR2 (60 min), H2O2 concentration (4 mM)) (Figure S3), differential pulse voltammetry (DPV) peak currents increased with increasing concentration of VEGFR2 protein from 0.4 pM to 86 pM (Figure 5A). As shown in Figure 5B, the reduction peak current was proportional to the logarithm of the VEGFR2 concentration. The linear regression equation of the calibration curve was Ipc (μA) = 5.946 + 3.446 lg[VEGFR2] (pM) in the range from 0.4 pM to 86 pM with a correlation coefficient of 0.999. The detection limit for VEGFR2 was 0.28 pM at a signal-to-noise ratio of 3. VEGFR2 quantification using ELISA was served as a comparison (The detail of ELISA assay procedure was described in Supporting Information). The ELISA kit showed a linear range from 0.9 pM to 43 pM with a detection limit of 0.6 pM for VEGFR2 (Figure S4). This demonstrated that our proposed electrochemical biosensor was more sensitive and had wider detection range than those of the ELISA results. Besides the above, our proposed electrochemical detection method for VEGFR2 was low-cost and much simpler, comparing with the ELISA.

A) DPV curves for detection of different concentrations of VEGFR2 (a) 0, (b) 0.3, (c) 0.4, (d) 0.9, (e) 4.3, (f) 8.6, (g) 43, (h) 65, (i) 86, (j) 103 pM) by using the proposed biosensor and B) calibration curve.
The concentration of thionine was 30 μM.
Specificity, reproducibility and stability of the assay
In order to assess the specificity of the proposed biosensor for VEGFR2 protein, some related proteins including BSA, recombinant human VEGF165, recombinant human VEGF121, recombinant human VEGFR1 and recombinant human VEGFR3 were used as the possible interferences. The electrochemical signals of 8.6 pM (1 ng mL−1) VEGFR2 was compared with that of BSA (100 ng mL−1) or 50 ng mL−1 other interferential substance (Figure 6). The peak currents showed negligible difference between the blank solution and the incubation solution contained interfering agents, indicating the excellent specificity of the proposed biosensor. Therefore, the proposed biosensor could be further used for detection of VEGFR2 in real samples.

Selectivity analysis of the biosensor tested in blank solution, 8.6 pM (1 ng mL−1) VEGFR2 protein, 100 ng mL−1 BSA, 50 ng mL−1 VEGF165, 50 ng mL−1 VEGF121, 50 ng mL−1 VEGFR1 and 50 ng mL−1 VEGFR3, respectively.
The coefficient variations of intra- and inter-assays were used to assess the reproducibility of the proposed biosensor. Both the inter-assay and intra-assay precisions of the biosensor were examined with two samples containing 0.9 pM and 43 pM of VEGFR2 five times. The intra-assay coefficient variation was valued by detecting each sample for five times using five parallel prepared biosensors. The intra-assay coefficient variations were 4.2% for 0.9 pM VEGFR2 and 3.9% for 43 pM VEGFR2, respectively. The inter-assay precision was calculated by measuring each sample with five biosensors using the same GCE. The inter-assay coefficient variations were 5.8% for 0.9 pM VEGFR2 and 4.3% for 43 pM VEGFR2, respectively. The above results showed acceptable reproducibility and precision of the proposed biosensor. In addition, when the biosensor was stored in PBS at 4°C for 10 days, it remained 91.9% of initial responses, suggesting the acceptable stability of the proposed biosensor.
VEGFR2 analysis in real samples
The VEGFR2 electrochemical assay was further applied to a complex biological matrix to examine its practicality. The average VEGFR2 concentration in cell culture supernatant of three parallel N1 (0 h) samples were detected to be 4.0 pM in average, which was in good agreement with 3.8 pM obtained with the ELISA method. VEGFR2 real samples were prepared by spiking 0.9 pM, 8.6 pM and 43 pM of purified VEGFR2 in N1 (0 h) cell lysate, respectively. The peak current increased proportionally with the increasing concentration of the added VEGFR2, which was coincident with the calibration curve obtained with standard VEGFR2 solutions. According to the calibration curve, the VEGFR2 content in the N1 (0 h) cell lysate was obtained to be 4.2 pM, which was consistent with the above measure results (4.0 pM and 3.8 pM). The recoveries were in the range of 92.12–114.7%. Thus, these results indicated that the electrochemical sensing approach was reliable and could precisely quantify the expression of protein in complex biological matrixes with little matrix effect.
As all the above results have definitely illuminated the applicability of the proposed biosensor in the practical samples, the changes of VEGFR2 expression induced by different irritants' treatments were detected. The effects of identical concentration of Sorafenib and Vandetanib on the total VEGFR2 protein expression were tested using RF/6A cells. VEGFR2 had a low expression in normal RF/6A cells, which could be seen through N1 sample (Figure 7). It is reported that VEGF up-regulated VEGFR2 expression through binding and activation of the VEGFR2 receptor tyrosine kinase39, which was validated herein by our work (N2 of Figure 7). As shown in N2 of Figure 7, when cells were co-incubated with 25 ng mL−1 VEGF165, the expression of VEGFR2 increased rapidly and drastically compared with the blank control (N1). Thus we chose VEGF165 to stimulate the expression of VEGFR2 to simulate pathological circumstance. Then we hypothesized VEGFR2 tyrosine kinase inhibitors would affect the role of VEGF. To test this hypothesis, 5 μM Sorafenib and 5 μM Vandetanib were added in the cell cultures containing 25 ng mL−1 VEGF165, respectively. Their influences on the VEGFR2 expression were also determined by the proposed electrochemical biosensor. As shown in N3 of Figure 7, Sorafenib effectively blocked the influence of VEGF165 on the VEGFR2 expression immediately and steadily. Meanwhile, in N4 of Figure 7, comparing with Sorafenib, Vandetanib took a slower effect but seemed to be more effective on inhibiting VEGFR2 expression.

Mapping of the functional dependence of the measured concentrations of VEGFR2 on the incubation time and different cases.
Serum-starved RF/6A cells were incubated with 25 ng mL−1 VEGF165, 5 μM Sorafenib and 5 μM Vandetanib in cases number 1–4 (N1–4). These cases were marked along the z axis and the details were shown in the table below the histogram. Samples were taken from each case at different time intervals: 0, 4, 8 and 12 h, respectively. The concentration of VEGFR2 in each sample's cell lysate was detected by using the proposed electrochemical biosensor.
Discussion
To understand why different inhibitors caused different VEGFR2 expression levels, herein, some information about properties and interaction with VEGFR2 tyrosine kinase domain of Sorafenib and Vandetanib was further discussed briefly (Figure S5 and Figure 8). Comparing their physical property, Sorafenib had higher lipid solubility than that of Vandetanib. The rate of membrane permeability usually increased with increasing fat solubility. In the meantime, considering their interaction with VEGFR2 (Figure 8), they were stabilized mainly via hydrophobic interaction as shown in Figure 7A2 and B2. And some polar residues played an important role in stabilizing drug via H-bonds and electrostatic interactions. As shown in Figure 8A3 and B3, there were more hydrogen bonds between Sorafenib and VEGFR2 tyrosine kinase region, indicating that Sorafenib had better specificity and selectivity. Combining the above two factors, the reason why Sorafenib played its function on blocking the influence of VEGF on the VEGFR2 expression much quicker (Figure 7 N3–4 h) than Vandetanib (Figure 7 N4–8 h) might be easy to explain. The binding free energy of Sorafenib with VEGFR2 (ΔG°binding(Sorafenib)) was found to be −62.71 kJ mol−1, while ΔG°binding(Vandetanib) was −90.41 kJ mol−1, showing that Vandetanib bound much stronger with VEGFR2 than Sorafenib. This might account for the phenomenon shown in Figure 7 that Vandetanib blocked VEGFR2 up-expression more efficiently than Sorafenib.

Chemical formulas for A1) Sorafenib and B1) Vandetanib.
Binding models of A2), A3) VEGFR2-Sorafenib and B2), B3) VEGFR2-Vandetanib. These binding models showed the interaction information about Sorafenib or Vandetanib with the amino acids residues around 11 Å of VEGFR2. In A2) and B2), hydrophobic and hydrophilic amino acid residues were shown in red and white lines, respectively. Hydrogen bonds and intermolecular bumps were separately marked with green and purple dotted lines and labeled with names.
In summary, an electrochemical VEGFR2 biosensor was successfully designed and its high practical usability was demonstrated in the sensitive, selective and accurate quantification of total VEGFR2 protein in cell lysates. The immunoassay was successfully used to monitor the changes of VEGFR2 expression levels induced by different irritants. Furthermore, the mechanism of inhibitors' effects had been elucidated reasonably using theoretical simulation at the molecular level. The designed strategy has an expansive and promising perspective of application in drug screening and clinical diagnosis for the construction of versatile electrochemical assay platforms.
Methods
Reagents and materials
The human VEGFR2 ELISA kit including VEGFR2 capture antibody, VEGFR2 antigen, biotinylated VEGFR2 detection antibody and streptavidin-HRP were purchased from R&D Systems Co., Ltd. BSA, aprotinin, leupeptin, chitosan, thionine, fetal bovine serum (FBS), dimethyl sulfoxide (DMSO), 3,3′,5,5′-tetramethylbenzidine (TMB) were acquired from Sigma-Aldrich. 96 well microplates were obtained from Costar Co., Ltd. GO was produced from Nanjing XF Nano Materials S&T Co., Ltd. Sorafenib and Vandetanib were from Nanjing Chemlin Chemical Industry Co., Ltd, whose purities were more than 99% (HPLC). RF/6A cells were purchased from Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences and cell culture medium F12K were from Wuhan Boster Biological Technology Co., Ltd. Recombinant human VEGF165 were purchased from ZhongKeWuYuan Biotechnology Co., Ltd. Other chemicals regents were analytical reagents and used without further purification. All solutions were prepared with double-distilled water, unless indicated otherwise.
Apparatus
Electrochemical experiments, including CV and DPV were performed with an electrochemical analyzer CHI 660D (CH Instruments). The morphology of the chitosan-RGO was observed using a transmission electron microscope (TEM, JEM-200CX) with an accelerating voltage of 80 kV. UV-vis measurements were carried out at room temperature on a Cary 50 UV-vis absorption Spectrometer (Varian, America). Raman spectra were recorded on a Labram HR800 (Jobin Yvon Co., France). All experiments were conducted at ambient temperature. Optical density (OD) at 450 nm which subtracted readings at 540 nm was determined by a Safire microplate reader (TECAN, America). The EIS was carried out with an Autolab potentiostat/galvanostat PGSTAT302N (Eco chemie, BV, The Netherlands) and controlled by Nova 1.8 software with a three-electrode system under open circuit conditions. The voltage frequencies ranged from 105 Hz to 0.1 Hz and the AC voltage amplitude was 5 mV. The supporting electrolyte was 10 mM [Fe(CN)6]3−/4− solution containing 0.1 M KCl.
Synthesis of chitosan-RGO composite
Briefly, GO suspension (5 mL, 0.5 mg mL−1) was gradually added into chitosan solution (5 mL, 0.5% w.t. in 1% acetic acid) with stirring. Then L-ascorbic acid (23.6 mg) was added and the mixture was held at 60°C for 5 h. At last, the chitosan-RGO was filtered over a cellulose membrane (0.22 μm), washed with water and then redispersed in acetic acid solution (10 mL, 1%). The upper homogeneous dispersion was taken for future use.
Fabrication of the biosensor
The fabrication of the biosensor was illustrated in Figure 1. Before modification of the GCE, it was polished to a mirror finish with alumina slurry (0.3 and 0.05 μm, respectively), followed by ultrasonication for 30 s to remove the alumina particles and then rinsed with water and dried under nitrogen gas. Thionine (7 μL, 30 μM) was placed on the GCE (d = 3 mm) at room temperature for 2 h to dry. Then chitosan-RGO (7 μL) was dropped to coat the thionine film and dried at room temperature. After thoroughly washing with water, the electrode was immediately followed by incubation with glutaraldehyde (7 μL, 2.5%) for 2 h. After washing, Ab1 (10 μL, 7.2 μg mL−1) in phosphate buffered saline (PBS) (8.1 mM Na2HPO4, 1.5 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) was applied to the electrode and then incubated for 3 h at room temperature. After washing with 0.05% Tween-20 and PBS, the Ab1/chitosan-RGO/thionine/GCE was incubated in BSA (6 μL, 3%) and PBS at room temperature for 1 h to block excess active groups and nonspecific binding sites on the surface. Finally, the electrode was washed with 0.05% Tween-20 and PBS before use.
Measurement procedures
A classical sandwich immunoassay was used for determination of VEGFR2 protein. First, the BSA/Ab1/chitosan-RGO/thionine/GCE was incubated with 15 μL of different concentrations of VEGFR2 standard antigen (Ag) for 60 min at 37°C, followed by washing with 0.05% Tween-20 and PBS. Subsequently, the electrode (Ag/BSA/Ab1/chitosan-RGO/thionine/GCE) was incubated with 10 μL of Ab2 for 60 min at 37°C and washed. The diluted streptavidin-HRP (10 μL, 1:50) solution was dropped on the electrode and incubated for 30 min at 37°C, then washed. At last, the electrochemical detection was performed under nitrogen atmosphere in PBS containing 4 mM H2O2 (the solution was deaerated with nitrogen gas for 15 min before use).
Cell treatment and sample preparation
The different effects of drugs on regulation the expression of VEGFR2 were invested by incubating cells with VEGF165, Sorafenib and Vandetanib. Sorafenib and Vandetanib were dissolved in DMSO and then diluted by culture media. DMSO was added to culture medium at 0.1% (v/v) final concentration as a vehicle control. Cells were cultured until the fifth passage at 37°C and 5% CO2 in medium (90% F12K and 10% FBS). After seeding 24 hours, cells were serum-starved for 24 hours and incubated with VEGF165, Sorafenib and Vandetanib at the indicated concentrations at different time intervals: 0 h, 4 h, 8 h and 12 h, respectively. Then cells were rinsed two times with PBS. Cells were solubilized at 1×107 cells mL−1 in lysis buffer (1% NP-40 Alternative, 20 mM pH 8.0 Tris-HCl buffered saline, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM activated sodium orthovanadate, 10 μg mL−1 Aprotinin, 10 μg mL−1 Leupeptin) and allowed samples to sit on ice for 15 min. Assay immediately or store at −70°C. Before use, samples were centrifuged at 2000g for 5 min and the supernate was transferred to a clean test tube.
Molecular modeling of interactions between VEGFR2 and its irritants
Molecular simulations were performed using the Discovery Studio 2.1 software package (Accelrys). The flexible docking between the VEGFR2 and Sorafenib or Vandetanib was carried out with Sorafenib or Vandetanib as a ligand and VEGFR2 protein as a receptor. The crystal structure of VEGFR2 tyrosine kinase domain (PDB entry code 2OH4) was downloaded from the Protein Data Bank and the potential 3D structures of Sorafenib and Vandetanib was assigned to the CHARMm force field. During the docking process, a maximum of 10 conformers were chosen for the final energy minimization. The conformation with the lowest energy was selected for final analysis. Crystallographic water molecules and heteroatoms were removed from VEGFR2 and the structure of VEGFR2 was assigned according to the CHARMm force field. The optimal ligand-receptor complex was obtained by choosing the highest score of scoring function PMF. The binding free energy between VEGFR2 and inhibitors was calculated by considering the Poisson-Boltzmann Surface Area as the implicit solvent model for the whole system.
References
Tugues, S., Koch, S., Gualandi, L., Li, X. J. & Claesson-Welsh, L. Vascular endothelial growth factors and receptors: Anti-angiogenic therapy in the treatment of cancer. Mol. Asp. Med. 32, 88–111 (2011).
Ferrara, N. & Kerbel, R. S. Angiogenesis as a therapeutic target. Nature 438, 967–974 (2005).
Freeman, R. et al. Optical aptasensors for the analysis of the vascular endothelial growth factor (VEGF). Anal. Chem. 84, 6192–6198 (2012).
Lee, S. et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703 (2007).
Steeg, P. S. Tumor metastasis: mechanistic insights and clinical challenges. Nat. Med. 12, 895–904 (2006).
Kalishwaralal, K., BarathManiKanth, S., Pandian, S. R. K., Deepak, V. & Gurunathan, S. Silver nano—a trove for retinal therapies. J. Control. Release 145, 76–90 (2010).
Ferrajoli, A. et al. High levels of vascular endothelial growth factor receptor-2 correlate with shortened survival in chronic lymphocytic leukemia. Clin. Cancer Res. 7, 795–799 (2001).
Fabris, L. et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology 43, 1001–1012 (2006).
Spannuth, W. A. et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int. J. Cancer 124, 1045–1053 (2009).
Taylor, M. et al. High levels of circulating VEGFR2+ bone marrow-derived progenitor cells correlate with metastatic disease in patients with pediatric solid malignancies Clin. Cancer Res. 15, 4561–4571 (2009).
Liu, D. H., Liu, F. X., Liu, Z. H., Wang, L. L. & Zhang, N. Tumor specific delivery and therapy by double-targeted nanostructured lipid carriers with anti-VEGFR-2 antibody. Mol. Pharm. 8, 2291–2301 (2011).
Pajares, M. J. et al. Expression of tumor-derived vascular endothelial growth factor and its receptors is associated with outcome in early squamous cell carcinoma of the lung. J. Clin. Oncol. 30, 1129–1136 (2012).
Birkhahn, M. et al. Predicting recurrence and progression of noninvasive papillary bladder cancer at initial presentation based on quantitative gene expression profiles. Eur. Urol. 57, 12–20 (2010).
Mitra, A. P. & Cote, R. J. Molecular pathogenesis and diagnostics of bladder cancer. Annu. Rev. Pathol.-Mech Dis. 4, 251–285 (2009).
Youssoufian, H., Hicklin, D. J. & Rowinsky, E. K. Review: Monoclonal Antibodies to the Vascular Endothelial Growth Factor Receptor-2 in Cancer Therapy. Clin. Cancer Res. 13, 5544s–5548s (2007).
Ivy, S. P., Wick, J. Y. & Kaufman, B. M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 6, 569–579 (2009).
Dev, I. K. et al. Antitumour efficacy of VEGFR2 tyrosine kinase inhibitor correlates with expression of VEGF and its receptor VEGFR2 in tumour models. Br. J. Cancer 91, 1391–1398 (2004).
Meissner, M. et al. Down-regulation of vascular endothelial growth factor receptor 2 is a major molecular determinant of proteasome inhibitor-mediated antiangiogenic action in endothelial cells. Cancer Res. 69, 1976–1984 (2009).
Benedito, R. et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF–VEGFR2 signalling. Nature 484, 110–114 (2012).
Nacev, B. A., Grassi, P., Dell, A., Haslam, S. M. & Liu, J. O. The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking and signaling in endothelial cells. J. Biol. Chem. 286, 44045–44056 (2011).
Ji, Z. et al. 3-Amino-benzo[d]isoxazoles as novel multitargeted inhibitors of receptor tyrosine kinases. J. Med. Chem. 51, 1231–1241 (2008).
Tong, S., Ren, B. B., Zheng, Z. L., Shen, H. & Bao, G. Tiny grains give huge gains: nanocrystal-based signal amplification for biomolecule detection. ACS Nano 7, 5142–5150 (2013).
Lou, X. H., Zhao, T., Liu, R., Ma, J. & Xiao, Y. Self-assembled DNA monolayer buffered dynamic ranges of mercuric electrochemical sensor. Anal. Chem. 85, 7574–7580 (2013).
Han, C. et al. A multiwalled-carbon-nanotube-based biosensor for monitoring microcystin-LR in sources of drinking water supplies. Adv. Funct. Mater. 23, 1807–1816 (2013).
Chai, X. L. et al. A two-channel ratiometric electrochemical biosensor for in vivo monitoring of copper ions in a rat brain using gold truncated octahedral microcages. Angew. Chem. 125, 8287–8291 (2013).
Wen, Y. L. et al. DNA Nanostructure-based Interfacial engineering for PCR-free ultrasensitive electrochemical analysis of microRNA. Sci. Rep. 2, 867 (2013).
Deng, W. P. Diagnosis of schistosomiasis japonica with interfacial co-assembly-based multi-channel electrochemical immunosensor arrays. Sci. Rep. 3, 1789 (2013).
Martic, S., Labib, M., Freeman, D. & Kraatz, H. B. Probing the role of the linker in ferrocene-ATP conjugates monitoring protein kinase catalyzed phosphorylations electrochemically. Chem. Eur. J. 17, 6744–6752 (2011).
Huang, Y. X., Cai, D. & Chen, P. Micro- and nanotechnologies for study of cell secretion. Anal. Chem. 83, 4393–4406 (2011).
Chen, D., Feng, H. B. & Li, J. H. Graphene oxide: preparation, functionalization and electrochemical applications. Chem. Rev. 112, 6027–6053 (2012).
McTigue, M. et al. Molecular conformations, interactions and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. U. S. A. 109, 18281–18289 (2012).
Zhang, J. L. et al. Reduction of graphene oxidevia L-ascorbic acid. Chem. Commun. 46, 1112–1114 (2010).
Li, D., Muller, M. B., Gilje, S., Kaner, R. B. & Wallace, G. G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 3, 101–105 (2008).
Stankovich, S. et al. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 45, 1558–1565 (2007).
Guo, H. L., Wang, X. F., Qian, Q. Y., Wang, F. B. & Xia, X. H. A green approach to the synthesis of graphene nanosheets. ACS Nano 3, 2653–2659 (2009).
Fang, M., Long, L. A., Zhao, W. F., Wang, L. W. & Chen, G. H. pH-responsive chitosan-mediated graphene dispersions. Langmuir 26, 16771–16774 (2010).
Du, D. et al. Functionalized graphene oxide as a nanocarrier in a multienzyme labeling amplification strategy for ultrasensitive electrochemical immunoassay of phosphorylated p53 (S392). Anal. Chem. 83, 746–752 (2011).
Yang, M. H., Yang, Y. H., Yang, Y., Shen, G. L. & Yu, R. Q. Bienzymatic amperometric biosensor for choline based on mediator thionine in situ electropolymerized within a carbon paste electrode. Anal. Biochem. 334, 127–134 (2004).
Adya, R., Tan, B. K., Punn, A., Chen, J. & Randeva, H. S. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 78, 356–365 (2008).
Acknowledgements
This work was supported by the National Natural Science Foundation of China for the project (21175069, 21171096 and 21205061), Foundation of the Jiangsu Education Committee (11KJA150003). We appreciate the financial support from the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
Z.D. and T.W. designed the experiments and wrote the main manuscript text, T.W., W.T., B.Z. and Y.L. performed experiments, J.B. and Z.D. provided sampels. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wei, T., Tu, W., Zhao, B. et al. Electrochemical monitoring of an important biomarker and target protein: VEGFR2 in cell lysates. Sci Rep 4, 3982 (2014). https://doi.org/10.1038/srep03982
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03982
This article is cited by
-
An Overview of Carbon Nanotubes and Graphene for Biosensing Applications
Nano-Micro Letters (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
