
Abstract
Small molecules with dithieno[3,2-b;2′,3′-d]thiophene as central building block and octyl cyanoacetate and octyl cyanoacetamide as different terminal building blocks have been designed and synthesized. The amide containing small molecule can form intermolecular hydrogen bonding between N-H…O = C of the amide group. The photovoltaic properties and active layer morphologies of the two molecules in bulk heterojunction solar cells are compared to study the influence of hydrogen bonding on the active layer morphology. New methanofullerene compound containing amide group has also been synthesized and compared with conventional fullerene electron acceptors.
Similar content being viewed by others

Introduction
Active layer morphology is of key importance in bulk heterojunction (BHJ) solar cells. Bi-continuous nanoscale domains of donor and acceptor materials are required for efficient charge separation and transport1,2,3,4,5,6,7. To achieve the optimum active layer morphology, various methods, such as thermal annealing, solvent annealing, solvent additives, have been used in the process of solar cells fabrication1,2,3,4,5,6,7. From a molecular point of view, chemists want to use self assembly or self organization to control the morphology by molecular design8,9,10,11,12,13. In this respect, hydrogen bonding is the first choice due to its strong intermolecular interaction, high selectivity and directionality14,15,16,17. In some cases, improved device performance had been demonstrated when hydrogen bonding was employed in BHJ solar cells18,19,20,21,22. Here, we describe a case study in which the performance of two similar small molecular donor materials, one with amide group to form intermolecular hydrogen bonding, the other with ester group, are compared in BHJ solar cells. In addition, an amide containing fullerene derivative was also synthesized and used in conjunction with the donor materials in devices.
A common design rule for the small molecule donor material is connecting central building blocks and terminal electron withdrawing units with an oligothiophene linker23,24,25,26,27,28. Based on our previous work29, dithieno[3,2-b;2′,3′-d]thiophene (DTT) was used as the central building block and 3′,3″-dihexyl-2,2′:5′,2″-tertthiophene13 was used as the linkage. Octyl cyanoacetate and octyl cyanoacetamide served as electron withdrawing groups to end cap the small molecules (Figure 1). Octyl cyanoacetamide will lead to a Knoevenagel condensation product which is capable of strong hydrogen bonding. Synthesis of the targeted small molecules started from 3′,3″-dihexyl-2,2′:5′,2″-tertthiophene-5-carbaldehyde13, compound 1. Bromination of compound 1 with NBS provided compound 2 which reacted with bistrimethyltin DTT (compound 3) by Stille coupling reaction to afford compound 4. Then compound 4 was converted to M1 and M2 by Knoevenagel condensation with octyl cyanoacetate and octyl cyanoacetamide respectively (Figure 2). It should be noted that M1 can be obtained in the presence of triethylamine, but M2 can only be synthesized in the presence of stronger base such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). The detailed synthetic procedures for M1 and M2 are described in Supplementary Information. Both products showed good solubility in common organic solvents such as tetrahydrofuran, dichloromethane, chloroform, chlorobenzene and dichlorobenzene. M1 and M2 were fully characterized with 1H and 13C NMR spectroscopy, mass spectrometry and elemental analysis.

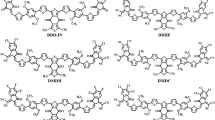
Chemical structures of M1, M2, PC61BM and PC61MBA.

Synthesis of small molecules M1 and M2.
The presence of hydrogen bonding moieties on the electron acceptor fullerene derivatives may provide further insights into the effect of hydrogen bonding on BHJ solar cell devices. As with the electron donor material, the acceptor should ideally phase separate on a suitable length scale to allow maximum ordering and thus charge can transport effectively in continuous pathways to the electrodes and the recombination of free charges can be minimized6. In this regard, an analogue of [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) with an amide group was also designed and synthesized (see SI for details), resulting in [6,6]-phenyl-C61-N-methyl butyramide (PC61MBA, Figure 1)19.
Results and Discussion
The intermolecular hydrogen bonding of M2 and PC61MBA
The amide group of the small molecule M2 made it possible for the formation of intermolecular hydrogen bonding which was first observed in solution by 1H NMR dilution experiment (Figure 3). As the concentration increased, the chemical shift of the amide proton as well as the adjacent protons shifted indicating the aggregation of M2. This phenomenon was not observed in M1. In the solid film infrared (IR) spectrum of M2, the N-H and C = O stretching frequencies were at 3358 cm−1 and 1664 cm−1 respectively. These IR stretching frequencies indicated hydrogen bonding in the solid state sample of M230,31. Thermal behavior of M1 and M2 was examined using differential scanning calorimetry (DSC). Interestingly, the melting point (Tm = 168°C) and the crystallization temperature (Tc = 117°C) for M1 was higher than the corresponding values for M2 (Tm = 148°C and Tc = 96°C; Figure S2). Although the hydrogen bonding of the amide groups on M2 was expected to increase the temperature of thermal processes, these interactions could also have an adverse effect on the π − π intermolecular association of these conjugated compounds. Further evidence for the disruption of π − π association by hydrogen bonding can be found in the UV-vis absorption and X-ray diffraction data (vide infra).

Partial 1H NMR spectra of M2 in CDCl3 at different concentration.
(From bottom to top: 1, 14 mM; 2, 10 mM; 3, 7 mM; 4, 5 mM; 5, 3.5 mM; 6, 1.8 mM, respectively).
The morphologies of the films deposited from chloroform solution of M1 and M2 were studied by atomic force microscopy (AFM) (Figure 4). M2 showed fibrous structure due to the intermolecular hydrogen bonding while the film of M1 did not. All these experiments indicated that the amide group of M2 has significant effects on the aggregation behavior of the dye and thus the film morphology. The nanowire-like fibrous structures of M2 might be advantageous for BHJ solar cells6. 1H NMR dilution experiment of PC61MBA also exhibited slight chemical shift change of the amide proton (Figure S1). The IR spectra of PC61MBA showed a N-H stretching frequency at 3299 cm−1 which was distinctive for hydrogen bonded amide groups19.

AFM images of spin coated films from 10 mg mL−1 chloroform solution of M1 (a and c) and M2 (b and d).
(a) and (b) are height images; (c) and (d) are phase images. Images are 2 × 2 μm.
The optical and electrochemical properties
The UV-vis absorption spectra of the small molecules M1 and M2 are shown in Figure 5a. M1 and M2 had very similar absorption in chloroform except that the absorption of M1 was a little red shifted due to the more electron withdrawing property of the ester group. However, the absorption in solid films was quite different for M1 and M2 (Figure 5a). The maximum absorption of M2 in solid film is at 560 nm which was only red shifted 60 nm compared to solution absorption (Table 1). M1 displayed maximum absorption at 640 nm in solid film which is red shifted 130 nm compared to the solution absorption maximum. The difference in solid film absorption could be attributed to different aggregation resulting from the presence or absence of hydrogen bonding15,16,31. It is likely that the amide hydrogen bonding in compound M2 is disrupting close π − π stacking between the conjugated planar molecules resulting in less red shifted absorption compared to the UV-vis absorption characteristics of compound M1. This is in agreement with data from the grazing incidence wide-angle X-ray scattering (GIWAXS) experiment which showed stronger π − π stacking molecular order for compound M1 compared with M2 (vide infra). The UV-vis absorption spectra of PC61BM and PC61MBA are shown in Figure 5b. The amide derivative showed sharp peak at 328 nm which was identical to that of PC61BM. Broad peaks ranging from 450 to 650 nm were also observed.

UV-Vis absorption spectra of (a) M1 and M2, (b) PC61BM and PC61MBA in chloroform solution (solid line) and in solid films (dash line).
The solution and thin film photoluminescence (PL) spectrum of the electron donor materials M1 and M2 were recorded (Figure S3a). The emission maximum for M1 and M2 in chloroform solution were 665 nm and 640 nm respectively with excitation wavelength at 500 nm (Table 1). Both M1 and M2 showed very weak emission in solid state with 550 nm excitation (Figure S3 and Table 1). The weak fluorescence of these compounds in thin films is probably a result of self-quenching with both materials showing strong intermolecular association. The photophysical properties of blend films of M1 and M2 with fullerene acceptors were also examined (Figure S3b and S4). No emission was detected in blend films of M1 and M2 with fullerene acceptors indicating charge transfer between the electron donor and electron acceptor materials upon excitation of the blend films.
Cyclic voltammetry (CV) was performed to determine the HOMO energy levels of M1 and M2 (Figure S5). From the value of the onset oxidation potential, the HOMO energy levels were determined to be −5.09 eV and −5.01 eV for M1 and M2, respectively. The HOMO and LUMO level of PC61MBA are −5.74 and −3.80 eV, respectively, which were nearly the same as that of PC61BM (Table 1). The similarity of the electrochemical properties of the two donors as well as the two acceptors made them very suitable for comparing the influence of hydrogen bonding on solar cells performance in which the influence of different frontier orbital energy levels can be excluded.
The photovoltaic performance of the BHJ solar cells
The photovoltaic properties of the two small molecular donors were investigated in BHJ solar cells with conventional device architecture [ITO/PEDOT:PSS/active layer/ZnO/Al] (Figure S7). Chloroform was chosen as the processing solvent due to its good solvent properties23. The weight ratios of small molecules and acceptor were varied to balance the absorbance and the charge transporting network of the photoactive layer. Common optimization parameters such as spinning rates (film thickness) were also investigated (SI, Table S1–S3). The devices were tested under the illumination of AM 1.5 G, 100 mW cm−2. The current density–voltage curves are displayed in Figure 6a and the photovoltaic performance parameters are listed in Table 2.

(a) Current density vs. voltage data and (b) external quantum efficiency (EQE) spectrum for devices containing compounds M1 and M2.
With the optimized weight ratio of M1 to PC61BM at 1:2, a power conversion efficiency (PCE) of 0.65% was obtained with an open circuit voltage (Voc) of 0.64 V, a fill factor (FF) of 40% and a short circuit current density (Jsc) of 2.55 mA cm−2. The active layers were also investigated by using PC71BM as the electron acceptor, which has a higher absorption coefficient in visible range32. The device with M1:PC71BM (1:2) as active layer exhibited improved PCE of 1.15% with a higher Jsc of 3.90 mA cm−2, a higher FF of 47% and a Voc of 0.64 V. It is clear from the external quantum efficiency (EQE) spectrum that there was a significant increase in contribution to the photocurrent from the fullerene acceptor as a result of using PC71BM (Figure 6b). In fact, the EQE spectrum for all the devices indicate that the fullerene acceptor was providing a large portion of the photocurrent contribution. The amide containing small molecule M2 was also tested and a maximum PCE of 0.30% was recorded with the optimized weight ratio of M2 to PC61BM at 1:4. Compared with the M1 based device, M2 based device showed lower Jscof 1.10 mA cm−2 and lower FF which could be attributed to the unfavorable morphology induced by the intermolecular hydrogen bonding of M2 as evidenced by morphological studies (vide infra). The replacement of PC61BM with PC71BM also yielded a higher PCE (0.40%) mainly due to an improved Jsc (1.30 mA cm−2). However, the performance of the M2:PC71BM based device was lower than that of the M1:PC71BM based device. The hole mobility values for donor compounds M1 and M2 were measured using space charge limited current technique (see Supplementary Information for details). Hole-only devices were assembled with blends of the donor compounds with PC61BM. The calculated hole mobilities were 6.3 × 10−6 cm2V−1s−1 and 1.1 × 10−6 cm2V−1s−1 for M1 and M2 respectively. These mobility values are in agreement with the UV-vis absorption and GIWAXS data as well as the observed solar cell device performance. The UV-vis absorption and GIWAXS data indicate compound M1 showed stronger π − π stacking molecular order in solid state. Closed packing of conjugated organic semiconductor molecules often lead to better charge transport properties of the material. Better charge transport in the active layer blends of solar cell devices often result in improved device performance especially in combination with favorable blend film morphology.
The amide containing acceptor, PC61MBA, was then used to replace its PCBM analogues. Interestingly, the PCE of devices based on M1: PC61MBA (1:2) and M2: PC61MBA (1:2) decreased sharply to 0.012% and 0.004%, respectively. Extremely low Jsc and Voc were observed even though PC61MBA has very similar absorption and LUMO energy level to PC61BM. Optimization of donor-acceptor blend ratios did not provide any significant improvement in device performance (SI, Table S3).
Morphological studies
Previous literature reported that amide fullerene derivatives induced large-scale phase separation and serious aggregations19. It was likely that the poor performance of PC61MBA was also related to the undesirable active layer morphology. Morphology of the active layers was investigated using tapping mode AFM. The height as well as phase images of M1:PC61BM, M2:PC61BM, M1:PC61MBA and M2:PC61MBA blends (donor/acceptor ratio 1:2) at optimized conditions are shown in Figure 7. From the phase images of the PC61BM based blend, interpenetrating networks can be observed indicating well-defined nanoscale phase separation and efficient percolation channels. Compared to the active layer of M2:PC61BM which gave low Jsc and low FF, M1:PC61BM active layer also displayed smoother surface morphology (for M1:PC61BM blend, rms = 0.41 nm; for M2:PC61BM blend, rms = 0.46 nm) and smaller domain sizes (typical domain size for M1:PC61BM blend, 17–30 nm; M1:PC61BM blend, 28–50 nm). Larger phase domain sizes in the M2:PC61BM blend was likely a result of the intermolecular hydrogen bonding and strong aggregation of M2. This result was also consistent with previous reports in which smoother blend films and smaller domain sizes gave better performance in bulk heterojunction solar cells23,32. Large-scale phase separation and aggregation were observed when PC61MBA was used in the BHJ solar cells. Sectional analysis of Figure 7c and 7d verified the aggregation with size up to hundreds of nanometers in width and several tens of nanometers in height. The aggregation was attributed to the strong hydrogen bonding between PC61MBA itself as well as the hydrogen bonding between PC61MBA and the small molecule donor since the active layer were spin casted from homogenous solution19. The low performance of PC61MBA devices was a direct result of large scale phase separation which was unfavorable for exciton diffusion and charge separation.

AFM images of films spin coated from M1:PC61BM (a and e), M2:PC61BM (b and f), M1:PC61MBA (c and g) and M2:PC61MBA (d and h).
(a), (b), (c) and (d) are height images; (e), (f), (g) and (h) are phase images. All images are 1 × 1 μm.
Grazing incidence wide angle X-ray scattering (GIWAXS) studies
GIWAXS is a widely used technique to determine crystal packing and structural orientation of donor and acceptor blend films33. From the GIWAXS patterns presented in Figure 8a, the crystalline nature of M1 in the pristine film is evident which exhibited strong (100) Bragg reflection peak at q = 0.43 Å−1 and (200) Bragg reflection peak at q = 0.86 Å−1 corresponding to d-spacing values of 14.6 Å and 7.3 Å, respectively. The reflection at q = 1.80 Å−1 was attributed to the π − π stacking distance. In great contrast, the pristine film of M2 displayed very weak reflection peak at q = 0.42 Å−1 indicating a lower degree of order. Similar (100) and (200) reflection peak was observed in the M1:PC61BM (1:2) blend film indicating the addition of PC61BM did not change the crystalline nature of M1. The broad diffraction ring centered at q = 1.35 Å−1 was arising from PC61BM28. For the M2:PC61BM (1:2) blend film, only the reflection of PC61BM (broad reflection centered at q = 1.35 Å−1) was observed suggesting that the amide group of M2 has interaction with PC61BM and thus changed the packing mode of the small molecular donor (Figure 8b). The M2:PC61BM blend films displayed different scattering patterns probably due to the aggregation of PC61BM and also the interaction of M2 and PC61BM. Presumably the introduction of hydrogen bonding can decrease orientation order and lead to increased packing frustration relative to M1:PC61BM.

GIWAXS images of (a) pristine film of M1, (b) pristine film of M2, (c) M1:PC61BM blend film and (d) M2:PC61BM blend film.
Performed at the Australian Synchrotron (Beam centre: 215.1, 968.2) at an incident angle of 0.10°.
Conclusion
Two small molecular donor materials, one end capped with ester group and the other end capped with amide group, were synthesized and fully characterized. The amide group of the donor material M2 formed intermolecular hydrogen bonds which led to fibrous aggregates in solid state. Application of the two small molecular donor materials in BHJ solar cells revealed that the amide group containing donor material showed lower performance due to the hydrogen bonding induced aggregation and disadvantageous morphology. Replacement of PCBM with its amide analogue further increased aggregation and large scale phase separation was observed in the blend films. Well defined morphology34,35,36, enhanced stability22,35, improved performance18,19,20,21,22,34,37,38,39 have been discussed in the literature for materials with hydrogen bonding moieties. In this work, the results clear showed that a cautious approach must be taken when using strong intermolecular forces such as hydrogen bonding in organic solar cell materials.
Methods
Materials
Small molecules M1 and M2 are synthesized following the procedure described in SI (Scheme S1). PC61MBA was synthesized following a modified literature procedure (Scheme S2). PC61BM and PC71BM were purchased from Nano-C, Inc.
BHJ solar cells fabrication
BHJ solar cells were processed on pre-patterned indium tin oxide (ITO) coated glass substrates with a sheet resistance of 15 Ω per square. First a thin layer (30 nm) of poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS; Baytron AI 4083 from HC Starck) was spin-coated on a UV-Ozone cleaned ITO substrates, followed by baking on a hot plate at 140°C for 10 min. An active layer of the device was deposited by spin coating a chloroform solution containing 10 mg of donor and 20 mg of acceptor. The thickness of the active layer was 75 nm as determined by Veeco Dektak 150+ Surface Profiler. A thin layer of ZnO nanopaticle was deposited on the active layer by spin-coating (3000 rpm) to form a 25 nm thick ZnO layer. The films were then transferred to a metal evaporation chamber and aluminum (100 nm) were deposited through a shadow mask (active area was 0.1 cm2) at approximately 1 × 10−6 torr. Film thickness was determined by Veeco Dektak 150+ Surface Profiler. The current density-voltage measurements of the devices were carried out using a 1 kW Oriel solar simulator with an AM 1.5 G filter as the light source in conjunction with a Keithley 2400 source measurement unit. Solar measurements were carried out under 1000 W/m2 AM 1.5 G illumination conditions. For accurate measurement, the light intensity was calibrated using a reference silicon solar cell (PV measurements Inc.) certified by the National Renewable Energy Laboratory. Device fabrication and characterizations were performed in an ambient environment without any encapsulation.
References
Yu, G., Gao, J., Hummelen, J. C., Wudl, F. & Heeger, A. J. Polymer photovoltaic cells: Enhanced efficiencies via a network of internal donor-acceptor heterojunctions. Science 270, 1789–1791 (1995).
Nagarjuna, G. & Venkataraman, D. Strategies for controlling the active layer morphologies in OPVs. J. Polym. Sci., Part B: Polym. Phys. 50, 1045–1056 (2012).
Benanti, T. L. & Venkataraman, D. Organic solar cells: An overview focusing on active layer morphology. Photosynthesis Research 87, 73–81 (2006).
Bavel, S., Veenstra, S. & Loos, J. On the importance of morphology control in polymer solar cells. Macromol. Rapid Commun. 31, 1835–1845 (2010).
Yang, X. & Loos, J. Toward high-performance polymer solar cells: The importance of morphology control. Macromolecules 40, 1353–1362 (2007).
Thompson, B. C. & Frechet, J. M. J. Polymer–fullerene composite solar cells. Angew. Chem. Int. Ed. 47, 58–77 (2008).
Coakley, K. M. & McGehee, M. D. Conjugated polymer photovoltaic cells. Chem. Mater. 16, 4533–4542 (2004).
Kang, S. J. et al. Using self-organization to control morphology in molecular photovoltaics. J. Am. Chem. Soc. 135, 2207–2212 (2013).
Mayer, A. C. et al. Bimolecular crystals of fullerenes in conjugated polymers and the implications of molecular mixing for solar cells. Adv. Funct. Mater. 19, 1173–1179 (2009).
Kennedy, R. et al. Self-assembling fullerenes for improved bulk-heterojunction photovoltaic devices. J. Am. Chem. Soc. 130, 17290–17292 (2008).
Wong, W. W. H. et al. Fluorenyl hexa-peri-hexabenzocoronene-dendritic oligothiophene hybrid materials: synthesis, photophysical properties, self-association behaviour and device performance. Chem. Eur. J. 17, 5549–5560 (2011).
Wong, W. W. H. et al. Self-assembling thiophene dendrimers with a hexa-peri-hexabenzocoronene core-synthesis, characterization and performance in bulk heterojunction solar cells. Chem. Mater. 22, 457–466 (2010).
Kumar, R. J. et al. Hierarchical self-assembly of semiconductor functionalized peptide α-helices and optoelectronic properties. J. Am. Chem. Soc. 133, 8564–8573 (2011).
Emsley, J. Very strong hydrogen bonding. Chem. Soc. Rev. 9, 91–124 (1980).
Prins, L. J., Reinhoudt, D. N. & Timmerman, P. Noncovalent synthesis using hydrogen bonding. Angew. Chem. Int. Ed. 40, 2382–2426 (2001).
Schenning, A. P. H. J. et al. Photoinduced electron transfer in hydrogen-bonded oligo(p-phenylene vinylene)–perylene bisimide chiral assemblies. J. Am. Chem. Soc. 124, 10252–10253 (2002).
Glowacki, E. D. et al. Hydrogen-bonds in molecular solids – from biological systems to organic electronic. J. Mater. Chem. B 1, 3742–3753 (2013).
Lin, Y. et al. Morphology control in TiO2 nanorod/polythiophene composites for bulk heterojunction solar cells using hydrogen bonding. Macromolecules 45, 8665–8673 (2012).
Liu, C. et al. New methanofullerenes containing amide as electron acceptor for construction photovoltaic devices. J. Phys. Chem. C 113, 21970–21975 (2009).
Patra, D. et al. Enhancement of photovoltaic properties in supramolecular polymer networks featuring a solar cell main-chain polymer H-bonded with conjugated cross-linkers. Polymer 53, 1219–1228 (2012).
Sahu, D. et al. Synthesis and application of H-Bonded cross-linking polymers containing a conjugated pyridyl H-Acceptor side-chain polymer and various carbazole-based H-Donor dyes bearing symmetrical cyanoacrylic acids for organic solar cells. Polymer 51, 6182–6192 (2010).
Hsu, S. L. et al. New carbazole-based conjugated polymers containing pyridylvinyl thiophene units for polymer solar cell applications: Morphological stabilization through hydrogen bonding. J. Polym. Sci., Part A: Polym. Chem. 49, 603–611 (2011).
Zhou, J. et al. Small molecules based on benzo[1,2-b:4,5-b′]dithiophene unit for high-performance solution-processed organic solar cells. J. Am. Chem. Soc. 134, 16345–16351 (2012).
Takacs, C. J. et al. Solar cell efficiency, self-assembly and dipole–dipole interactions of isomorphic narrow-band-gap molecules. J. Am. Chem. Soc. 134, 16597–16606 (2012).
van der Poll, T. S. et al. Non-basic high-performance molecules for solution-processed organic solar cells. Adv. Mater. 24, 3646–3649 (2012).
Liu, Y. et al. Solution-processed small molecules using different electron linkers for high-performance solar Cells. Adv. Mater. 25, 4657–4662 (2013).
Fitzner, R. et al. Correlation of π-conjugated oligomer structure with film morphology and organic solar cell performance. J. Am. Chem. Soc. 134, 11064–11067 (2012).
Wang, H. et al. The role of additive in diketopyrrolopyrrole-based small molecular bulk heterojunction solar cells. Adv. Mater. 10.1002/adma.201301716 (2013).
Kwon, T. H. et al. Dithienothiophene (DTT)-based dyes for dye-sensitized solar cells: synthesis of 2,6-dibromo-DTT. J. Org. Chem. 76, 4088–4093 (2011).
Stals, P. J. M. et al. Asymmetrically substituted benzene-1,3,5-tricarboxamides: self-assembly and odd–even effects in the solid state and in dilute Solution. Chem. Eur. J. 15, 2071–2080 (2009).
Kajitani, T. et al. Liquid crystalline amides: linear arrangement of rod-like molecules by lateral intermolecular hydrogen bonding and molecular shape effect. J. Mater. Chem. 14, 3449–3456 (2004).
Liang, Y. Y. et al. Highly efficient solar cell polymers developed via fine-tuning of structural and electronic properties. J. Am. Chem. Soc. 131, 7792–7799 (2009).
Rivnay, J. et al. Quantitative determination of organic semiconductor microstructure from the molecular to device scale. Chem. Rev. 112, 5488–5519 (2012).
Yao, K. et al. Cooperative assembly donor acceptor system induced by intermolecular hydrogen bonds leading to oriented nanomorphology for optimized photovoltaic performance. J. Phys. Chem. C 116, 714–721 (2012).
Lin, Y. et al. Cooperative assembly of hydrogen-bonded diblock copolythiophene/fullerene blends for photovoltaic devices with well-defined morphologies and enhanced stability. Chem Mater. 24, 622–632 (2012)
Lin, Y. H. et al. Supramolecular conjugated block copolymers. Macromolecules 45, 6571–6579 (2012).
Fang, H. P. et al. Synthesis of novel dithienothiophene- and 2,7-carbazole-based conjugated polymers and H-bonded effects on electrochromic and photovoltaic propertie. J. Polym. Sci., Part A: Polym. Chem. 50, 5011–5022 (2012).
Kumar, R. J. et al. Enhancement of efficiency in organic photovoltaic devices containing self-complementary hydrogen-bonding domains. Beilstein J. Org. Chem. 9, 1102–1110 (2013).
Ruiz-Carretero, A. et al. Stepwise self-assembly to improve solar cell morphology. J. Mater. Chem. A 1, 11674–11681 (2013).
Acknowledgements
This work was made possible by support of the Victorian Organic Solar Cell Consortium, with funding provided by the Victorian State Government Department of State Development, Business and Innovation (Energy Technology Innovation Strategy and Victorian Science Agenda) and the Australian Renewable Energy Agency (ARENA Project 2-A018). Dr W. H. H. Wong is supported by an Australian Research Council Future Fellowship (FT130100500). We thank Andrew B. Holmes for helpful discussion. We thank the Australian Synchrotron for the SAXS/WAXS beam time to perform the GIWAXS experiments.
Author information
Authors and Affiliations
Contributions
Z.X. synthesized and characterized the materials. Z.X. and W.W.H.W. prepared the manuscript. K.S. performed morphological studies and device fabrication. J.S. and S.J. did device fabrication. D.J.J. and W.W.H.W. supervised the project. All authors analyzed the data, discussed the results and contributed to revisions.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Xiao, Z., Sun, K., Subbiah, J. et al. Hydrogen bonding in bulk heterojunction solar cells: A case study. Sci Rep 4, 5701 (2014). https://doi.org/10.1038/srep05701
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05701
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
