
Abstract
Single nucleotide polymorphisms (SNPs) are a prime source of genetic diversity. Discriminating between different SNPs provides an enormous leap towards the better understanding of the uniqueness of biological systems. Here we report on a new approach for SNP discrimination using toehold-mediated DNA strand displacement. The distinctiveness of the approach is based on the combination of both 3- and 4-way branch migration mechanisms, which allows for reliable discrimination of SNPs within double-stranded DNA generated from real-life human mitochondrial DNA samples. Aside from the potential diagnostic value, the current study represents an additional way to control the strand displacement reaction rate without altering other reaction parameters and provides new insights into the influence of single nucleotide substitutions on 3- and 4-way branch migration efficiency and kinetics.
Similar content being viewed by others


Introduction
Specificity of nucleic acid hybridization underpins the precise transfer of genetic information in living organisms through the generations. At the same time, diversity and evolution of life is closely related to DNA sequence alterations. In medical diagnostics and human identification, precise nucleic acid hybridization and sequence discrimination between single nucleotides (or a single nucleotide polymorphism (SNP)) is of great importance and significance. Much effort has been made in order to increase the discrimination power of currently available nucleic acid tests (for review see Ref. 1). Among these attempts are the developments of molecular beacons2,3,4,5,6,7,8, conformationally constrained binary probes9,10,11, probes modified with artificial nucleotides such as peptide nucleic acids (PNAs)8,12,13,14 and locked nucleic acids (LNAs)14,15,16,17 etc. Thermodynamic approaches such as an elevated hybridization temperature (close to the duplex melting point)18,19 or high concentrations of chaotropic agents in hybridization buffers19,20,21 are often used to weaken non-complementary strand interactions in order to increase specificity.
Modern achievements in dynamic DNA nanotechnology have also significantly contributed to the development in DNA genotyping approaches. These advances are predominantly based on toehold-mediated strand displacement reactions22,23,24 enabling unidirectional DNA re-hybridization between a double stranded DNA duplex (dsDNA) and a displacing single stranded oligonucleotide (displacing sequence (DS)) via a 3-way branch migration mechanism (3-WBM). The vast majority of genotyping research published in recent years has described the application of toehold-mediated strand exchange for SNP discrimination25,26,27,28,29,30,31,32,33,34,35,36,37,38. The publications in this field showed successful discrimination of a single nucleotide substitution where the nucleotide in question was located within a toehold sequence (“external” SNP), thereby preventing docking of the DS to the overhanging part of the dsDNA25,26,27,28,29,30,31. Zhang et al. have investigated a large variety of ‘internal’ SNPs, including both deletions and insertions within short DNA sequences and showed that during displacement the thermodynamic gain of many correctly paired bases can often override the thermodynamic penalty of a few mismatches; thus leading to complete “false-positive” displacement26,32. In order to minimize non-specific displacement, Zhang et al. proposed truncating the displacing sequence by several nucleotides from the opposite end of the toehold termini32, which allowed for full thermodynamic control of the displacement outcome. A similar approach with double-stranded toehold exchange and transition through a Holliday junction structure (4-way branch migration, 4-WBM) was shown to be successful in the detection of point mutations in the E. Coli rpoB gene33.
SNP discrimination using a 3-WBM mechanism can be significantly improved by introducing steric hindrance during the displacement process. For example, reliable discrimination of ‘internal’ SNPs through a 3-WBM strand displacement has been achieved using a unique DNA origami scaffold by Subramanian et al.34 They observed non-specific displacement (up to 40%) for the interactions resulting in a dT-dG mismatched base pair. Other reactions leading to the formation of a mismatched nucleotide pair in the displaced product showed non-specific displacement at significantly lower level (<20%). The likely reason for this lack of displacement specificity in these experiments is the anchoring of the dsDNA duplexes to a semi-rigid DNA origami scaffold, which restricted flexible rotation (i.e., provided conformationally constrained rotation) of the interacting DNA molecules. Another example of conformationally constrained toehold-mediated SNP sensing was demonstrated by Xiao et al.35 The authors used a combination of molecular beacons (MBs) with binary probes, which in turn resulted in the creation of a single sterically hindered triple-stem DNA structure capable of SNP specific discrimination.
Previous work by our group has shown a simple method for producing toehold-PCR products (TPPs) amplified from real-life DNA samples36. The dsPCR products have an overhanging toehold sequence and therefore are perfectly suitable for 3-WBM toehold-mediated strand displacement reactions. Successful discrimination between two closely related 106 and 112 bp long toehold-PCR products, differing by a 6-nt insertion, as well as 7 single nucleotide substitutions, was shown. However, discrimination between 80 bp long samples with only one nucleotide difference was unpredictable and kinetically compromised36.
Here, we present an approach for the reliable discrimination of SNPs using real-time PCR instrumentation within 80 bp real-life dsDNA sequences, in this case TPPs, using a combination of both 3-WBM and 4-WBM mechanisms. This approach (3/4/3-WBM) uses the 4-WBM stage as a topological filter for enhanced mismatch discrimination. The present research also reveals new insights into how SNPs influence 3- and 4-way branch migration efficiency and kinetics. Moreover, performing melting curve analysis of the displacement reaction products allows for visual monitoring of the foregoing displacement.
Results
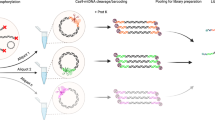
Figure 1 shows the idealized molecular mechanism of the proposed method. The first two stages, namely the docking of the DS to a TPP followed by 3-WBM exchange, are the standard steps for toehold-mediated strand displacement. Then the mechanism enters its 4-WBM stage. This is simply achieved by initial hybridization of the DS with an auxiliary short oligonucleotide (‘protector’). The protector sequence is fully complementary to the region of the DS carrying an internal discriminatory nucleotide (Fig. 1  ), i.e., a nucleotide responsible for SNP discrimination (which in turn is complementary to the nucleotide in question (Fig. 1
), i.e., a nucleotide responsible for SNP discrimination (which in turn is complementary to the nucleotide in question (Fig. 1  ) in the TPP). Finally, the displacement finishes with another 3-WBM step, resulting in the formation of two double-stranded displacement products (the main displacement product representing a fully complementary DNA duplex and the displacement ‘side’ product).
) in the TPP). Finally, the displacement finishes with another 3-WBM step, resulting in the formation of two double-stranded displacement products (the main displacement product representing a fully complementary DNA duplex and the displacement ‘side’ product).

Simplified representation of the approach consisting of 3-WBM and 4-WBM mechanisms for SNP discrimination.
F and Q are fluorophore and fluorescent quencher, respectively, used for real-time strand displacement kinetic monitoring.
The choice of protecting sequence length was determined by two main constraints: 1) the minimal length of the sequences was restricted by the integrity/stability of the double-stranded structure formed between the displacing sequence and the protector at 30°C (reaction temperature); and 2) the longer the protector sequence the slower the reaction rate of the 4-WBM step since the elementary step for 4-WBM process is at least one order of magnitude slower than that for the 3-WBM process41,42. The protecting sequences of 19, 15 and 11 nucleotide length were therefore evaluated. In all cases the protecting sequences were designed in such a way that the discriminatory nucleotide (Fig. 1  ) in each DS was localized in the middle of the protector sequence.
) in each DS was localized in the middle of the protector sequence.
Kinetic discrimination
Figure 2 shows the results of the toehold-PCR products (TPP(C), TPP(T), TPP(A) and TPP(G)) discrimination using four displacing sequences (DS(A), DS(T), DS(G) and DS(C)), where the letter in brackets stands for the nucleotide in question for TPPs and the discriminatory nucleotide for DSs, respectively) with and without protectors.

Toehold-mediated strand displacement kinetic curves showing the influence of the protecting sequence length on (A) TPP(C), (B) TPP(T), (C) TPP(A) and (D) TTP(G) discrimination with the displacing sequences DS(A), DS(T) DS(G) and DS(C).
Among the four TPPs, the easiest to discriminate using non-protected DS(A)np, DS(T)np, DS(G)np and DS(C)np (subscripted “np” stands for no protector) was TPP(C) (Fig. 2A(I–IV) blue traces). The complementary reaction of TTP(C) with DS(C)np reached the 70% plateau of displacement in ≈25 min (Fig. 2AIV, blue trace), having a rate constant of 1.47 × 103 M−1 s−1 (Fig. 3II, Table S2). In all cases for the non-complementary reactions of TPP(C) with DS(A)np, DS(T)np and DS(G)np (Fig. 2A(I–III), blue traces) there was a similar degree of conversion of 25–40% over a reaction time of 1 h. These three non-complementary processes showed an average constant rate of 1.62 × 102 M−1 s−1 (varying in a range from 7.88 × 101 to 2.23 × 102 M−1 s−1, Fig. 3II, Table S2). Thus, Rks of 6.6, 7.9 and 18.6 were calculated for the reactions of the TPP(C) with non-protected DS(A)np, DS(T)np and DS(G)np, respectively (Fig. 2A(I–III) blue traces, Fig. 3II, Table S3). It is worth noting that the mismatch pairs being made during these non-complementary reactions are dC-dT, dC-dA and dC-dC, which in turn are the three weakest mismatches according to SantaLucia et al.43 Among these values the largest Rk of 18.6 (TPP(C)-DS(G)np, Fig 3II, Table S3) is related to the formation of the weakest (dC-dC) mismatch, supporting the finding that the dC nucleotide is the most discriminative43.

Influence of protector length on rate constants of toehold-mediated strand displacement reactions.
Brackets on the plots show the discrimination ratios (Rk) between a complementary reaction rate constant (kcomp) and a non-complementary reaction constant (knon-comp).
As can be seen, the discrimination of TPP(T), TPP(A) and TPP(G) using non-protected DSs (Figs. 2B(I–IV), 2C(I–IV) and 2D(I–IV) blue traces) can barely be performed. The reactions of TPP(T), TPP(A) and TPP(G) with their complementary non-protected DSs reached the 80–90% level of conversion in just 25–30 minutes, with rate constants of 2.8 × 103 M−1 s−1, 2.13 × 103 M−1 s−1 and 2.67 × 103 M−1 s−1, respectively (Fig. 3I, 3III, 3IV, Table S2). On the other hand, for all cases of non-complementary reactions a high degree of conversion up to 80% was also achieved over 1 h of reaction. Among the three non-complementary processes the reaction of TPP(T) with DS(A)np (Fig. 2BI, blue trace) was the fastest, showing a rate constant of 1.14 × 103 M−1 s−1 (Fig. 3I, Table S2). Rk for TPP(T)-DS(A)np was determined to be 2.5 (Fig. 3I, Table S3). The rate constant for the reaction of the TPP(A) with the DS(C)np (Fig. 2CIV blue trace) was fitted to be 6.74 × 102 M−1 s−1, which was only 3.2 (Rk) times slower than the TPP(A)-DS(A)np complementary reaction (Fig. 2CI blue trace, Fig. 3III). An Rk of 2.4 (Fig. 3IV) was calculated for the fastest non-complementary reaction of TPP(G) and DS(A)np (1.12 × 103 M−1 s−1, Fig. 2DI blue trace, Table S2).
In order to increase the kinetic discrimination ratios for non-complementary reactions, short auxiliary oligonucleotide ‘protector’ sequences complementary to the displacing sequence were employed. This modification introduces an additional internal slow 4-WBM step. Given that both the toehold length and sequence are identical through all the strand displacements carried out, it was believed that the introduction of the protector sequence would significantly reduce the overall rate of the strand displacement. Moreover, such a modification of the traditional 3-WBM mechanism would decrease the rate of the non-complementary reaction much more than that of the complementary. This would be achieved through the formation of two mismatched base pairs in the main and ‘side’ displacement products, whereupon there would be an increase in the energetic barrier to overcome during the 4-WBM step.
As expected, a monotonic decrease of the rate constant of the reaction between TPPs and complementary ‘protected’ displacing sequences was observed along with an increase in the protector length (Fig. 3). The decrease of the non-complementary reaction rate constants was even more significant. This therefore allowed for better kinetic discrimination between the complementary and non-complementary reactions. For example, TPP(C) was discriminated at least twice as well as better using DS(A)15 (where the subscripted number shows the length of the protector sequence) than with non-protected DS(A)np, with Rks of 12.8 vs 6.6, respectively (Fig. 2AI red and blue traces, respectively, Fig. 3II, Table S3). Better performance in kinetic discrimination was shown for the TPP(A), where the Rk of 3.2 for the reaction with DS(C)np was improved up to 14.5, 33.0 and 53.9 using DS(C)11, DS(C)15 and DS(C)19, respectively (Fig. 2CIV, green, red and black traces, Fig. 3III, Table S3).
The introduction of the protector sequences greatly affected the rate constant ratios for the reaction of TPP(T) with either DS(G) or DS(C) (Fig. 2BIII and Fig. 2BIV) and increased Rk from 6.8 (DS(G)np) and 5.0 (DS(C)np) up to 23.1 (DS(G)15) and 22.1 (DS(C)11) (Fig. 3I, Table S3). However, the discrimination ratio for the reaction between TPP(T) with the protected displacing sequences DS(A)11, DS(A)15 and DS(A)19 (Fig. 2BI) was not significantly improved compared with the non-protected DS(A)np (Fig. 2BI blue trace). Thus, the largest Rk of 5.0 was achieved for DS(A)19, which was twice as high as for DS(A)np (Fig. 2BI black and blue traces, respectively, Fig. 3I, Table S3). A similar situation was observed for the discrimination of TPP(G) with the protected displacing sequences DS(A)11, DS(A)15 and DS(A)19 (Fig. 2DI). Two protected displacing sequences were shown to be able to efficiently discriminate TTP(G) with a Rk of at least 26.6 and 32.7 for DS(T)19 and DS(C)11 (Fig. 2DII black trace and Fig. 2DIV red trace, respectively, Table S3). However, discrimination of TPP(G) using DS(A)19 (Fig. 2DI black trace) gave the largest Rk of only 3.6 (Fig. 3IV, Table S3), which was 1.5 times higher than that for the non-protected DS(A) sequence (Fig. 2DI, blue trace). It is notable that in both cases (TPP(T) and TPP(G)) the lowest discrimination ratios were observed for the reactions with DS(A) regardless of protector length (Fig. 2DI, 2DI, 3I, 3IV), which form relatively strong dT-dT and dG-dT mismatches43 in the respective main displacement products.
Thermodynamics of DNA base pairing and kinetic discrimination
The correlation between the level of kinetic discrimination and the thermodynamics of DNA base pairing is illustrated in Fig. 4, in which the logarithm of the kinetic discrimination ratio for all of the reactions is plotted as a function of the standard free energy change for the strand displacement process relative to that of the complementary reaction at 30°C, ΔG°30, calculated from the SantaLucia nearest-neighbor model of DNA hybridization thermodynamics43. The figure shows that, in general, a larger thermodynamic penalty for base-pairing mismatches in the displacement process leads to a greater discrimination ratio between complementary and non-complementary reactions. For this reason, the reactions using protector sequences and 4-WBM are generally more discriminatory, as the thermodynamic penalty for forming two base-pair mismatches is typically greater than that for forming a single mismatch; this is as discussed above and as shown in the figure. The correlation between the kinetic discrimination ratio and DNA hybridization thermodynamics is not perfect, as a one-to-one correspondence between the energetic barriers for the 3- and 4-WBM steps and the free energy differences between the stable states that are connected by these branch migration steps does not necessarily exist. Also the stability of the nucleotide mismatch within 4-WBM migration point does likely depend on the structure of the mismatch and, even more crucially, on its interaction with flanking nucleotides44. In addition, although the discrimination ratios in figure 4 for the 19-nt protector show a generally increasing trend with ΔG°30, the slope of the trend is significantly greater than that for the other protector lengths. The changes in the ‘dynamic’ interactions between the four DNA strands during the 4-way branch migration steps are not well understood and complex, involving the concerted interaction of four different strands. It is therefore difficult to attribute the changes in the discrimination ratios to specific interactions between the bases in the four strands. To capture the variation in the discrimination ratio with protector length would require a more sophisticated kinetic model that accounts for branch migration steps other than those involving the mismatch.

Correlation between the level of kinetic discrimination and the thermodynamics of DNA base pairing.
Discrimination using melting analysis
In addition to real-time monitoring of the kinetics of the displacement reaction, a second approach based on fluorescent DNA melting analysis of the strand displacement products was used. We have previously reported that if any displacement reaction between the displacing sequence and the TPP occurs, a ‘positive’ melting peak (pointing up) appears when the change of the fluorescence signal intensity is plotted as a positive derivative with respect to temperature (dF/dT) against the temperature (T)45. If displacement does not occur the melting peak points down (‘negative’ melting peak or trough). We therefore used this approach to assess the reaction products obtained in the current work. Figure 5 shows the results of the melting analysis of the displacement products. Melting analysis of the TPPs and any non-protected DSs, including non-complementary sequences, resulted in only ‘positive’ melting peaks (Fig. 5, blue traces). Thus, melting curve-based discrimination using non-protected DSs is unfeasible. Interestingly, only ‘positive’ melting peaks were observed for the melting analysis of the displacement products obtained in the reactions between TPPs and their complementary protected DSs regardless of the protector length (Figs. 5AIV, 5BII, 5CI, 5DIII black, red and green traces). In contrast, the melting curves associated with non-complementary protected DSs irrespective of protector length, had a sine-like curve shape, with a peak and a trough clearly visible (e.g., Figs. 5AI, 5BIII, 5CIII, 5DIV black, red and green traces) except for the reaction of TPP(G) with DS(A)11 and DS(A)15 (Fig. 5DI green and red traces), which showed only “positive” melting peaks.

Melting temperature analysis of toehold-mediated strand displacement products produced by the reaction of (A) TPP(T), (B) TPP(C), (C) TPP (A) and (D) TPP(G) with both non-protected (blue traces) and protected (black, red and green traces) DS(A), DS(T), DS(G) and DS(C).
The trough is likely explained by completion of the displacement reaction at the elevated temperatures during the melting analysis. The use of a protector sequence slows down the whole strand displacement process. This is particularly the case for the reaction of a TPP with non-complementary ‘protected’ DSs, in which two mismatched nucleotide pairs form (one in the main displacement product and the other in the ‘side’ displacement product). It has been shown previously that a temperature increase facilitates the strand displacement reaction36. Thus we assume that the increase in the temperature during the melting analysis makes the slow “protected” displacement occur regardless of the mismatches formed. This may be due to dehybridization of the protector sequence, which leads to completion of the displacement reaction. Subsequent melting of the formed displacement product results in the positive peak on the melting analysis plot. The reaction between a TPP with its complementary “protected” DS always gives a single positive peak due to the displacement reaching completion long before melting starts. This data is in full accordance with the kinetic data obtained for the reactions, again proving that formation of relatively strong mismatched nucleotide pairs for TPP(G) with DS(A)11, DS(A)15 and DS(A)19 allows nonspecific displacement to occur. In the case of the 19 nt protector sequence (Fig. 5DI black trace) the preliminary displacement reaction was significantly slowed down, which in turn allowed the displacement to occur only at elevated temperature.
Calculation of the normalized area under the curve (AUC) allows easy numerical assessment of the shape of the melting curve. Thus, the AUC for strictly positive melting curves always tends to give a positive value approaching 1 (for normalized data). On the other hand, the AUC for melting curves with a peak and a trough approaches zero. Figure 6 shows an example of the discrimination of the TPPs using the melting curve shape analysis approach. As stated previously the discrimination of TPP(T), TPP(A) and TPP(G) with non-protected displacing sequences was not possible since all the areas under the melting curves nearly equal 1 (Figs. 6I, 6III and 6IV, respectively). Introducing protector sequences allowed for a significant decrease of the AUC value with increasing protector length, down to 0.1 for the reaction of TPP(T) with DS(A) (Fig. 6I, black bars) and even slightly negative values for non-complementary displacement reactions with DS(C) and DS(G) (Fig. 6I, blue and green bars, respectively). The normalized AUC values for the complementary reactions of TPP(T) with DS(T)np, DS(T)11, DS(T)15 and DS(T)19 varied from 1 to 0.78 (Fig. 6A, red bars). Similar trends were observed for the rest of the toehold-PCR products (Fig. 6II, 6III and 6IV), so setting up a cut off value of 0.3 allows for reliable discrimination using all displacing sequences with 19 nt long protectors. Non-parametric Spearman correlation analysis showed a statistically significant relatedness between the reaction rate constants and the normalized AUC (ρ = 0.71, p < 1 × 10−3).

Normalized area under the curve for the curves obtained during the melting temperature analysis of the toehold-mediated strand displacement reaction of (I) TPP(A), (II) TPP(T), (III) TPP(G), (IV) TTP(C) with both protected and non-protected DSs.
Discussion
We have demonstrated a new method for SNP identification and discrimination based on a toehold- mediated strand displacement reaction proceeding through both 3- and 4-way branch migration mechanisms. Additional short oligonucleotide sequences of various lengths were hybridized to single-stranded displacing sequences, which introduced a 4-way branch migration step into the overall reaction mechanism.
Our results suggest for the first time that, for most base pairs, there is a reasonable correlation between sequence-dependent 3- and 4-WBM migration barriers and DNA hybridization free energies. If the rate-determining step for the displacement process is branch migration through the base mismatch (as values of the kinetic discrimination ratio generally a factor of 5 or more would suggest) and if there is much greater variation between the rates of complementary versus non-complementary reactions than between complementary reactions for a given protector length (as the experimental data indicates), the discrimination ratio would be expected to scale approximately with exp(ΔG*/kBT), where ΔG* is the activation free energy difference between the non-complementary and complementary reactions for the SNP branch migration step. A linear correlation between the activation free energy difference, ΔG* and the difference between the free energy change of strand displacement, ΔG°30, would give a linear relationship between the logarithm of the kinetic discrimination ratio and ΔG°30, as Fig. 4 clearly shows (except for a few outlying points). The slope of the linear fit to the data in Fig. 4 is 0.25, indicating that the activation free energy differences due to base mismatches for 3- and 4-WBM are approximately 1/4 the thermodynamic free energy differences. This result is expected, as interactions between bases are partially formed or broken in the transition state for the branch migration step and therefore likely to be less sensitive to base mismatches than the free energies of the stable states. The outliers below the linear regression line in Fig. 4 correspond to the TPP(T) and TPP(G) reactions with protected DS(A), while the outliers above the line correspond to TPP(T) and TPP(C) reactions with protected DS(C). The lack of correlation suggests that simple thermodynamic considerations are not completely sufficient for describing the energetics of these processes and that further investigation will be necessary to elucidate the mechanisms of 3- and 4-WBM transition through sequence heterology.
In summary, the method presented here allows for reliable kinetic SNP discrimination. Among the four toehold-PCR products of 80 bp, which carried four different SNPs at the same position, the easiest to discriminate was the one with the SNP dC, which forms the weakest and the most destabilizing 4-WBM junction structure with mismatched pairs43,44. Conversely, the poorest discriminative displacing sequence was the one carrying a dT discriminatory nucleotide. This is likely due to the smaller size of the dT nucleotide, which results in less steric hindrance during the branch migration transition through the SNP site and moderate stability of the newly formed mismatches. Also, the use of the protector sequence represents an additional way to controllably decrease the strand displacement rate without altering other reaction parameters such as the strength of toehold binding26,46 or the reaction temperature36,42,46. It is worth to note that the control of the displacement rate with no changing the reaction conditions was also previously shown using the remote toehold approach of Genot et al.39
In addition, performing melting analysis of the displacement products allows simple visual discrimination of all tested SNPs based only on the assessment of the shape and area of the obtained melting curves.
Methods
Nucleic acid isolation and PCR amplification
Human genomic and mitochondrial DNA were co-extracted from the authors' own blood samples using a QIAamp DNA blood mini kit (Qiagen, Germany), according to the recommendations of the manufacturer. PCR primers, including dU-modified and fluorescently labeled primers (FAM) (see, Table s1), were purchased from IDT DNA Technology, USA and used at a concentration of 0.2 μM.
In order to evaluate the proposed method for SNP discrimination, in addition to the SNPs found in the authors' own DNA samples (C16223 (SNP-C) and T16223 (SNP-T) within the Hyper Variable Region 1 (HVR-1) of the mtDNA), two recombinant plasmid DNA carrying SNP-A and SNP-G were engineered (for more details see SI).
PCRs were performed using a HotStar Taq DNA polymerase (Qiagen, Germany) within 1× HotStar Taq PCR buffer with a final MgCl2 concentration of 2.5 mM. The concentration of each of the standard deoxynucleotide triphosphates (dATP, dTTP, dGTP, dCTP) was 0.2 mM. A PCR amplification regime of 95°C for 15 min, 30 cycles of 94°C for 15 s, 61°C for 15 s, 72°C for 25 s and a final elongation of 72°C for 1 min was used. A template of 5 ng of the extracted human genomic DNA or 1 ng of the plasmid DNA carrying PCR product with either SNP-A or SNP-G was used. For the detailed protocol of the plasmid DNA preparation see SI.
Preparation of the toehold-PCR products (TPPs)
Directly after the PCR, Uracil-DNA glycosylase (NEB, USA) (2.5 U) was added to the PCR solution and the entire mixture was then gently mixed via pipetting. After incubation at room temperature for 5 min the mixture was heated to 95°C for 5 min and then cooled back to room temperature; this resulted in the formation of a 9 nucleotide long toehold sequence. The PCR-UDG mixtures were purified using an Amicon Ultra-0.5 30K (Millipore, USA) according to the recommendations of the manufacturer. The nucleic acids were then eluted with a displacement 1× TEM buffer (pH 8), consisting of Tris·HCl (10 mM), EDTA (1 mM) and MgCl2 (12.5 mM). Quantification of the eluted PCR products was carried out using a NanoDrop 1000 spectrophotometer (Thermo Scientific, USA).
Strand displacement reaction
Displacing sequences DS(A), DS(T), DS(C) and DS(G) (Table S1), 80 nt in length and labelled at the 3′ termini with the fluorescent quencher carboxytetramethylrhodamine (TAMRA), were purchased from Eurogentec, Belgium.
Displacing sequences were mixed with corresponding protector sequences in stoichiometric ratios and then annealed from 95 to 20°C at a constant rate of 0.5°C/min.
All displacement reactions were carried out with 5 times excess of the DS over the TPP at 30°C in a reaction volume (15 μL) using a RotorGene Q 6-plex real-time PCR thermocycler (Qiagen, Germany). Prior to the displacement reaction the toehold-PCR product (2 pmol, dissolved in 1× TEM buffer) was placed in a thin-wall PCR tube (0.2 mL) and briefly centrifuged. Then, the displacing sequence (10 pMole, dissolved in 2 μL of 1× TEM buffer) was carefully applied onto the lid of the same PCR tube. The tube lid was then carefully closed and the tube placed into the thermocycler avoiding mixing of the TPP and the DS before the run protocol started.
Acquisition of fluorescent signals was performed within the SYBR Green/FAM channel at a gain of 7 within a time interval of 20 sec between fluorescent reads. Normalization of the raw fluorescence was made by dividing the signal values by the initial signal value, as described previously39.
Melting curve analysis was performed according to the RotorGene Q 6-plex built-in protocol, which included a temperature ramp from 30 to 95°C with an increase of 1°C each step of 5 s waiting time.
Curve-fitting of the kinetic data was carried out using the non-linear curve fitting function in OriginPro 9 software (Origin Corporation, USA). Assuming that an excess of the displacing sequence shifts the displacement kinetics towards the first order behavior the obtained data were fit to the pseudo-first-order kinetic equation as described by Baker et al.40,

where F is the fluorescence intensity at time t, F0 is the initial fluorescence intensity, F1 is the intensity after reaction completion, k is the rate constant, [DS]o is the initial concentration of the displacing sequence and the t is the time in seconds.
The ratio between a complementary reaction rate constant (kcomp) and a non-complementary reaction constant (knon-comp) with no protector sequence, or with a protector of the same length as that of the complementary reaction, was used as a kinetic discrimination ratio,

References
Knez, K., Spasic, D., Janssen, K. P. F. & Lammertyn, J. Emerging technologies for hybridization based single nucleotide polymorphism detection. Analyst 139, 353–370 (2014).
Yang, C. J. Molecular Beacons. (Springer Berlin Heidelberg: Berlin, Heidelberg, 2013). 10.1007/978-3-642-39109-5.
Li, Y.-Q. et al. Simultaneous detection of dual single-base mutations by capillary electrophoresis using quantum dot-molecular beacon probe. Biosensors & bioelectronics 26, 2317–2322 (2011).
Ryazantsev, D. Y. et al. Design of molecular beacons: 3′ couple quenchers improve fluorogenic properties of a probe in real-time PCR assay. Analyst 139, 2867–2872 (2014).
Jacroux, T., Rieck, D. C., Cui, R., Ouyang, Y. & Dong, W.-J. Enzymatic amplification of DNA/RNA hybrid molecular beacon signaling in nucleic acid detection. Anal. Biochem. 432, 106–114 (2013).
Bengtson, H. N. & Kolpashchikov, D. M. A differential fluorescent receptor for nucleic acid analysis. Chembiochem 15, 228–231 (2014).
Frutos, A. G., Pal, S., Quesada, M. & Lahiri, J. Method for detection of single-base mismatches using bimolecular beacons. J. Am. Chem. Soc. 124, 2396–2397 (2002).
Kam, Y. et al. Detection of a long non-coding RNA (CCAT1) in living cells and human adenocarcinoma of colon tissues using FIT-PNA molecular beacons. Cancer Lett. 1–7 (2013). 10.1016/j.canlet.2013.02.014.
Nguyen, C., Grimes, J., Gerasimova, Y. V. & Kolpashchikov, D. M. Molecular-beacon-based tricomponent probe for SNP analysis in folded nucleic acids. Chem. 17, 13052–13058 (2011).
Cornett, E. M., O'Steen, M. R. & Kolpashchikov, D. M. Operating Cooperatively (OC) sensor for highly specific recognition of nucleic acids. PloS ONE 8, e55919 (2013).
Vieregg, J. R., Nelson, H. M., Stoltz, B. M. & Pierce, N. A. Selective nucleic acid capture with shielded covalent probes. J. Am. Chem. Soc. 135, 9691–9 (2013).
Kam, Y., Rubinstein, A., Nissan, A., Halle, D. & Yavin, E. Detection of endogenous K-ras mRNA in living cells at a single base resolution by a PNA molecular beacon. Mol. Pharmaceutics 9, 685–693 (2012).
Zanoli, L. M. et al. Peptide nucleic acid molecular beacons for the detection of PCR amplicons in droplet-based microfluidic devices. Anal. Bioanal. Chem. 405, 615–624 (2013).
Briones, C. & Moreno, M. Applications of peptide nucleic acids (PNAs) and locked nucleic acids (LNAs) in biosensor development. Anal Bioanal. Chem. 402, 3071–3089 (2012).
Astakhova, I. V., Ustinov, A. V., Korshun, V. A. & Wengel, J. LNA for optimization of fluorescent oligonucleotide probes: improved spectral properties and target binding. J. Bioconjugate Chem. 22, 533–539 (2011).
Karmakar, S. & Hrdlicka, P. J. DNA strands with alternating incorporations of LNA and 2′-O-(pyren-1-yl)methyluridine: SNP-discriminating RNA detection probes. Chem. Sci. 4, 3447–3454 (2013).
Kumar, T. S., Myznikova, A., Samokhina, E. & Astakhova, I. K. Rapid genotyping using pyrene-perylene locked nucleic acid complexes. Artificial DNA, PNA & XNA 4, 58–68 (2013).
Yang, W. & Lai, R. Y. Effect of diluent chain length on the performance of the electrochemical DNA sensor at elevated temperature. Analyst 136, 134–139 (2011).
Vasiliskov, V. A. et al. Separate production of single-stranded DNA is not necessary: circuit denaturation of double-stranded DNA followed by hybridization of single strands on oligonucleotide microchips. J. Biomol. Struct. Dyn. 27, 347–360 (2009).
Gryadunov, D. et al. Hepatitis C virus genotyping using an oligonucleotide microarray based on the NS5B sequence. J. Clin. Microbiol. 48, 3910–3917 (2010).
Zimenkov, D. V. et al. Detection of second-line drug resistance in Mycobacterium tuberculosis using oligonucleotide microarrays. BMC Infect. Dis. 13, 240–248 (2013).
Zhang, D. Y. & Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 3, 103–113 (2011).
Srinivas, N. et al. On the biophysics and kinetics of toehold-mediated DNA strand displacement. Nucleic Acids Res. 41, 10641–58 (2013).
Zhang, D. Y. Cooperative hybridization of oligonucleotides. J. Am. Chem. Soc. 133, 1077–1086 (2011).
Zhang, Z. et al. A DNA-Origami chip platform for label-free SNP genotyping using toehold-mediated strand displacement. Small 6, 1854–1858 (2010).
Zhang, D. Y. & Winfree, E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 38, 4182–97 (2010).
Wang, D. et al. Highly Selective Detection of Single-Nucleotide Polymorphisms Using a Quartz Crystal Microbalance Biosensor Based on the Toehold-Mediated Strand Displacement Reaction. Anal. Chem. 84, 708–714 (2012).
Picuri, J. M., Frezza, B. M. & Ghadiri, M. R. Universal translators for nucleic acid diagnosis. J. Am. Chem. Soc. 131, 9368–9377 (2009).
Byrom, M., Bhadra, S., Jiang, Y. S. & Ellington, A. D. Exquisite allele discrimination by toehold hairpin primers. Nucleic Acids Res. 1–13 (2014). 10.1093/nar/gku558.
Tang, W., Wang, D., Xu, Y., Li, N. & Liu, F. A self-assembled DNA nanostructure-amplified quartz crystal microbalance with dissipation biosensing platform for nucleic acids. Chem. Comm. 48, 6678–6680 (2012).
Gao, Z. F. et al. Detection of single-nucleotide polymorphisms using an ON-OFF switching of regenerated biosensor based on a locked nucleic acid-integrated and toehold-mediated strand displacement reaction. Anal. Chem. 86, 2543–2548 (2014).
Zhang, D. Y., Chen, S. X. & Yin, P. Optimizing the specificity of nucleic acid hybridization. Nat. Chem. 4, 208–214 (2012).
Chen, S. X., Zhang, D. Y. & Seelig, G. Conditionally fluorescent molecular probes for detecting single base changes in double-stranded DNA. Nat. Chem. 5, 782–789 (2013).
Subramanian, H., Chakraborty, B., Sha, R. & Seeman, N. C. The Label-Free Unambiguous Detection and Symbolic Display of Single Nucleotide Polymorphisms on DNA Origami. Nano Lett. 11, 910–913 (2011).
Xiao, Y. et al. Fluorescence detection of single-nucleotide polymorphisms with a single, self-complementary, triple-stem DNA probe. Angew. Chem., Int. Ed. Engl. 48, 4354–4358 (2009).
Khodakov, D. A., Khodakova, A. S., Linacre, A. & Ellis, A. V. Toehold-mediated nonenzymatic DNA strand displacement as a platform for DNA genotyping. J. Am. Chem. Soc. 135, 5612–5619 (2013).
Song, T. et al. An Efficient DNA-Fueled Molecular Machine for the Discrimination of Single-Base Changes. Adv. Mater. (2014). 10.1002/adma.201402314.
Deng, R. et al. Toehold-initiated rolling circle amplification for visualizing individual microRNAs in situ in single cells. Angew. Chem., Int. Ed. Engl. 53, 2389–2393 (2014).
Genot, A., Zhang, D., Bath, J. & Turberfield, A. Remote toehold: a mechanism for flexible control of DNA hybridization kinetics. J. Am. Chem. Soc. 133, 2177–2182 (2011).
Baker, B. A. & Milam, V. T. Hybridization kinetics between immobilized double-stranded DNA probes and targets containing embedded recognition segments. Nucleic Acids Res. 39, e99 (2011).
Biswas, I., Yamamoto, A. & Hsieh, P. Branch migration through DNA sequence heterology. J. Mol. Biol. 279, 795–806 (1998).
Panyutin, I. G. & Hsieh, P. The kinetics of spontaneous DNA branch migration. Proc. Natl. Acad. Sci. U. S. A. 91, 2021–2025 (1994).
SantaLucia, J. & Hicks, D. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 33, 415–440 (2004).
Duckett, D. R. & Lilley, D. M. Effects of base mismatches on the structure of the four-way DNA junction. J. Mol. Biol. 221, 147–161 (1991).
Khodakov, D. A., Khodakova, A. S., Linacre, A. & Ellis, A. V. Amelogenin locus typing using toehold-assisted fluorescent DNA melting analysis. Forensic Sci. Int.: Genet. 4, e119–e120 (2013).
Zhang, D. Y. & Winfree, E. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 131, 17303–17314 (2009).
Acknowledgements
The authors kindly acknowledge the Australian Research Council Future Fellowship Fund (Grant No. FT130100211) for funding.
Author information
Authors and Affiliations
Contributions
D.A.K. and A.S.K. conceived, designed and performed the experiments; D.A.K. and D.M.H. analyzed the data; D.A.K., A.S.K. and D.M.H. wrote the paper draft; A.L. and A.V.E. edited the paper draft; A.V.E. guided the research.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Khodakov, D., Khodakova, A., Huang, D. et al. Protected DNA strand displacement for enhanced single nucleotide discrimination in double-stranded DNA. Sci Rep 5, 8721 (2015). https://doi.org/10.1038/srep08721
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08721
This article is cited by
-
A nanoflow cytometric strategy for sensitive ctDNA detection via magnetic separation and DNA self-assembly
Analytical and Bioanalytical Chemistry (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
