
Abstract
Selective targeting of cancer cells employing multiple combinations as co-drug holds promise for new generation therapeutics. Betulinic acid (BA), a plant secondary metabolite kills cancer cells and Dichloroacetate (DCA) is capable of reversing the Warburg phenotype by inhibiting pyruvate dehydrogenase kinase (PDK). Here, we report synthesis, characterization and tumoricidal potential of a co-drug Bet-CA, where a DCA molecule has been appended on C-3 hydroxyl group of BA to generate an ester derivative for increased solubility and subsequent cleavage by internal esterase(s) to release one unit each of BA and DCA. In vitro studies revealed pronounced synergistic cytotoxicity of Bet-CA against a broad spectrum of cancer cells and it selectively killed them when co-cultured with human fibroblasts. Bet-CA treatment increased reactive oxygen species (ROS) production, significantly altered mitochondrial membrane potential gradient (ΔΨm); followed by the release of cytochrome c (Cyt c) which prompted cells to undergo mitochondria mediated apoptosis. In vivo experimentation expectedly exhibited tumor inhibitory potential of Bet-CA and clinically achievable doses did not produce any apparent toxicity. Taken together, results suggestively raise an important corollary hypothesis stating that Bet-CA selectively and synergistically combats cancer without producing toxic manifestations and emerges to be the prospect for the new generation therapeutics.
Similar content being viewed by others


Introduction
One of the most threatening conditions afflicting human health in today's world is cancer. The propensity of tumor cells to avidly consume glucose in a hypoxic environment is known as Warburg phenotype1. Although no etiological significance can be drawn with carcinogenesis and much remains unproven about this phenomenon, this acidic environment promotes angiogenesis facilitating metastasis2. The glycolytic phenotype of cancer cells and its ability to evade apoptosis takes them to an advantageous high where proliferation predominates death3. The global burden of cancer tends to ascend due to incorporation of cancer causing habits and there is a parallel decline in the survival rate among patients from developing countries4. Armed with its own risks the option continues to be chemotherapy, when it comes to curbing down cancer, but its prime disadvantages remain as patients develop variety of side effect. Most of the present chemotherapeutic drugs are cytotoxic and do not specifically target cancer cells. Therefore, there is an urgent need to actualize a novel therapy that would provide a possible juncture to trigger targeted killing of cancer cells and confer an effective treatment regimen to reduce the side effects and increase the response to therapy. Cytotoxicity associated with chemotherapy is a major impediment in the field of cancer therapeutics for which we were fuelled with conviction to concentrate on developing a co-drug that would selectively and synergistically target cancer cells and orchestrate a benefit to cure.
One highly promising class of natural compound, betulinic acid (3β, hydroxyl-lup-20(29)-en-28-oic acid), is a prominent representative from the class of the pentacyclic triterpenoid5,6,7. In-vitro studies recognized this molecule as a promising candidate to potentially act against a wide variety of cancer cell lines including therapy rebound tumors but strikingly it is ineffective towards normal cells8. Numerous studies have shown that BA induces subsequent alteration in mitochondrial membrane potential (MMP) and induces apoptosis via the mitochondrial pathway9,10. Unfortunately, poor solubility of BA is the stumbling block in its routine medical practice11. It possesses three sites that are amenable to modification including the C-3 hydroxyl, C-20 alkene and C-28 carboxylic acid positions to convey and fine-tune the desirable properties such as selective killing, lipophilicity, solubility and increased cellular uptake.
It is a well established fact that two drugs when added in combination at defined doses can inhibit cancer in a synergistic way by changing the characteristic metabolic signatures of cancer cells, driving them towards apoptosis12,13. There is recently an emerging evidence for the effect of co-drug against cancer cell signalling and given the momentum that the co-drug theory against cancer is gaining interest in developing targeted metabolic modulators as cancer therapies, we introduced DCA and appended it to BA on its C-3 hydroxyl end for potentiating activity.
Dichloroacetate (DCA) is used regularly against lactic acidosis14,15. DCA inhibits the activity of pyruvate dehydrogenase kinase (PDK) by alleviating its inhibitory effect over pyruvate dehydrogenase (PDH) and as a result flux of pyruvate is channelized to the mitochondria for resuming the TCA cycle regenerating large amount of ATP. Thus, by inhibiting PDK16, DCA steers the cellular metabolism from glycolysis to glucose oxidation, which presents the cancer cells a proliferative disadvantage and subsequent descend in MMP17,18. This results in opening of the mitochondrial inner membrane pores and facilitates translocation of Cyt c with other apoptotic mediators from mitochondria to cytosol triggering apoptosis19. DCA reverses the malignancy making cancer cells vulnerable towards programmed cell death20,21. Avalanche of preclinical studies and discoveries for DCA using both in vitro and in vivo models claim that it might be used as an attractive sensitizer for chemotherapy and radiotherapy22,23. Being an orphan drug it is readily available and many cancer patients opt for it although not yet approved for cancer therapy24.
The idea of fabricating Bet-CA was envisioned to manifest a boost in tumor killing potential, to reduce the subtoxic doses (IC50) of both the molecules and to bring about a prominent, equimolar and simultaneous surge of BA and DCA under physiological condition post cleavage by intracellular esterase(s). Our tactic was therefore to use an established, effective, natural anticancer compound BA alongwith a small molecule DCA that inhibits lactic acidosis to generate a derivative, Bet-CA that would not only target but demolish cancer cells leaving normal cells unharmed. We successfully esterified C-3 hydroxyl group of BA with DCA to form a co-drug that would not only possess increased activity but also enhanced solubility and therefore postulated that the derivatized molecule would upgrade the tumor killing potential in a selective and synergistic manner. BA and DCA released upon cleavage would lead to the increased production of ROS, prompt the alteration in MMP and induce mitochondria mediated apoptosis. Here, we present for the first time successful synthesis and characterisation and have triangulated new and meaningful insights about the tumorigenic potential of Bet-CA that possess complementary synergistic and selective capability to stall malignancy.
Results
Synthesis and characterisation of Bet-CA
Bet-CA was synthesized by treating betulinic acid with dichloroacetylchloride in dry dichloromethane (DCM) under N2-atmosphere with high yield of about 75% (Fig. 1). Formation of Bet-CA was evidenced by the disappearance of C-3 O–H stretching frequency at 3450 cm−1 of BA and appearance of an ester C = O stretching band at 1745 cm−1 in Infrared spectra. Structure of Bet-CA was confirmed by ESI-MS, 1H-NMR and 13C-NMR. ESI-MS (M-Na) calculated 589.29, found 589.28. 1H-NMR (600 MHz, CDCl3) δ: 0.802, 0.817, 0.888, 0.901, 0.944, 1.738 (all s, each 3H, H-23, H-24, H-25, H-26, H-27, H-30), 4.744 (s, 2H, H-29), 5.933 (s, 1H, H-32). 13C-NMR (600MHz, CDCl3), δ: 20.8, 22.7, 23.2, 24.9, 25.4, 27.8, 29.1, 29.3, 29.6, 30.5, 30.9, 31.9, 32.1, 34.1, 37.0, 37.1, 38.2, 38.3, 40.7, 42.4, 46.8, 49.2, 50.4, 55.3, 56.2, 64.8, 65.1, 76.8, 84.9, 109.7, 150.3, 164.3, 179.4. See supplementary Fig. S1–S4. Melting Point: 269°C.

Synthetic scheme of Bet-CA.
Bet-CA inhibits proliferation and survival of cancer cells
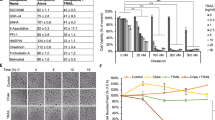
With background information on BA and DCA, we speculated that our derivatized Bet-CA might have a better cytotoxic activity and selective animosity against cancer cells and thereby sought to determine whether this compound can inhibit cancer cell viability and proliferation. Actualizing this assumption, we screened Bet-CA, BA, DCA and equimolar proportions of BA and DCA on MCF7, MDA-MB-231, MDA-MB-468, DU 145, PC-3, B16-F10, WI-38 and NIH/3T3 cell lines. Cells were incubated with these compounds at concentrations ranging from 5 to 70 μM for 72 h and preliminary cancer cell inhibitory effects were testified using MTT assay. Table 1 depicts that the concentration of Bet-CA required to confer 50% cancer cell viability (IC50) and as anticipated the dose is significantly lower when compared with its individual counterparts and/or their mixture and therefore Bet-CA might be considered a potential candidate to inhibit cancer cell proliferation and survival.
In vitro cellular pharmacokinetic analysis of Bet-CA
The demonstrable cellular killing potential of Bet-CA based on aforementioned results further hinted us to scrutinise the intuitive postulation raised for the cleavage of Bet-CA by intracellular esterases. In principle one simple way to depict the phenomenology would be to enumerate the amount of Bet-CA entering the cells at a time point and its subsequent degradation to BA and DCA. Thereby MCF7 cells were exposed to subtoxic doses of Bet-CA for varying time points and extracted using t-butyl methyl ether and rationalized using HPLC. Graphical depictions of the uptake milieu and intracellular cleavage are shown in Fig. 2A and the representative HPLC chromatogram is charted out in Supplementary Fig. S5. Observations readout that at 4 h there was accumulation of Bet-CA within the cells and when studied for the next 6 and 8 h, a gradual cumulative increase of BA was noted. Significantly within 16 h, 98.7% of Bet-CA breakdown to BA was registered. The eluents collected at 9.2 and 10.2 min suggestive of being BA and Bet-CA respectively from the retention time of pure BA and Bet-CA, were further confirmed by ESI-MS (see Supplementary Fig. S6). Observations draw a reasonably complete picture for the fact that BA loads up gradually within the cells to provocatively point out the fact that Bet-CA enters the cells and gets cleaved afterwards. To rule out the supposed pretentions of non-specific Bet-CA cleavage due to esterases present in FBS, Bet-CA was incubated for varying time point upto 72 h in complete media. Clarifying the premonitions, results depict a significant maximal 2.5% of Bet-CA cleavage to BA (Fig. 2B) and the representative HPLC chromatograms (see Supplementary Fig. S7) significantly state the absence of nonspecific extracellular cleavage. Implicit to this notion the observations register a reasonably complete picture relating to the fact that Bet-CA specifically enters the cells and gets cleaved to release BA and DCA as documented from these collective corroborative HPLC analyses. Thus, Bet-CA is endowed with the ability to enter the cells and invoke a comprehensive tumor killing response.

Pharmacokinetic uptake study of Bet-CA.
(A) Graphical representation demonstrating cleavage of Bet-CA by intracellular esterases of MCF7 cells and increased accumulation of BA over time points. (B) Graph depicts the percent of nonspecific extracellular cleavage of Bet-CA when incubated with DMEM+10% FBS at the mentioned time points. (* p < 0.05, ** p < 0.01, compared to Bet-CA cleavage at 0 h time point). In all panels error bars represent mean ± SD.
Bet-CA mediates selective killing and forgoes cytotoxicity
Based on the observation that Bet-CA has an obvious toxic effect on cancer cells, we promptly went on to decipher its toxicity towards normal cells. WI-38 (human lung normal fibroblast) and NIH/3T3 (mouse embryonic fibroblast) were treated with Bet-CA upto 70 μM for 72 h and MTT assay was carried out. Figure 3A represents comparative depiction of the effect of Bet-CA on both cancer and normal cells lines and delineates that it has practically no effect on normal cells. We further performed flow cytometric analysis using Annexin-V-FITC and propidium iodide (PI) on NIH/3T3 cells. The readout held clear as about 80% of fibroblasts were alive even after 50 μM of Bet-CA treatment for 24 h whereas this concentration poses to be the subtoxic dose for the neoplastic variety (Fig. 3B).

Preferential effect of Bet-CA.
(A) Viability of WI-38 and NIH/3T3 cell lines were analyzed using MTT assay and compared with that of the cancerous cell lines. Bars represent % of cell viability after 72 h. (B) Graph represents flow cytometric analysis of NIH/3T3 cells after 24 h of Bet-CA treatment using Annexin V/PI dual staining protocol. Stacked bars exhibit the % of cell that are viable, early apoptotic, in the late apoptotic phase or that are dead. (C) Graph demonstrating the % of TUNEL positive cells after 24 h of Bet-CA treatment in the co-culture of malignant DU 145 cells and normal WI-38 fibroblastic cell line (D) The co-culture of WI-38 cells and DU 145 cells were treated with Bet-CA for 24 h. Arrows indicate apoptotic DU 145 cells. (E) The graph shows % of micronuclei formation and chromosomal aberration in human peripheral lymphocytes after 72 h Bet-CA treatment. (* p < 0.05, ** p < 0.01, compared to the vehicle and positive control sets). In all panels error bars represent mean ± SD.
Additionally to reaffirm the preferential treatment of Bet-CA towards cancer cells, a straightforward method of study was extended to the culture of WI-38 and DU 145 cells. WI-38 cells were seeded on 3 μm transwell chambers and cancerous DU 145 cells on 24 well plates. The plate chambers with hanging inserts were treated with 50 μM of Bet-CA for 24 h and the responses of both these cell lines were evaluated using TUNEL assay. Graph showcases that after treatment with 50 μM of Bet-CA there is an absolute maximal increase of 3.36% TUNEL positive WI-38 cells whereas the same challenge fated a staggering 38.8% of DU 145 cells to death as implied with the increase in the number of TUNEL positive staining (Fig. 3C). Furthermore the co-culture of WI-38 and DU 145 cells was treated with 10, 30 and 50 μM of Bet-CA for 24 h and the responses of both these cell lines to Bet-CA were evaluated and morphology analysed by visualizing them under confocal laser scanning microscopy (CLSM) as the two types of cells are clearly distinguishable due to their differences in morphology. Figure 3D reveals that Bet-CA has no significant effect on the viability of the fibroblasts whereas it selectively killed the cancerous cells. Arrows indicate cancer cells exhibiting apoptotic morphology whereas the fibroblasts appear unaffected by Bet-CA.
A further experimentation followed close on the heels of the above observations and we deduced the cytotoxic potential of Bet-CA by determining its ability to induce chromosomal aberration and micronuclei (MN) formation in primary culture of human lymphocytes in presence of 100 μM of Bet-CA for 72 h. The results propose a significantly low percentage of chromosomal aberration and micronuclei formation upto 100 μM of Bet-CA treatment when compared to that of positive control (Fig. 3E). Taken together underpinnings of these results attribute to the fact that Bet-CA resoundingly manipulates its potential to spare the normal cells and mediate selective killing of cancer cells.
Bet-CA induces loss in MMP and mitochondria mediated cell death
To explore the effectiveness of Bet-CA in targeting mitochondria of cancer cells, alterations in the mitochondrial transmembrane potential were determined. Cancerous MCF7 cells were treated with 50 μM of Bet-CA for 16 h and subsequently stained with 5, 5′, 6, 6′-tetrachloro-1, 1′, 3, 3′- tetraethyl benzimidazolyl carbo-cyanine iodide (JC-1), a surrogate marker extensively used to detect damages in the mitochondrial potential25. As with previous experiments, the effect of Bet-CA was apparent as shown in (Fig. 4A). Control MCF7 cells depicted a heterogeneous staining pattern exhibiting both green and red fluorescence whereas treatment with Bet-CA reflected a sharp decrease in red fluorescence analogous with an increase in green fluorescence as noted via CLSM. Actualizing the selective aptitude concept of Bet-CA, NIH/3T3 cells were challenged with 50 μM of Bet-CA for 16 h and alterations of the red and green fluorescence intensities were quantitatively determined. Changes in their ratio was analysed and graphically documented to significantly decipher and establish the presumed postulation (Fig. 4B). Cytometric data conferred no effective alteration in MMP of the fibroblastic mitochondria factually drawing us to a conceptual paradigm regarding the preferential potential of Bet-CA to limit the deterioration of MMP within the tumorigenic population. MCF7 cells were additionally treated with 50 μM each of Bet-CA, BA, DCA and BA+DCA (1:1) to understand the dynamic behaviour of Bet-CA that becomes crucial to establish its synergistic effect. Flow cytometric data suggestively depict that BA and DCA alone or their 1:1 stoichiometric mixture fails to mount a demonstrable destruction of the neoplastic mitochondria. Thereby, Bet-CA presents a double benefit in that it is selective and at the same time endowed with synergistic ability to mitigate cancer (Fig. 4C).

Bet-CA promotes mitochondrial damage and initiates the caspase cascade.
(A) Representative confocal images demonstrating MMP alteration using JC-1 dye. Control MCF7 cells (upper panel) show heterogeneous staining pattern of red and green fluorescence whereas treatment with Bet-CA (lower panel) causes increase in green and decrease in red fluorescence intensities. (B) Graph demonstrates quantitative determination of red: green fluorescence intensity ratio in NIH/3T3 cells after 16 h of Bet-CA treatment. (C) Graph represents quantitative determination of alterations in MMP. Bet-CA, BA, DCA and BA+DCA (1:1) treated MCF7 cells were stained with JC-1, subjected to flow cytometry and analyzed. Quantitative comparative analysis depicts a significant decrease in red: green fluorescence intensity ratio with respect to the other counterparts. (D) Graph shows flow cytometric analysis of MPTP. After Bet-CA treatment for 16 h, control and treated MCF7 cells were loaded with calcein-AM and quencher CoCl2 and green fluorescence intensity of the cells were estimated. Fluorescence of treated cell mitochondria is highly reduced to that of control. (E) Figure represents AFM images of control and treated MCF7 mitochondria. Change in surface morphology and swelling of mitochondria with respect to that of control is obvious after treatment as indicated. (F) MCF7 cells were treated for 24 h with Bet-CA and subjected to flow cytometric analysis for determination of Bcl-xl levels. Results depict a subsequent decrease in Bcl-xl expression in a dose dependent manner. (G) Bet-CA treated MCF7 cells were subjected to immunoblotting for determination of Bax expression where β-actin was used as loading control. (H) Representative fluorescence micrographs of control (upper panel) and Bet-CA treated (lower panel) MCF7 cells and WI-38 cells depicting Cyt c release after 16 h. (I) Representative CLSM images showing increase in the levels of cleaved caspase-3. MDA-MB-231 cells were treated with Bet-CA for 24 h, fixed and stained with anti-cleaved caspase-3 (PE conjugate) antibody. (* p < 0.05, ** p < 0.01, compared to the vehicle control sets). In all panels error bars represent mean ± SD.
Mitochondrial potential dilapidation leads to stress accompanied by calcium overload and ATP depletion that induces mitochondrial permeability transition (MPT) with formation of nonspecific MPT pores (MPTP) in the inner mitochondrial membrane. Pore opening assertively induces mitochondrial dysfunction eventually leading to cell death26,27. Stress prone mitochondria allow the passage of various molecules28 and in analogy with the same phenomenon we considered that Bet-CA might as well promote MPTP and this was validated using the fluorescent dye calcein acetoxymethyl (calcein-AM) and its quencher CoCl229. Figure 4D depicts that under normal conditions, without the quencher, MCF7 cells exhibited high levels of green intensity which expectedly declined when CoCl2 was added. 50 μM of Bet-CA treatment for 16 h resulted in a distinct fall in intensity suggesting that CoCl2 entered via the MPTP generated by Bet-CA.
MPTP promotes swelling of the mitochondria and this was validated employing atomic force microscopy (AFM). Isolated mitochondria from vehicle control and Bet-CA treated MCF7 cells were analyzed for subsequent changes in size along with surface morphology. Figure 4E represents ~1.6 fold increase in size in treated sets in contrast to that of control, leading us to infer that mitochondria are robustly affected due to Bet-CA treatment.
MPTP and mitochondrial swelling insisted us further to check the status of the Bcl-2 family proteins along with Bax that actively takes part in the pro-survival programme of tumor cells30. Results clearly ascertain that Bet-CA treatment leads to the decline of Bcl-xl expression and enhancement of Bax expression in MCF7 cells (Fig. 4F and 4G).
In context with Bet-CA induced enhanced Bax expressions, we investigated whether this event is succeeded by Cyt c release from the mitochondria31,32. MCF7 and WI-38 cells were treated with 50 μM of Bet-CA for 16 h and Cyt c release from the mitochondria was studied using immunofluorescence assay. Control MCF7 and WI-38 cells were characterized for their distinct punctuate mitochondrial immunostaining pattern whereas treatment for 16 h resulted in Cyt c efflux from that of the neoplastic mitochondria as evidenced by the diffuse green cytoplasmic immunofluorescence staining pattern. The phenomenology of Cyt c efflux was not observed in fibroblast cell line (Fig. 4H). MCF7 cells were further treated with 50 μM each of BA, DCA and BA+DCA (1:1) and their effects were evaluated further (see Supplementary Fig. S8). Presumably none of them garnered a significant impact albeit in the aforementioned concentration hinting on the synergistic potential of Bet-CA.
Cyt c release from the mitochondria might lead to the activation of the caspase cascade33,34. To address this possibility, we determined the status of caspases employing immunofluorescence assay (Fig. 4I). Staining showed higher levels of cleaved caspase-3 in Bet-CA treated MDA-MB-231 cells and no prominent generation of the same in WI-38 cell line (see Supplementary Fig. S9). These results indicate the activation of the caspase cascade prior to the apoptotic event in malignant cells. Hitherto, Bet-CA appears to damage the mitochondria and specifically recruit caspases in mediating apoptosis.
Bet-CA induces oxidative stress and cell death
The major feature of cancer cells when compared to normal cells is their propensity to manifest a pro-oxidative state by generation of superoxide radical which provides them the advantage to cope with various stresses35,36. Higher levels of ROS are the general phenotype of the cancer cells, however, when exposed to excessive ROS their viability is challenged leading them towards mitochondria mediated apoptotic death37,38. To dissect the relative contribution of ROS in mitochondria mediated cell death we exemplified the ability of Bet-CA to produce ROS using 2′, 7′-dichlorodihydrofluorescein diacetate (H2 DCF-DA). To test this, MCF7 and WI-38 cells were treated with 50 μM of Bet-CA for 0, 2, 4, 6 and 8 h, stained with H2 DCF-DA and analyzed using CLSM. Highest level of mean fluorescence intensity was observed at 6 h time point in case of cancerous cells (Fig. 5A) whereas there was no counterintuitive existence of ROS in fibroblasts (see Supplementary Fig. S10A). Furthermore, MCF7 cells were treated with 50 μM of Bet-CA for 0, 2, 4, 6, 8 and 16 h time points and the amount of DCF fluorescence was quantified employing flow cytometry. Graphical depictions of the cytometric data is in absolute concordance with the microscopic images and thereby helps us to infer and unequivocally cement that maximum ROS accumulation occur at 6 h time point (Fig. 5B). Additionally, to evaluate the synergistic capability of Bet-CA, MCF7 cells were treated with 50 μM each of Bet-CA, BA, DCA and BA+DCA (1:1) and ROS generation was quantitatively notified adopting flow cytometry. Supplementary Fig. S10B represents the comparative analogous ROS generation after treatment with the above mentioned regimens at varying time points. To obtain a clearer understanding we chalked out the fold increase in green fluorescence at 6 h time points from each treatment regimens. Data depicts the fruition of a significant 1.8 fold elevation in the mean fluorescence intensity of Bet-CA treated sets at 6 h time point which significantly out levelled the other sets (Fig. 5C) demonstratively allowing us to conclusively justify the supposed synergistic potential of Bet-CA to generate ROS.

Bet-CA induces ROS generation resulting in apoptosis.
(A) Representative confocal micrographs depicting ROS generation in MCF7 cells after treatment with Bet-CA for mentioned time points. (B) Graph shows quantification of DCF fluorescence intensity in MCF7 cells at indicated time points after Bet-CA addition employing flow cytometric analysis. (C) MCF7 cells were treated with 50 μM each of Bet-CA, BA, DCA and BA+DCA (1:1) stained with H2 DCF-DA and DCF fluorescence intensity was analyzed using flow cytometry. Graph depicts comparative quantitative fold increase in DCF fluorescence intensity at 6 h time point. (D) TUNEL assay showing inter-nucleosomal DNA breakage in MCF7 cells after 24 h of Bet-CA treatment. Cells were stained using FITC labelled BrdU antibody and counterstained with PI. Graph shows % of TUNEL positive cells in vehicle control and treated sets. (E) Apoptosis was analysed by flow cytometry using Annexin-V-FITC and PI. Stacked bar graph represents the comparative results obtained after treatment of MCF7 cells with varying doses of Bet-CA, BA, DCA and BA+DCA (1:1) treatment at 24 h time point. (* p < 0.05, ** p < 0.01, compared to the vehicle control sets). In all panels error bars represent mean ± SD.
Reports provide evidence that ROS is involved with the induction of apoptosis39,40. Since Bet-CA treated neoplastic cells experience mitochondrial damage and a subsequent avalanche of ROS, we sought to determine whether these responses are finally coupled with DNA degradation and apoptosis. MCF7 cells were treated with 10, 30 and 50 μM of Bet-CA for 24 h and DNA strand breakage was assessed using the Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay technique. Bars represent the number of TUNEL positive cells which increased in an escalating manner indicating a dose dependent effect of the compound on the cancer cells (Fig. 5D). To further add impetus and quantify apoptosis in cancer cells, annexin V assay was performed using flow cytometry. With this analysis the numbers of apoptotic cells were determined at 24 h after exposure to 10, 30 and 50 μM each of Bet-CA, BA, DCA and BA+DCA (1:1). A significant 43% of the cell death was noted after 50 μM of Bet-CA treatment whereas the same concentration for BA, DCA and BA+DCA (1:1) proved ineffective as 22.1, 12 and 28.5% of cellular sacrifice was observed respectively (Fig. 5E). These data exemplify the role of Bet-CA in enhancing ROS and inducing apoptotic death in malignant cells and stipulate Bet-CA as a potential candidate to synergistically inhibit survival and proliferation of cancer cells.
Bet-CA inhibits tumor formation and pulmonary metastasis in syngeneic model of mouse melanoma
Prompted by the in vitro observations depicting the manifestations of Bet-CA we examined whether it also exhibited the tumor abrogating effect in vivo. Syngeneic 106 B16-F10 cells were injected subcutaneously on the right flank of immuno-competent female BALB/c mice. After three days, intravenous (i.v.) injections of 2.5 mg/kg body weight of Bet-CA, BA, DCA, BA+DCA (1:1) and vehicle control were administered according to the schedule via lateral tail vein during the next 25 days (Fig. 6A). Mice were regularly monitored for tumor formation in control and treated sets. On the 25th day, control and experimental mice were sacrificed and accordingly tumor volume was measured. Representative images of the excised tumors from that of the vehicle control and treated mice and comparative graphical analysis of the tumor volumes depict a suggestive decrease in the parameter, evidently exemplifying the increased effectiveness of Bet-CA in comparison to the other treatment regimens (Fig. 6B).

Reduction of melanoma tumor growth and pulmonary metastasis post Bet-CA treatment.
Six tumor bearing BALB/c mice were taken for each of Bet-CA, BA, DCA and BA+DCA (1:1) treated and vehicle control groups and six normal BALB/c mice were also taken as untreated control. (A) Outline of the dosing schedule used in primary melanoma model. 2.5 mg/kg each of Bet-CA, BA, DCA and BA+DCA (1:1) was directed to the tumor challenged mice on the respective days as indicated. (B) Inhibition of primary melanoma tumor growth after Bet-CA treatment. Mice were sacrificed on day 25, tumor excised, volume assessed and photographed. Graph outlines comparative quantification and reduction of tumor growth rate in treated mice sets with respect to that of vehicle control. Bet-CA significantly hinders tumor growth rate when compared to that of other treated sets. (C) Outline of the dosing schedule used in pulmonary metastatic model of BALB/c mice. Intravenous injections each of 2.5 mg/kg of Bet-CA, BA, DCA and BA+DCA (1:1) was administered to the tumor challenged mice as designated in the schedule. (D) Representative images of lungs obtained from untreated, vehicle control, Bet-CA, BA, DCA and BA+DCA (1:1) treated mice on day 14. Graph represents quantification of metastatic lesions on both dorsal and ventral sides of lungs after excision. Tumor nodules of Bet-CA treated animals when assessed appear to be much less in number than that of vehicle control or other treated sets. Bet-CA responsibly limits the number of pulmonary metastatic lesions. (* p < 0.05, ** p < 0.01, compared to that of vehicle control sets). In all panels error bars represent mean ± SD.
We next evaluated the comparative and synergistic role of Bet-CA in inhibiting metastasis in syngeneic model of metastatic B16-F10 mouse melanoma. Intravenous injection of B16-F10 cells yields highly reproducible number of pulmonary tumor metastasis41. Accordingly, female BALB/c mice were injected intravenously with 106 B16-F10 cells. 3 days post tumor challenge, i.v. doses of 2.5 mg/kg each of Bet-CA, BA, DCA and BA+DCA (1:1) were administered once every alternate day for 14 days (Fig. 6C). Mice were sacrificed on the 14th day and images of representative mice from each group are shown. The pulmonary metastatic lesions were assessed and graph portrays the number of metastatic nodules in the treatment groups compared to that of vehicle control (Fig. 6D). As anticipated, a significant reduction of lesions was observed in Bet-CA treated sets as compared with that of the other sets, provocatively implying its synergistic effect. Additionally toxic effects of Bet-CA within organs were analysed using haematoxylin/eosin staining of representative tissue sections (see Supplementary Fig. S11). Histological sections of lung, liver, spleen and kidney when visualized showed that Bet-CA does not manifest any toxicity as treated tissue sections appeared absolutely at par to that of control with normal nucleation and epithelium. Evidently Bet-CA not only stalls primary tumor growth but also effectively controls the metastatic spread. Therefore, we infer that Bet-CA regulates the spread of primary tumor and slows the progression of life threatening metastasis without any aversive side effects.
Discussion
Here we describe a therapeutically unique strategy to combat cancer, sidelining the cytotoxicity towards normal cells. Ushering this concept itself is challenge to the establishment but uncorking the bottleneck in the drug development industry requires novel concepts to increase the drug spectrum to unprecedented level. The co-drug Bet-CA was developed by esterifying BA, a plant secondary metabolite possessing anticancer property alongwith DCA, used against lactic acidosis18,19,20. There is an emerging evidence for the co-drug concept against cancer cell signalling42 and such knowledge may inturn lead to opportunities to develop novel therapies that understand and exploit the differences in order to selectively target tumor cells. Given the impetus for the co-drug theory against cancer this intriguing mechanism may provide an unmatchable agility and flexibility to combat dynamic malignancy and therefore we gained renewed interest in developing targeted metabolic modulators as cancer therapies. Derivatization of such co-drug molecules using BA and DCA is by itself a novel concept and its actualization fuelled us to evaluate the therapeutic impact of Bet-CA on cancer, which has so far not been studied. A single step synthetic procedure benefitted us with a high yield of Bet-CA and we hereby extend the evidence justifying our postulation that this compound can selectively promote apoptosis mechanistically in cancer cells directly to a criterion in which they can have additional beneficial effects, decreasing the toxicity towards normal cells and increase the response to therapy.
In this study we accentuated the effect Bet-CA deliberates on cancer cells and furthermore the molecular mechanisms by which it induces cell death. It potently inhibits proliferation and induces apoptosis on a wide spectrum of cell lines. Equally importantly, applying discreet yet novel experimental designs we confer Bet-CA spares normal fibroblasts and additional data produced promising evidence for the fact that Bet-CA is purportedly a non toxic compound. Thus, the theme emerging from the studies is that Bet-CA selectively targets the neoplastic cells and impairs their survival and proliferation.
Cellular uptake studies were performed to document the proposed postulation regarding the cleavage of Bet-CA by intracellular esterase. Astoundingly it was noted that Bet-CA being an ester derivative can bypass the barriers of extracellular breakdown, offer itself for cleavage by cellular esterases to present a prominent damage to the malignant variety. Therefore it can be convincingly claimed that Bet-CA is fortuitously endowed with the potential to escape non-specific degradation and synergistically act for benefit.
Suppressed glucose oxidation/glycolysis ratio, mitochondrial hyperpolarization and increased levels of handling stress contribute to the resistance of apoptosis. As cancer cells already have elevated levels of intrinsic ROS it is quite apparent that these malignant cells will become vulnerable when exposed to further oxidative insults43. Exposure of cancer cells to subtoxic doses of Bet-CA leads to the significant enhancement of ROS in a temporal fashion. Furthermore, Bet-CA caused no significant generation of ROS in the fibroblastic cell line clarifying the ambiguity and ascertaining the paradigm for the selective killing property of Bet-CA. Of equal importance when cancer cells were challenged with a similar dose of BA, DCA or its stoichiometric mixture and analysed, there was no significant accumulation of intracellular ROS to immediate apoptosis and this phenomenology uncovered suggestively yet another simplifying notion and unequivocally concreted the synergistic potential of Bet-CA.
Cells cannot gain a growth of advantage by losing a mitochondrion and thereby damages inflicted upon it prove to be fatal44. True to this fact, Bet-CA confers the degeneration of MMP, opening of MPTP and swelling of the mitochondria. Furthermore, our studies establish that Bet-CA treatment leads to the downregulation of Bcl-xl and activation Bax expression. We have critically determined that Cyt c efflux is followed by apoptosis via the caspase cascade in our experimentations. TUNEL assay documented DNA strand breakage and the phenomenon of apoptosis was further confirmed by dual colour mode flow cytometric analysis. These data are in general agreement with the observation that Bet-CA regulates the expression of apoptosis related proteins, leads to the release of Cyt c and initiates caspase mediated cell death. To detail the granularity for the selective potential of Bet-CA, its effect was studied on the normal cell line. Opposed to the effect Bet-CA poses on the neoplastic cells, there was no alteration in MMP, neither release of Cyt c nor increase in the levels of cleaved caspase-3. Additionally equivalent doses of BA, DCA or BA+DCA (1:1) failed to confer a degradation of ΔΨm or the release of Cyt c and observations readout a similar phenomenon straightway pointing the obvious hypothesis for the synergistic potential of Bet-CA. Therefore, what was a rather provocative hypothesis initially, gained a solid basis with ultimate precision factually obviating the selective and synergistic potential of Bet-CA to establish it as the futuristic version of medicine for tackling the nettlesome problems associated with therapy.
In vitro observations enticed us to explore and evaluate whether Bet-CA poses similar barriers to the growth of cancer cells in vivo. Our observations were further corroborated in syngeneic mice melanoma model and pulmonary metastatic melanoma model of BALB/c mice. Expectedly, Bet-CA treatment significantly limited tumor growth and metastatic nodule formation in lungs. The results clearly authenticate the apparent synergistic antitumor behaviour of this compound in vivo. Histological appearances of the organs in treated sets were normal depicting no level of toxicity of Bet-CA.
From the above studies we present an important consequential agenda that Bet-CA is endowed with ability to target and kill cancer cells. Our data clearly indicate that Bet-CA might be considered a capable drug as it enforces repression of cancer cell proliferation in vitro and tumor initiation and growth in vivo without any toxic manifestations. The conjunction of a plant secondary metabolite with a small molecule presents a promising synergistic and encouraging selective strategy to circumvent malignancy and obliterate cytotoxicity. Based on our findings further elucidation of detailed course of action of Bet-CA is a riveting area and likely to resonate a substantial promise for the future.
Methods
Chemicals
Betulinic acid (>90% pure), dichloroacetylchloride, N,N-dimethylaminopyridine (DMAP), dimethylsulphoxide (DMSO), paraformaldehyde, Dulbecco's modified eagles medium (DMEM), bovine serum albumin (BSA) and Histopaque-1077 were purchased from Sigma (St. Louis, Missouri, USA). RPMI 1640, PenStrep, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazoliumbromide), Hoechst 33342 (bisbenzamide trihydrochloride), 5,5′,6,6′-tetrachloro-1,1′,3,3′- tetraethyl benzimidazolyl carbocyanine iodide (JC-1), fetal bovine serum (FBS), Calcein-AM was obtained from Life Technologies (Carlsbad, CA, USA). Dichlorofluorescein diacetate (DCFDA) and Protein-A HRP were obtained from Calbiochem/Merck-Millipore (Darmstadt, Germany). The antibodies viz., anti-Bcl-2, anti-Bcl-xl, anti-Bax, anti-Cyt c, anti-cleaved caspase3, anti-β-actin was purchased from Cell Signalling Technologies (Beverly, MA). Other reagents were of analytical grade.
Synthesis and characterisation of Bet-CA
Bet-CA was obtained by treating betulinic acid (500 mg, 1.1 mmole) with dichloroacetylchloride (194 mg, 1.3 mmole) in presence of pyridine and DMAP (pinch) in dry CH2Cl2 (30 ml). After stirring at room temperature for 18 h under nitrogenous atmosphere, the reaction was stopped and the reaction volume was reduced in rotor evaporator. The reaction mass was initially purified using silica gel (60–120 mess) column chromatography (ethyl acetate: petroleum ether: 3:7) followed by RP C-18 HPLC column (X-Terra, 300 × 10 mm, 5 μm, Waters) chromatography using 209 nm UV detector. Purity of Bet-CA (yield: 78%, 390 mg) was checked using 1H-NMR (600 MHz), 13C-NMR (600 MHz), FT-IR and ESI-MS.
Cell culture
MCF7, MDA-MB231, MDA-MB468 (breast cancer), DU 145, PC-3 (prostate cancer), B16-F10 (mouse skin cancer), WI-38 (lung fibroblast) and NIH/3T3 (mouse embryonic fibroblast) were cultured in DMEM or RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in 5% CO2 at 37°C. Cells from exponentially growing cultures were used for all the experiments.
Bet-CA, BA was dissolved in DMSO, DCA in PBS and accordingly used for experimentations.
Cellular uptake study
50 μM of Bet-CA was added to DMEM containing 10% FBS or to complete culture media containing MCF7 cells for varying time points. In case of measurement for cellular uptake and subsequent specific intracellular esterase cleavage of Bet-CA, cells were harvested using trypsin/EDTA, washed with PBS and processed for HPLC analysis. Additionally, to determine the non-specific extracellular cleavage of Bet-CA, if any, the media containing 10% FBS was processed for subsequent analysis. Bet-CA was extracted from the cells and the media by vortexing with t-butyl methyl ether for 2 min and repeated twice. From the respective experimental sets organic and aqueous layers were separated through centrifugation (8000 × g, 10 min), organic layers were pooled, evaporated to dryness and dissolved in acetonitrile. Standard was prepared by dissolving Bet-CA in DMSO, diluted in acetonitrile and subjected to HPLC (Waters Corporation 1525 binary pump, 2487 dual UV detector) analysis using 100% acetonitrile as mobile phase before analysing the experimental batches. The column was re-equilibrated at initial conditions for 30 min before next injection and the corresponding analytes were monitored using UV detector at 209 nm and the peak area was quantified using Empower Pro 2006 Waters Corporation Empower 2 software.
Measurement of MMP using JC-1
Mitochondrial depolarization was assessed by CLSM (Andor) and flow cytometry using the cationic carbocyanine dye JC-1. Cells were cultured on coverslips overnight, treated with 50 μM of Bet-CA for 16 h at 37°C, washed twice with PBS and incubated with 10 μg/ml of JC-1 at 37°C for 15 min. Finally cells were washed and analysed by CLSM at 40×. For FACS, 50 μM of Bet-CA, BA, DCA and BA+DCA (1:1) pre-treated cells were harvested by centrifugation, suspended in PBS and incubated with 10 μg/ml of JC-1 at 37°C for 15 min. Stained cells were washed twice, suspended in PBS and analysed using BD LSRFortressa II flow cytometer.
MPTP assay using Calcein-AM
Mitochondrial permeability transition pore (MPTP) opening was assessed using calcein-AM. Neoplastic cells were cultured in presence or absence of Bet-CA (50 μM, 16 h), harvested and incubated with calcein-AM (1 mM) at 37°C in the dark. After 30 min, CoCl2 (1 mM) was added and the cells were incubated for another 10 min at 37°C in the dark. The fluorescence signal of mitochondria-trapped calcein in control and treated cells were analyzed using a BD LSRFortessa II flow cytometer at the excitation wavelength of 488 nm. For each experiment, fluorescence from 10,000 cells was acquired and analyzed using the FACS Diva 6.2 software.
Atomic force microscopy of mitochondria
Cells were seeded on a 6-well plate and treated with 50 μM Bet-CA or vehicle for 16 h at 37°C and 5% CO2. Mitochondria were isolated using Qproteome mitochondria isolation kit (Qiagen) according to the manufacturer's protocol. Mitochondria were deposited onto freshly cleaved muscovite Ruby mica sheet (ASTM V1 Grade Ruby Mica from MICAFAB, Chennai) and dried. Samples were gently washed and further dried. The ultrastructures of the mitochondria were detected by an AFM (Agilent Technologies USA) at ~20 to 24°C. AFM probes with resonance frequency of 275.1 kHz and spring constant of 21e98 N/m were used with a scan rate of 0.499 lines/sec or 0.474 μm/sec. Images were processed and length, height and width of mitochondria were measured using Pico view 1.10.1 software.
Immunofluorescence assay
Cells were seeded on coverslips, incubated at 37°C, 5% CO2 and treated as mentioned. The coverslips were washed several times in PBS before fixing with 4% (w/v) paraformaldehyde for 20 min at room temperature and subsequently permeabilized with 0.2% Triton X-100 for 15 min on ice. After blocking in 1% BSA for one hour at room temperature, cells were incubated overnight with primary antibodies against Cyt c and cleaved caspase-3 (PE conjugate) at 4°C. In case of Cyt c, coverslips were incubated with secondary antibody (Alexa Fluor-488 conjugate) in 1% BSA at room temperature for 2 h. Nucleus was stained using Hoechst 33342. Finally, coverslips were washed in PBS, mounted on glass slides and examined under microscope. Cyt c images of MCF7 and WI-38 cells were captured using fluorescence microscope (Nikon Eclipse Ti) at 60× and processed using NIS-Elements AR 4.20.00 software. Caspase-3 images were captured employing CLSM at 40× magnification and processed using Andor iQ2 software.
Estimation of intracellular ROS
To examine the comparative effect of Bet-CA, BA, DCA and BA+DCA (1:1) on the generation of ROS, H2 DCF-DA was used. Cells were treated with 50 μM of each for six different time points of 0, 2, 4, 6, 8, 16 h. After treatment, cells were washed in PBS twice and incubated with H2 DCF-DA (10 μM) for 15 min at 37°C and immediately analyzed using CLSM or harvested for flow cytometry. Green fluorescence of 2, 7-dichlorofluorescein (DCF) was measured using CLSM as well as flow cytometer (BD LSRFortressa II) with excitation at 488 nm and emission at 530 nm. Mean fluorescence intensity was estimated using BD FACS DIVA 6.2 software and CLSM images were analyzed using ImageJ software. Images were captured at 40× magnification.
Flow cytometry (live-dead)
Bet-CA, BA, DCA and BA+DCA (1:1) induced apoptosis was determined using flow cytometric analysis. Cells were treated for 24 h, washed with PBS, resuspended in 100 μl of binding buffer and further incubated with Annexin-V FITC and PI for 15 min in dark at room temperature. Prior to flow cytometric analysis, 400 μl of binding buffer was added and immediately subjected for quantification of the number of apoptotic cells. Data was generated using BD LSRFortressa II and analyzed using BD FACS DIVA 6.2 software.
Animal maintenance and melanoma tumor model
Adult female BALB/c mice (20–25 gm) used in this study were received from the animal house of the institute and maintained under laboratory conditions (12: 12, dark: light cycle), fed with standard pellet diet and water ad libitum. All experiments were performed in accordance with the national guidelines and as recommended by the institute's animal ethics committee. All possible efforts were made to minimize the animals' suffering and to reduce the number of animals used. The institutional animal care committee of CSIR-Indian Institute of Chemical Biology reviewed and approved the animal experimentations.
Female virgin BALB/c mice were challenged with 106 melanoma cells subcutaneously to form primary tumor or via i.v. administration of cells to achieve pulmonary metastatic lesions. The mice were treated intravenously with 2.5 mg/kg of Bet-CA, BA, DCA and BA+DCA (1:1) according to the dosing schedule after day 3 of tumor challenge. The mice were sacrificed on the respective day and tumors were excised for further studies.
Statistical analysis
All experiments were performed in triplicates and data are representative of three independent experiments. For continuous normally distributed data, one-way ANOVA was used to determine differences among different treatments. If the test was significant, (P < 0.01/0.05), pairwise two-sided Student's t-test was used to compare between treatments. When measurements not normally distributed a Kruskal-Wallis rank test was used and if found significant (p < 0.05), Mann-Whitney test was performed for comparisons between different treatments. All statistical calculations were carried out using Origin 6.0 Professional software.
References
Warburg, O. On The Origin of Cancer Cells. Science 123, 309–314 (1956).
Gatenby, R. A. & Gillies, R. J. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 4, 891–899 (2004).
Fantin, V. R., St-Pierre, J. & Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology and tumor maintenance. Cancer Cell. 9, 425–434 (2006).
Jemal, A. et al. Global cancer Statistics. CA Cancer J Clin. 61, 69–90 (2011).
Whibley, C. E. et al. Reactive oxygen species mediated apoptosis of esophageal cancer cells induced by marine triprenyl toluquinones and toluhydroquinones. Mol Cancer Ther. 6, 2535–43 (2007).
Pisha, E. et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nature Medicine. 1, 1046–1051 (1995).
Wick, W., Grimmel, C., Wagenknecht, B., Dichgans, J. & Weller, M. Betulinic acid-induced apoptosis in glioma cells: A sequential requirement for new protein synthesis, formation of reactive oxygen species and caspase processing. J Pharmacol Exp Ther. 289, 1306–1312 (1999).
Zuco, V. et al. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 175, 17–25 (2002).
Fulda, S. & Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Disc Today. 14, 885–890 (2009).
Fulda, S. et al. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J Biol Chem. 273, 33942–33948 (1998).
Rajendran, P., Jaggi, M., Singh, M. K., Mukherjee, R. & Burman, A. C. Pharmacological evaluation of C-3 modified Betulinic acid derivatives with potent anticancer activity. Invest New Drugs. 26, 25–34 (2008).
DeVita, V. T., Jr, Young, R. C. & Canellos, G. P. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer. Cancer. 35, 98–110 (1975).
Frei, E., 3rd Combination cancer therapy: Presidential address. Cancer Res. 32, 2593–2607 (1972).
Stacpoole, P. W. et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 117, 1519–1531 (2006).
Stacpoole, P. W., Lorenz, A. C., Thomas, R. G. & Harman, E. M. Dichloroactate in the Treatment of Lactic Acidosis. Ann Inter Med. 108, 58–63 (1998).
Sutendra, G. & Michelakis, E. D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 3, 57–63 (2013).
Bonnet, S. et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell. 11, 37–51 (2007).
Sun, R. C. et al. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 120, 253–260 (2010).
Michelakis, E. D., Webster, L. & Mackey, J. R. Dichloacetae (DCA) as a potential metabolite- targeting therapy for cancer. BJC. 99, 989–994 (2008).
Stacpoole, P. W. The pharmacology of dichloroacetate. Metabolism. 38, 1124–1144 (1989).
Wong, J. Y., Huggins, G. S., Debidda, M., Munshi, N. C. & De Vivo, I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 109, 394–402 (2008).
Dhar, S. & Lippard, S. J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc Natl Acad Sci USA. 52, 22199–22204 (2009).
Cao, W., Yacoub, S., Shiverick, K. T., Namiki, K., Sakai, Y. & Porvasnik, S. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 68, 1223–1231 (2008).
Pearson, H. Cancer patients opt for unapproved drug. Nature 446, 474–475 (2007).
Smiley, S. T. et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 88, 3671–3675 (1991).
Martinou, J. C., Desagher, S. & Antonsson, B. Cytochrome c release from mitochondria: all or nothing. Nat Cell Biol. 2, E41–3 (2000).
Bernardi, P. et al. Modulation of the mitochondrial permeability transition pore. Effect of and divalent cations. J Biol Chem. 267, 2934–2939 (1992).
Halestrap, A. P., McStay, G. P. & Clarke, S. J. The permeability transition pore complex: another view. Biochimie. 84, 153–166 (2002).
Katoh, H., Nishigaki, N. & Hayashi, H. Diazoxide Opens the Mitochondrial Permeability Transition Pore and Alters Ca2+ Transients in Rat Ventricular Myocytes. Circulation. 105, 2666–2671 (2002).
Precht, T. A. et al. The permeability transition pore triggers Bax translocation to mitochondria during neuronal apoptosis. Cell Death Differ. 12, 255–265 (2005).
Jurgensmeier, J. M. et al. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 95, 4997–5002 (1998).
Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev 15, 2922–2933 (2001).
Ow, Y. P., Green, D. R., Hao, Z. & Mak, T. W. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 9, 532–542 (2008).
Riedl, S. J. & Salvesen, G. S. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 8, 405–413 (2007).
Szatrowski, T. P. & Nathan, C. F. Production of Large Amounts of Hydrogen Peroxide by Human Tumor Cells. Cancer Res. 51, 794–798 (1991).
Toyokuni, S., Okamoto, K., Yodoi, J. & Hiai, H. Persistent oxidative stress in cancer. FEBS Letters. 358, 1–3 (1995).
Gibellini, L. et al. Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin. Cancers 2, 1288–1311 (2010).
Tan, Y. M., Yu, R. & Pezzuto, J. M. Betulinic Acid-induced Programmed Cell Death in Human Melanoma Cells Involves Mitogen-activated Protein Kinase Activation. Clin Cancer Res. 9, 2866–2875 (2003).
Conde de la Rosa, L. et al. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: Involvement of JNK and ERK MAP kinases. J Hepatol. 44, 918–929 (2006).
Maillet, A., Yadav, S., Loo, Y. L. & Sachaphibulkij, K. A novel Osmium-based compound targets the mitochondria and triggers ROS-dependent apoptosis in colon carcinoma. Cell Death Dis. 4, e653; 10.1038/cddis.2013.185 (2013).
Vantyghem, S. A., Postenka, C. O. & Chambers, A. F. Estrous cycle influences organ-specific metastasis of B16F10 melanoma cells. Cancer Res. 63, 4763–4765 (2003).
Decker, M. Hybrid Molecules Incorporating Natural Products: Applications in Cancer Therapy, Neurodegenerative Disorders and Beyond. Curr Med Chem. 18, 1464–1475 (2011).
Trachootham, D., Lu, W., Ogasawara, M. A., Nilsa, R. D. & Huang, P. Redox regulation of cell survival. Antioxid Redox Signal. 10, 1343–1374 (2008).
Green, D. R. & Reed, J. C. Mitochondria and Apoptosis. Science 281, 1309–1312 (1998).
Acknowledgements
We thank the director for his continuous support in this work. T. Murganandan for AFM work and Diptadeep Sarkar for CLSM images. S.S. thanks CSIR (Ministry of Science and Technology, Govt. of India) and M.G. thanks UGC (Govt. of India) for the fellowships. This work was supported by CSIR Network Project No. 31-2 (112)/ NWP009/ 2007-2012.
Author information
Authors and Affiliations
Contributions
Conceived by S.K.D, designed the experiments: S.S., M.G. and S.K.D.; Analyzed the data: S.S., M.G. and S.K.D.; wrote the paper: S.S., M.G. and S.K.D.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Electronic Supporting Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Saha, S., Ghosh, M. & Dutta, S. A potent tumoricidal co-drug ‘Bet-CA’ - an ester derivative of betulinic acid and dichloroacetate selectively and synergistically kills cancer cells. Sci Rep 5, 7762 (2015). https://doi.org/10.1038/srep07762
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07762
This article is cited by
-
Tumor metabolism rewiring in epithelial ovarian cancer
Journal of Ovarian Research (2023)
-
Role of metabolic modulator Bet-CA in altering mitochondrial hyperpolarization to suppress cancer associated angiogenesis and metastasis
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
