
Abstract
Chickens are an invaluable model for studying human diseases, physiology and especially development, but have lagged in genetic applications. With the advent of Programmable Engineered Nucleases, genetic manipulation has become efficient, specific and rapid. Here, we show that the CRISPR/Cas9 system can precisely edit the chicken genome. We generated HIRA, TYRP1, DICER, MBD3, EZH2 and 6 other gene knockouts in two chicken cell lines using the CRISPR/Cas9 system, with no off-target effects detected. We also showed that very large deletions (>75 kb) could be achieved. We also achieved targeted modification by homology-directed repair (HDR), producing MEN2A and MEN2B mutations of the RET gene. We also targeted DGCR8 in neural cells of the chicken embryo by in vivo electroporation. After FACS isolation of transfected cells, we observed appropriate sequence changes in DGCR8. Wholemount and frozen section antibody labelling showed reduction of DGCR8 levels in transfected cells. In addition, there was reduced expression levels of DGCR8-associated genes DROSHA, YPEL1 and NGN2. We also observed morphological differences in neural tissue and cardiac-related tissues of transfected embryos. These findings demonstrate that precisely targeted genetic manipulation of the genome using the CRISPR/Cas9 system can be extended to the highly adaptable in vivo chicken embryo model.
Similar content being viewed by others


Introduction
Avian embryos, chiefly the chicken (Gallus gallus) and quail (Coturnix japonica), have been a mainstay of vertebrate embryologic research for over a century1. The avian embryo is easy to obtain cheaply in large numbers as a result of its commercial utility. Development of avian embryos is simple to synchronise with little individual variation and benefits from excellent tables of development. Because development is external, these embryos are accessible to experimental manipulation2. The similarity in the general ontogeny and gene expression patterns between avian embryos and those of other vertebrates including mouse embryos indicates that Aves can serve as an excellent model for the study of genetic, molecular and biochemical mechanisms in mammals including humans. Avian models have been especially useful in following developmental pathways, for example in tracking cell movements in morphogenesis, untangling inductive pathways and deciphering differentiation pathways. The results of these have been shown to be valid for other vertebrates3. Avian embryos have also been instructive in pathogenesis of embryonic diseases and also physiology, behaviour and toxicology4,5.
Avian embryology has lagged in genetic research partly because the commercial utility of poultry factored against collection of instructive mutants (as in the fly, fish and mouse) and partly because of the dearth of techniques to manipulate their genome. The introduction of in vivo transfection by electroporation and by viral vectors changed this dramatically and allowed a form of conditional mutagenesis that was cheap and rapid6. Great ingenuity has enabled up- and down-regulation of genes to be achieved in avian embryos7, but these techniques largely involved adding extra genetic information in a non-targeted way, in the form of plasmids or miRNAs, either episomally, or by means that randomly integrate into the host genome8.
Programmable engineered nucleases (PENs) are novel technologies developed to efficiently target and alter a specific allele in the genome. These PENs, the zinc finger nucleases (ZFNs), the transcription activator-like effector nucleases (TALENs) and the clustered regular interspaced palindromic repeats (CRISPR)/Cas9 system, have been used extensively in generating and correcting mutations in cells of plants9, humans10,11, rodents12,13, monkeys14,15, fish16,17,18, fly19,20 and worm21 in vitro and in vivo, to generate transgenic cells, animals and plants. Recently, Park et al. validated the efficiency of TALENs in generating knockout chicken primordial germ cells (PGCs)22 and showed that TALENs can be used to efficiently modify the genomes of chickens. Here we utilise the recently described CRISPR/Cas9 system, which is a very simple but powerful tool in the editing in vivo of the genomes of mice23,24, in knocking-out and knocking-in of sequences in chicken cells in vitro and in vivo.
Results
A recent report showed that the gene Pax7 could be modified by CRISPR/Cas9 in the chicken embryo in vivo25. To explore the general applicability of this technique, we investigated a range of chicken genes on both macrochromosomes and on the unusual avian microchromosomes. The genes chosen were DROSHA, DICER, MBD3, KIAA1279, CDKN1B, EZH2, HIRA, TYRP1, STMN2, RET and DGCR8 (Di George Critical Region8) (Supplementary Table 1). These genes have roles in embryonic development and the pathogenesis of embryonic diseases.
The CRISPR/Cas9 system mediates NHEJ and HDR gene disruptions in chicken cell lines
We validated the activity of the CRISPR/Cas9 system by designing sgRNAs (Supplementary Table 2) targeting the translational initiation region (start codon) of DROSHA, DICER, MBD3, KIAA1279, CDKN1B and EZH2. We generated NHEJ-induced mutation by co-transfecting the sgRNA CRISPR/Cas9 construct with or without a puromycin resistance-expressing construct into the chicken fibroblastic DF-1 cell line using Lipofectamine 3000 (Fig. 1a). Genomic DNA was isolated after 72–96 hrs and the frequency of induced mutation in the targeted locus was analysed using the T7E1 assay and DNA sequencing (Supplementary Table 3). In puromycin-resistant cells, cleavage bands ranging between 20–68% were visible in all target genes as calculated by Image J software. The mutation efficiency induced was, for example, 50–51% in KIAA1279, 26–49% in CDKN1B, 68% in MBD3 (Fig. 1a), 58% in DICER and 38% in EZH2 genes (Supplementary Fig. 1B). We further characterised cleavage by sequencing and this showed different indels detected at all the target sites with various mutation sizes (Supplementary Fig. 1D). We also targeted KIAA1279- and CDKN1B-with a different transfection method in a different cell line, using electroporation of the chicken B cell DT40 cell line, with similar results (Fig. 1b).

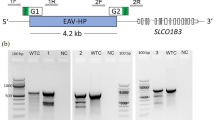
In vitro analysis of NHEJ and HDR genome modification (arrows) mediated by sgRNA-Cas9 system in chicken cell lines.
(a) Frequency (%) of NHEJ mutation mediated by KIAA1279, Cdkn1b and Mbd3-targeting sgRNA-Cas9 system in chicken DF-1 cells by PCR and T7E1 assay. 1kM- 1 kbp DNA ladder, M- 100 bp DNA ladder. (b) Frequency (%) of NHEJ mutation mediated by KIAA1279 and Cdkn1b-targeting sgRNA-CRISPR/Cas9 system in chicken lymphoma B DT40 cells by PCR and T7E1 assay. (c) Representative gel from DF-1 cells transfected with the RET-targeting sgRNA-Cas9 and the ssODN showing efficient integration of the HDR-based BamHI and EcoRV sequence. The frequency of HDR is represented in percentages. 1-No sgRNA, 2- MEN2B sgRNA #1 plus ssODN, 3- MEN2B sgRNA #1 and #2 plus ssODN and 4- MEN2A/HSCR sgRNA 1 plus ssODN. (d) Representative gel for single cell clones derived from DF-1 cells transfected with the RET-targeting sgRNA-Cas9 and the ssODN for the MEN2B and MEN2A/HSCR HDR modifications respectively. The table shows the ratio of the monoallelic and biallelic HDR-based mutations detected with single cell clones and the overall efficiency in percentage: N = 19 for MEN2B and N = 12 for MEN2A/HSCR.
The CRISPR/Cas9 system precisely edits genes in chicken cell lines
Next, to test whether specific gene editing through HDR could be generated by the CRISPR/Cas9 system, we chose exons 10 and 16 of the RET gene which harbour, respectively, the mutation causing Multiple Endocrine Neoplasia 2A (MEN2A) and Hirschsprung disease (MEN2A/HSCR: C620R in humans and C612R in chickens) and MEN2B (M918T in humans and M910T in chickens) (Supplementary Fig. 1A)26,27. We designed ssODNs with restriction enzyme site creation and disruption and co-transfected the sgRNA-CRISPR/Cas9 construct with the ssODN and the puromycin construct into the DF-1 cell line, using Lipofectamine 3000. Genomic DNA was isolated from 72–96 hrs post-transfected cells and the frequency of HDR-mediated genetic modification was analysed by digesting the PCR product with EcoRV and BamHI restriction enzymes. The digested bands indicate the frequency of HDR-mediated genetic modification from the ssODN template, which ranged between 34–66% of puromycin-resistant cells (Fig. 1c). Single clonal analysis shows the efficiency of biallelic and monoallelic HDR-mediated genetic modification by the CRISPR/Cas9 system as confirmed by gel and sequencing; 75% monoallelic with no biallelic for MEN2A/HSCR clones and 26% monoallelic and 21% biallelic for MEN2B clones (Fig. 1d).
The CRISPR/Cas9 system mediates larger genomic deletions in chicken cell lines
To see whether the CRISPR/Cas9 could also be used for large genomic fragment manipulation, we designed two sgRNAs targeting exon 1 and exon 3 of the STMN2 gene which spans >24 kilobase pairs (kbps), exon 10 and exon 18 of the RET gene which spans >11 kbp and exon 1 and 2 of DGCR8 gene and exon 1 of HIRA genes which spans >75 kbps of the chicken genome (Supplementary Table 2). We first co-transfected the two sgRNAs in DF-1 cells and analysed the targeted deletion by PCR after genomic DNA extraction. The PCR results (Fig. 2a) shows that the CRISPR/Cas9 system can mediate large genomic deletions (frequency 15%) in chicken cells in vitro which is in concordance with published data for other species18,28,29. We then further applied this approach to the chicken DT40 cell line, targeting the STMN2 locus and sequencing confirmed the >24 kbp deletion within this locus.

Targeted deletion of large genomic fragments and off-target effect analysis in chicken cells in vitro.
(a) Representative gels showing the large genomic deletions within the STMN2 locus (>24 kbp) in chicken DF-1 and DT40 lymphoma B cells and within the RET (>8 kbp) and HIRA-DGCR8 locus (>70 kbp). (b) Frequency of off-target effects mediated by RET-MEN2B and MEN2A/HSCR, CDKN1B, KIAA1279 and STMN2-targeting sgRNA-CRISPR/Cas9 system system in chicken DF-1 cells and CDKN1B and KIAA1279 -targeting sgRNA-CRISPR/Cas9 system in DT40 cells by PCR and T7E1 assay. ND-not detected, 1 kM- 1 kbp DNA ladder.
The CRISPR/Cas9 system produces no detectable off-target effects in chicken cell lines
Off-target mutagenesis remains a draw-back in the use of PENs and in human cells the CRISPR/Cas9 system has been reported to have relatively high off-target effects compared to other PENs30,31,32. Potential off-target sites with higher scores using the crispr.mit.edu software were selected and analysed by T7E1 assay in both the chicken DF-1 cells and the DT-40 cells of the KIAA1279 and CDKN1B-targeting sgRNA-CRISPR/Cas9 constructs and that of the HDR experiments targeting RET in DF-1 cells (Fig. 2b). There were no detectable off-target effects and these results suggest that chicken cells can potentially serve as a model for the use of the CRISPR/Cas9 nuclease, which is known for off-target effects in the human context.
The CRISPR/Cas9 system can act efficiently without selection in chicken cell lines
Drug selection cannot be used in in vivo applications so we investigated whether acceptable gene modification efficiency can be obtained without puromycin selection. sgRNA activity and hence efficiency of mutation-induction can be affected by target locus location, chromatin structure and nucleotide preferences33. Our results show that drug selection for the CRISPR/Cas9 system in chickens is not a necessity but can however improve efficiencies for some sgRNAs with low targeting efficiencies (Supplementary Fig. 1C).
CRISPR/Cas9 mediates somatic cell modification in chicken embryonic cells in vivo
Genetic engineering techniques in chicken have been the genomic modification of PGCs with a germ-line transmission capacity using the lentiviral system34,35 or Piggybac transposon vector34,36. Recent work by Park et al. showed that TALENs can efficiently generate knockout of targeted genes in chicken cells and in PGCs4,22. We then tested the efficiency of the CRISPR/Cas9 system in introducing NHEJ mutations into the avian embryo by in vivo electroporation. In vivo electroporation is a useful tool for the study of spatio-temporal gene functions, since the manipulation of genes can be used to study the roles of such genes in a restricted region during specific developmental stages4,6.
We injected and electroporated the DGCR8 exon 2-targeting sgRNA-CRISPR/Cas9 plasmid vectors incorporating mCherry marker in vivo into E1.5 embryo cranial neural tube, which transfects the brain and cranial neural crest (Supplementary Fig. 2A). DGCR8 is involved in miRNA processing and the targeted mutation should abrogate the gene function (Supplementary Fig. 2B). We also co-electroporated Tol2-GFP/transposase construct to indicate the trend and variability of transfection where neural crest cells migrate out of the neural tube to surround the brain and eye and migrate to the branchial arches and facial mesoderm (Supplementary Fig. 2A). Note that for this technique, the distribution and number of transfected cells, as shown by the extent of GFP expressing cells, is variable37.
After embryo electroporation with the CRISPR/Cas9 construct, we analysed the hindbrain and midbrain by whole mount immunofluorescence for DGCR8 expression. Since the mCherry plasmid is episomal, expression of mCherry is transient37,38, embryos were harvested after two days and immunostained with mCherry and DGCR8 antibodies (unelectroporated embryos, N = 8; electroporated embryos N = 24). Electroporated (i.e. mCherry-positive) cells in the midbrain, hindbrain and eye region showed a decrease in DGCR8 expression as determined by decrease in DGCR8 immunofluorescent intensity measured by pixel–count from confocal images (Fig. 3a,b). It is important to mention that DGCR8 expression in postnatal mouse brain has a major nuclear location (Supplementary Fig. 4A), but mainly cytoplasmic location was observed in embryonic chick brain cells (Fig. 3a). Subcellular heterogeneity of location has been reported for DGCR839. In addition DGCR8’s binding partner Drosha also shows cytoplasmic as well as nuclear localisation40. The neuronal RNA-binding proteins HUC/D also have different localisation patterns in rodent and chicken cells (compare figures in Hao et al.41 and Rollo et al.42). Western blotting of postnatal mouse and embryonic chick brain showed identical DGCR8-immunoreactive protein bands of appropriate molecular weight (Supplementary Fig. 4B), in accord with the high predicted sequence homology between the two species (NCBI database).

Protein expression 2 days after transfection with DGCR8 CRISPR/Cas9 construct.
(a) Immunofluorescence confocal images of single and merged channels of the indicated markers from whole mount staining of DGCR8 mutant embryos, indicating reduced to no DGCR8 expression in transfected (mCherry+) cells (shown by yellow arrows). (b) Histogram of pixel counts on control embryos and DGCR8 mutants embryos relative to DAPI. A total of 540 cells and 542 (> = 100 cells/embryo) were counted from 5 control and 6 electroporated embryos respectively. The low fluorescence in the mCherry waveband in controls is tissue autofluorescence. Scale bar: 5 μm. Error bars, mean ± s.e.m. *P < 0.05, ***P < 0.001.
In addition, electroporated embryos (N = 34) and control embryos (N = 85) were harvested after four days. The control embryos were used to gauge the background effects of the electroporation technique. The controls were sham-treated non-electroporated embryos (i.e. eggs opened, embryos visualised with India Ink, vitelline membrane nicked and eggs resealed; N = 23), embryos electroporated but without plasmids (i.e. non-transfected; N = 11), embryos electroporated and transfected with the benign Tol2-GFP/transposase plasmids (N = 29), embryos electroporated with the Cas9 construct targeting the unrelated STMN2 gene (N = 14) and embryos electroporated with the empty Cas9 construct (Cas9 vector with no cloned guide sequence; N = 8).
As expected, after 4 days mCherry fluorescence in wholemount embryos had declined to a level undetectable with the fluorescence stereomicroscope but we could FACs sort cells (mCherrylow and mCherryhigh) from the hindbrain. We performed T7E1 and qPCR analysis on these cells and showed a loss of DGCR8 mRNA levels in mCherryhigh sorted cells which was confirmed by sequence analysis (Supplementary Fig. 2B). DGCR8 sgRNA -CRISPR/Cas9 induced nucleotide deletions at the targeted locus with 1–5 bp deletions and insertions. In most clones this formed a stop codon which would result in nonsense-mediated decay of the DGCR8 mRNA (Supplementary Fig. 2B). The qPCR results coupled with the sequence data suggest a drastic decrease in DGCR8 gene expression in these brain cells during development, which could delay or disturb the growth of the midbrain as a whole. In addition the expression level of several other genes were analysed with findings consistent with previous findings (Supplementary Fig. 3A)43,44,45,46. Of the 34 DGCR8-targetted transfected embryos, 8 had a reduced head size and distorted morphology exemplified by major reduction of the midbrain (Fig. 4a). They also showed a reduction in their eyes: it has been reported earlier that miRNAs play an essential role in the differentiation of the retinal pigmented epithelium47. Furthermore, we compared the morphology of the hearts of the DGCR8-targetted embryos to the control embryos since in the DGCR8 mutant mouse, decreased DGCR8 expression results in a spectrum of malformations and reductions in cardiovascular development48. We observed deformations in the heart and outflow tract in 14 embryos (41.2%) of the transfected embryos. It is important to note that none of the 85 control embryos had these cranial, retinal and cardiac abnormalities (Fig. 4a,b).

Somatic targeted genetic modification by CRISPR/Cas9 system in chickens 4 days after in vivo electroporation.
(a) Frequency (%) of NHEJ mutation mediated by DGCR8-targeting sgRNA-CRISPR/Cas9 system in DF-1 cells by PCR. Red arrows indicate the NHEJ mutation created by the CRISPR/Cas9 system. M- 100 bp DNA ladder. Representative images of sham treated (con), electroporated untransfected embryos (electro.), Tol2 GFP transfected embryos (GFP transfect) with normal head development and DGCR8 CRISPR/Cas9 transfected embryos (D1 and D2) showing midbrain (open arrow) and eye (closed arrow) abnormalities. Graph shows the difference in the midbrain dimensions of DGCR8 mutant embryos compared to control embryos- N = 14. (b) Representative image of the hearts of unelectroporated embryos, electroporated with no construct embryos, Tol2 GFP and empty Cas9 transfected embryos, STMN2 transfected embryos (negative control) showing normal heart development and DGCR8 transfected embryos showing misshapen and reduced hearts. (c) qPCR analysis of cells isolated by FACS from DGCR8-targeted embryos demonstrating the reduced mRNA levels of DGCR8 in mCherry+ brain cells (M) relative to negatively sorted cells (N). Normalisation was done with ACTB and RPL32. ND-not detected. Error bars, mean ± s.e.m. ***P < 0.001.
These finding provide further support that the chicken embryo can be genetically modified in vivo in a targeted and sophisticated way to study disease models in developing embryos.
Discussion
The chick embryo is not only an excellent and reproducible system for embryonic developmental studies but also its accessibility and versatility makes it an alternative model in research directly relatable to humans and other animals. We have shown that the CRISPR/Cas9 system can modify multiple genes on both avian macro- and microchromosomes at acceptable efficiency with or without selection, with no detectable off-target effects, a previously mentioned drawback in the use of the CRISPR/Cas9 system49.
The function of genes can be spatio-temporally studied using CRISPR/Cas9 system in vivo provided that selection is not required. A recent example uses viral delivery to the adult mouse brain50. Moreover, refinements to increase the efficiency51 and limit the off-target errors52,53 are progressing rapidly. This means that for developmental studies, the advantages of the chick embryo as an accessible model and the convenience of in vivo electroporation at chosen developmental stages and locations can be combined with the power of CRISPR/Cas9 gene editing. We confirm this prediction here, extending the previous trial with Pax725.
In vivo transfection, including in vivo electroporation, affects a modest and variable proportion of cells37. This means that, as in many DGCR8 electroporated embryos, a gross phenotype will not be observed in every instance since gross phenotyppic change depends on high mutational load54,55. Despite this, functional effects of the genetic modification at the cellular level in vivo can be accurately gauged by imaging mutated cells and comparing with non-modified control cells in the same specimen. This requires markers of both the transfected cells and their otherwise similar control cells.
An interesting application of CRISPR/Cas9 editing would be the study of genes involved in nervous system development56, organogenesis and structural patterning. An important application would be to modify genes for growth factor response, proliferation or differentiation in neural crest cells. A specific clinically relevant example is the effect of MEN2 mutations which induce a variety of neurocristopathies and developmental cancers57. In addition, targeting of primordial germ cells by CRISPR/Cas9 offers the hope of genetically engineering avian models with any desired gene variant.
In conclusion, we have shown that transiently expressing the CRISPR/Cas9 construct can mediate genetic modification of avian embryonic somatic cells, reducing mRNA levels and generating phenotypes in the whole embryo. These results are in congruence with recent work also showing the efficiency of genome editing of postnatal mice using the CRISPR/Cas9 system58.
Methods
Ethics Statement
All experiments were performed with the official approval from the Murdoch Childrens Research Institute Animal Ethics Committee AEC650 and AEC677 and Institutional Biosafety Committee 226–2015 PC2 NLRD and in strict accordance with its guidelines and those of the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 7TH Edition 2004 and the Prevention of Cruelty to Animals Act, Victoria 1986.
sgRNA-CRISPR/Cas9 system design and construction
Potential target sites were predicted using crispr.mit.edu software in the chicken genome and two to three target sequences with lower predicted score for off-targets were chosen. To construct the sgRNA-CRISPR/Cas9 construct for each target gene, we annealed two complementary 24-bp oligonucleotides (Bioneer Company, South Korea) with the 20-bp target sequence to generate a double-strand DNA with 4-bp overhangs on both ends and cloned into BsaI-digested px330-IRES-mCherry. Oligonucleotides are listed in Supplementary Table 2.
DF-1 cell culture and transfection
The chicken DF-1 cell59 line was maintained and sub-passaged in DMEM (Thermo Scientific), supplemented with 10% fetal bovine serum (FBS; GIBCO) and 1× penicillin/streptomycin (GIBCO), at 37 °C in 5% CO2. Cells were seeded at 0.4–0.8 × 105 cells/well in 24-well plates, incubated for 4 hrs and then transfected with 1.5 μg CRISPR/Cas9 sgRNA targeting the specified gene or region and with or without 0.15 μg of puromycin expression vector using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s protocol with slight modifications. Briefly, 1 μL and 2 μL of Lipofectamine 3000 reagent was added to two different tubes with 25 μL of OPTI-MEM medium (Invitrogen) and briefly vortexed. Then a mixture of 1.5 μg of the CRISPR/Cas9-sgRNA plasmid (px330-IRES-mCherry), 0.15 ug puromycin expression vector and 3 μL of p3000 reagent (Invitrogen) in 50 μL OPTI-MEM was made and then 25 μL of the p3000-DNA complex was then added to each of the Lipofectamine 3000 complex tubes and incubated for 5 minute at room temperature. The complex mixture was then gently pipetted into a well of a 24-well plate with DF-1 cells at about 60–80% confluency. After 24 hrs post-transfection, the cells were treated with puromycin at a final concentration of 2 μg/mL for 2 days and the cells were allowed to recover for a day or two.
For HDR knock-ins, cells were transfected with 1.0 μg CRISPR/Cas9 sgRNA targeting the specified gene or region with 40 pmoles of ssODN and 0.15 μg of puromycin expression vector following the same protocol.
DT40 cell culture and transfection
Cells of the chicken B cell line DT4060 were cultured in chicken medium composed of RPMI-1640 medium (Sigma Aldrich), 10% FBS, 1% chicken serum (Sigma) and penicillin/streptomycin, at 39 °C in 5% CO2. A total of 1–2 × 107 cells were pelleted at 1500 rpm for 3 min at room temperature and the pellet was washed with PBS and pelleted again. The pellet was then resuspended in 600 μl PBS and after the addition of 30 μg of sgRNA-CRISPR/Cas9 plasmid and 3 μg of puromycin expression vector, the resuspension was placed in a BioRad 4 mm electroporation cuvette. Electroporation was done using BioRad Gene Pulser II at 250 V and 950 μF. After electroporation, cells were mixed with 10 ml of culture medium without penicillin/streptomycin and cultured for 12–24 hrs. The cells were treated with puromycin at a final concentration of 2 μg/mL for a day and the cells were allowed to recover for 2–3 days.
T7E1 mutation frequency analysis
Samples of cells and embryos were collected and digested in nuclear lysis buffer (Promega, Madison, WI). Genomic DNA was extracted from DF-1 cells or DT40 cells after transfection of each CRISPR/Cas9-sgRNA. The genomic DNA was extracted from the lysate by phenol-chloroform and recovered by isopropanol precipitation. The genomic region encompassing the CRISPR/Cas9 target site was amplified with a specific primer set for each gene (Supplementary Table 3). The amplicons were re-annealed to form a heteroduplex DNA structure after denaturation and then treated with 2.5 units T7E1 (New England Biolabs, Ipswich, MA) for 20 min at 37 °C and then analyzed by 2% agarose gel electrophoresis. Mutation frequencies were calculated as previously described61 based on the band intensities using Image J software and the following equation: mutation frequency (%) = 100 × (1 −(1 −fraction cleaved)1/2), where the fraction cleaved is the total relative density of the cleavage bands divided by the sum of the relative density of the cleavage bands and uncut bands. To confirm target locus mutation, PCR amplicons were cloned into a pGEM-T Easy vector (Promega, Madison, WI) and sequenced. Primers for the PCR analysis are listed in supplementary figures.
Restriction Enzyme digestion (RFLP)
Six μl of the PCR product was digested with 0.5 μl of the required restriction enzyme in a 20 μl reaction and incubated for 4–8 hrs at 37 °C. For MEN2B HDR templates, the PCR products were digested in BamHI restriction enzyme (New England Biolabs, Ipswich, MA) and in EcoRV restriction enzyme (New England Biolabs, Ipswich, MA) for MEN2A/HSCR HDR experiments. Digested products were then analysed by 1.5% agarose gel electrophoresis. Positive clones were cloned into the pGEM-T Easy vector (Promega, Madison, WI) and cloned products were sequenced.
Sequencing analysis
PCR products were cloned into the pGEM-T Easy vector (Promega, Madison, WI) and cloned products were sequenced using the T7 promoter primer (5′-TAATACGACTCACTATAGGG-3′).
Off-target prediction and analysis
Potential chicken KIAA1279, CDKN1B, STMN2 and RET gene off-targets were predicted using crispr.mit.edu software in the chicken genome. Off-target site with scores more than 1 or with 2 or more mismatches were chosen and amplified by PCR using the extracted genomic DNA as templates. The PCR products were first subjected to T7E1 cleavage assay14. Oligonucleotides are listed in Supplementary Table 4.
Single cell clonal analysis
Cells were trypsinized and plated in 96 well plates at average 0.3 cell/well and incubated at 37 °C for two weeks. Each well was then microscopically evaluated and single cell-derived clones were selected and expanded into 24 well plates. Genomic DNA from each clone was extracted and T7E1 assay was conducted following the above protocol. To confirm HDR of the ssODN, PCR amplicons were digested with 5 units of the restriction enzyme BamH1 (New England Biolabs) for more than 2 hrs at 37 °C and then analysed on 2% agarose gel by electrophoresis. PCR amplicons of BamH1 or T7E1 digested clones were cloned into a pGEM-T Easy vector (Promega, Madison, WI) and sequenced.
In vivo electroporation
Eggs from a cross breed White Leghorn x Black Australorp were commercially purchased (Research Hatchery, Victoria). In vivo electroporation were performed as previously described37,62,63. The CRISPR/Cas9 DGCR8 exon 2 targeting construct was co-electroporated with pT2K-CAAGGS-EGFP (termed Tol2-GFP) and pCAGGS-T2TP (transposase)38 at 6:6:1.5 μg/μL ratio respectively. The plasmid mixture was prepared and coloured with 2% Fast Green and then microinjected forward from the 3–4 somite level of the neural tube into the hind and midbrain of E1.5 chicken embryos. Electric pulses of 10.5 V, 50 ms duration were delivered 3 times bilaterally with 175-ms intervals. Chicken embryos were harvested 2 and 4 days post-electroporation and processed for immunostaining and cell sorting.
Fluorescence Activated Cell Sorting
Cell suspensions from harvested embryos were made using 0.5% w/v Dispase II (Roche, Switzerland) and 0.1% w/v CLSAFA Collagenase (Worthington, USA) at 37 °C in Hams F12 solution for 5–10 minutes. The digested cells were pelleted and resuspended in PBS containing 2% FBS and strained (40 μm mesh; BD Falcon; Becton, Dickinson and Co., Franklin Lakes, NJ) and FACS sorted using the BD Influx Cell Sorter, with separation based on GFP and mCherry fluorophores.
RNA extraction and SYBR Green qPCR
Total RNA was extracted from sorted cells using the Trizol reagent (Invitrogen) and lysate were purified by the acid-phenol chloroform and recovered by isopropanol/ethanol precipitation method. Extracted RNA was digested with DNaseI (Promega) following the manufacturer’s instructions to remove any residual DNA.
qPCR was performed to confirm the expression of DROSHA, Neurogenin 2 (Ngn2), Pax6, YPEL1 and DGCR8 genes in transfected modified embryonic cells from electroporated embryos. Briefly, 20 ng total RNA was converted into cDNA in the presence of SuperScript IV RT (Invitrogen) and random hexamers (Promega). Reactions were performed using cDNA converted from 10 ng of RNA, 250 nM of each primer and 2X SYBR Green qPCR Master Mix (Promega) in a total volume of 20 μl. Primers for qPCR analysis are listed in Supplementary Table 5. ACTB and RPL32 were used for data normalization. mCherry-/GFP- sorted cells from each embryo were used as a calibrator and relative fold changes were calculated using the 2−[Δ][Δ]Cq method.
Whole mount staining and immunohistochemistry
Embryos were harvested 2 days post electroporation (E3.5) and sagittally dissected, fixed in 4% paraformaldehyde in PBS at 4 °C overnight then washed in PBS three times. Embryos were blocked and permeabilised with 3% horse serum and 0.2% Triton-X100 in PBS/azide for 1 hr. Control cryostat 18 μm sections of chick embryo and post-natal mouse brain were also used. Rabbit anti-DGCR8 antibody (Abcam-ab82876) and mouse anti-mCherry (DSHB, Iowa City) at 1:200 and 1:100 respectively were applied in 1% horse serum and 0.1% Triton-X in PBS azide and incubated on a rocker at 4 °C overnight. The human DGCR8 immunogen (N-terminal amino acids 180–229) was 89% identical to the predicted chick amino acid sequence (NCBI database), with all changes conservative. Washing with PBS was done for 3 hrs with changes every 30 minutes on a rocker at 4 °C. Secondary antibodies were donkey anti-rabbit:Alexa Fluor 488 for mouse sections (Life Technologies-1:1000) and donkey anti-rabbit:Cy5 plus donkey anti-mouse:Alexa Fluor 568 for whole mounts (Jacksons Immunoresearch-1:500 and 1:1000 respectively) and 500 ng/mL (1 in 100 of 50 ug/mL stock) DAPI was applied and incubated for 3 hrs on the rocker at 4 °C. Embryos were washed with PBS three times and mounted using DABCO/glycerol mounting medium. Confocal microscopy was performed using the Zeiss LSM 780 confocal microscope.
For cryostat sections, fixed embryo heads (E5.5) were placed in 30% sucrose in PBS overnight, embedded in Tissue Tek OCT Compound Medium in Tissue Tek cryomoulds (both from ProSciTech, Thuringowa, Australia) and frozen in dry ice-cooled isopentane. Eighteen μm sections were cut transversely using a Leica CM 1900 cryostat microtome and collected on Superfrost microscope slides (Biolab Scientific, Auckland, NZ) coated with poly-l-lysine. Mouse post-natal brains were also fixed, sectioned, mounted and stained as above.
Western Blotting
Brains from chick embryos (E6) were homogenised in 2 mL of sample buffer for SDS-PAGE. Homogenates were sonicated for 40 sec and centrifuged at 13,000 rpm for 5 min. The resulting supernatants were divided into 100 mL aliquots and stored at −80 °C. Protein concentration of 3.3% homogenates (w/v) in PBS was determined by the Pierce BCA Protein Assay Kit (ThermoFisher Scientific) using bovine serum albumin as a standard. Forty micrograms of each sample were analyzed by Western blotting. The apparent molecular mass of DGCR8 was estimated by a prestained protein marker (Life Technologies). Control tissue was obtained from post-natal mouse brain.
Relative Pixel Quantification
Standard confocal images were selected and analysed using the Zeiss Image Analyser. Regions of interest were selected with the Free-Hand tool and choosing the DAPI channel, the cells in a field were counted and relative pixel quantification was calculated following a previous publication64. Relative fluorescence intensity was calculated by normalising the mCherry and DGCR8 expression to DAPI intensity.
Statistical analyses
Data were analyzed by the unpaired t test with Welch’s correction. Values were expressed as mean ± SEM. Changes were deemed significant if the p value was >0.05. Statistical significance is indicated as follows: *p > 0.05, **p > 0.01 and ***p > 0.001. Graphs were drawn using Microsoft Excel and GraphPad Prism.
Additional Information
How to cite this article: Abu-Bonsrah, K. D. et al. CRISPR/Cas9 Targets Chicken Embryonic Somatic Cells In Vitro and In Vivo and generates Phenotypic Abnormalities. Sci. Rep. 6, 34524; doi: 10.1038/srep34524 (2016).
References
Mason, I. The avian embryo: an overview. Methods Mol Biol 461, 223–230, 10.1007/978-1-60327-483-8_14 (2008).
Niswander, L. Methods in avian embryology experimental and molecular manipulation of the embryonic chick limb. Methods in cell biology 87, 135–152, 10.1016/S0091-679X(08)00207-0 (2008).
Le Douarin, N. M. & Dieterlen-Lievre, F. How studies on the avian embryo have opened new avenues in the understanding of development: a view about the neural and hematopoietic systems. Development, growth & differentiation 55, 1–14, 10.1111/dgd.12015 (2013).
Park, T. S., Kang, K. S. & Han, J. Y. Current genomic editing approaches in avian transgenesis. General and comparative endocrinology 190, 144–148, 10.1016/j.ygcen.2012.11.020 (2013).
Smith, S. M., Flentke, G. R. & Garic, A. Avian models in teratology and developmental toxicology. Methods Mol Biol 889, 85–103, 10.1007/978-1-61779-867-2_7 (2012).
Itasaki, N., Bel-Vialar, S. & Krumlauf, R. ‘Shocking’ developments in chick embryology: electroporation and in ovo gene expression. Nat Cell Biol 1, E203–207 (1999).
Sauka-Spengler, T. & Barembaum, M. Gain- and loss-of-function approaches in the chick embryo. Methods in cell biology 87, 237–256, 10.1016/S0091-679X(08)00212-4 (2008).
Takahashi, Y., Watanabe, T., Nakagawa, S., Kawakami, K. & Sato, Y. Transposon-mediated stable integration and tetracycline-inducible expression of electroporated transgenes in chicken embryos. Methods in cell biology 87, 271–280, 10.1016/S0091-679X(08)00214-8 (2008).
Shan, Q., Wang, Y., Li, J. & Gao, C. Genome editing in rice and wheat using the CRISPR/Cas system. Nature protocols 9, 2395–2410, 10.1038/nprot.2014.157 (2014).
Hou, Z. et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proceedings of the National Academy of Sciences of the United States of America 110, 15644–15649, 10.1073/pnas.1313587110 (2013).
Yang, L. et al. Optimization of scarless human stem cell genome editing. Nucleic acids research 41, 9049–9061, 10.1093/nar/gkt555 (2013).
Zhang, Y., Vanoli, F., LaRocque, J. R., Krawczyk, P. M. & Jasin, M. Biallelic targeting of expressed genes in mouse embryonic stem cells using the Cas9 system. Methods 69, 171–178, 10.1016/j.ymeth.2014.05.003 (2014).
Shao, Y. et al. CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nature protocols 9, 2493–2512, 10.1038/nprot.2014.171 (2014).
Niu, Y. et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843, 10.1016/j.cell.2014.01.027 (2014).
Shen, H. Precision gene editing paves way for transgenic monkeys. Nature 503, 14–15, 10.1038/503014a (2013).
Shin, J., Chen, J. & Solnica-Krezel, L. Efficient homologous recombination-mediated genome engineering in zebrafish using TALE nucleases. Development 141, 3807–3818, 10.1242/dev.108019 (2014).
Auer, T. O. & Del Bene, F. CRISPR/Cas9 and TALEN-mediated knock-in approaches in zebrafish. Methods 69, 142–150, 10.1016/j.ymeth.2014.03.027 (2014).
Ota, S., Hisano, Y., Ikawa, Y. & Kawahara, A. Multiple genome modifications by the CRISPR/Cas9 system in zebrafish. Genes to cells: devoted to molecular & cellular mechanisms 19, 555–564, 10.1111/gtc.12154 (2014).
Bassett, A. R. & Liu, J. L. CRISPR/Cas9 and genome editing in Drosophila. Journal of genetics and genomics = Yi chuan xue bao 41, 7–19, 10.1016/j.jgg.2013.12.004 (2014).
Gokcezade, J., Sienski, G. & Duchek, P. Efficient CRISPR/Cas9 plasmids for rapid and versatile genome editing in Drosophila. G3 4, 2279–2282, 10.1534/g3.114.014126 (2014).
Chen, C., Fenk, L. A. & de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic acids research 41, e193, 10.1093/nar/gkt805 (2013).
Park, T. S., Lee, H. J., Kim, K. H., Kim, J. S. & Han, J. Y. Targeted gene knockout in chickens mediated by TALENs. Proc Natl Acad Sci USA 111, 12716–12721, 10.1073/pnas.1410555111 (2014).
Long, C. et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403, 10.1126/science.aad5725 (2016).
Nelson, C. E. et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407, 10.1126/science.aad5143 (2016).
Veron, N., Qu, Z., Kipen, P. A., Hirst, C. E. & Marcelle, C. CRISPR mediated somatic cell genome engineering in the chicken. Developmental biology 407, 68–74, 10.1016/j.ydbio.2015.08.007 (2015).
Smith, D. P., Houghton, C. & Ponder, B. A. Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene 15, 1213–1217, 10.1038/sj.onc.1201481 (1997).
Gimm, O. et al. Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. The Journal of clinical endocrinology and metabolism 82, 3902–3904, 10.1210/jcem.82.11.4508 (1997).
He, Z. et al. Highly efficient targeted chromosome deletions using CRISPR/Cas9. Biotechnology and bioengineering 112, 1060–1064, 10.1002/bit.25490 (2015).
Xiao, A. et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic acids research 41, e141, 10.1093/nar/gkt464 (2013).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology 31, 827–832, 10.1038/nbt.2647 (2013).
Pattanayak, V. et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nature biotechnology 31, 839–843, 10.1038/nbt.2673 (2013).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology 31, 822–826, 10.1038/nbt.2623 (2013).
Doench, J. G. et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature biotechnology 32, 1262–1267, 10.1038/nbt.3026 (2014).
Park, S. H. et al. CpG methylation modulates tissue-specific expression of a transgene in chickens. Theriogenology 74, 805–816 e801, 10.1016/j.theriogenology.2010.04.005 (2010).
Motono, M. et al. Production of transgenic chickens from purified primordial germ cells infected with a lentiviral vector. Journal of bioscience and bioengineering 109, 315–321, 10.1016/j.jbiosc.2009.10.007 (2010).
Kubo, F., Takeichi, M. & Nakagawa, S. Wnt2b inhibits differentiation of retinal progenitor cells in the absence of Notch activity by downregulating the expression of proneural genes. Development 132, 2759–2770, 10.1242/dev.01856 (2005).
Simkin, J. E., Zhang, D., Ighaniyan, S. & Newgreen, D. F. Parameters affecting efficiency of in ovo electroporation of the avian neural tube and crest. Developmental dynamics: an official publication of the American Association of Anatomists 243, 1440–1447, 10.1002/dvdy.24163 (2014).
Sato, Y. et al. Stable integration and conditional expression of electroporated transgenes in chicken embryos. Developmental biology 305, 616–624, 10.1016/j.ydbio.2007.01.043 (2007).
Nakano, A., Onohara, Y., Yokota, S. & Fujita, H. DGCR8 Localizes to the Nucleus as well as Cytoplasmic Structures in Mammalian Spermatogenic Cells and Epididymal Sperm. Journal of Histology 2013, 11, 10.1155/2013/414891 (2013).
Link, S., Grund, S. E. & Diederichs, S. Alternative splicing affects the subcellular localization of Drosha. Nucleic acids research 44, 5330–5343, 10.1093/nar/gkw400 (2016).
Hao, M. M. et al. Enteric nervous system assembly: Functional integration within the developing gut. Developmental biology, 10.1016/j.ydbio.2016.05.030 (2016).
Rollo, B. N., Zhang, D., Simkin, J. E., Menheniott, T. R. & Newgreen, D. F. Why are enteric ganglia so small? Role of differential adhesion of enteric neurons and enteric neural crest cells. F1000Research 4, 113, 10.12688/f1000research.6370.1 (2015).
Fan, P. et al. miRNA biogenesis enzyme Drosha is required for vascular smooth muscle cell survival. PloS one 8, e60888, 10.1371/journal.pone.0060888 (2013).
Wang, Y., Medvid, R., Melton, C., Jaenisch, R. & Blelloch, R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nature genetics 39, 380–385, 10.1038/ng1969 (2007).
Boyer, L. A. et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353, 10.1038/nature04733 (2006).
Klein, M. E. et al. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nature neuroscience 10, 1513–1514, 10.1038/nn2010 (2007).
Ohana, R. et al. MicroRNAs are essential for differentiation of the retinal pigmented epithelium and maturation of adjacent photoreceptors. Development 142, 2487–2498, 10.1242/dev.121533 (2015).
Chapnik, E., Sasson, V., Blelloch, R. & Hornstein, E. Dgcr8 controls neural crest cells survival in cardiovascular development. Developmental biology 362, 50–56, 10.1016/j.ydbio.2011.11.008 (2012).
Park, T. S. & Han, J. Y. Genetic modification of chicken germ cells. Annals of the New York Academy of Sciences 1271, 104–109, 10.1111/j.1749-6632.2012.06744.x (2012).
Swiech, L. et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nature biotechnology 33, 102–106, 10.1038/nbt.3055 (2015).
Chu, V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nature biotechnology 33, 543–548, 10.1038/nbt.3198 (2015).
Kim, D. et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature methods 12, 237–243, 231 p following 243, 10.1038/nmeth.3284 (2015).
Kleinstiver, B. P. et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 10.1038/nature16526 (2016).
Shah, A. N., Davey, C. F., Whitebirch, A. C., Miller, A. C. & Moens, C. B. Rapid reverse genetic screening using CRISPR in zebrafish. Nature methods 12, 535–540, 10.1038/nmeth.3360 (2015).
Sung, Y. H. et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome research 24, 125–131, 10.1101/gr.163394.113 (2014).
Heidenreich, M. & Zhang, F. Applications of CRISPR-Cas systems in neuroscience. Nature reviews. Neuroscience 17, 36–44, 10.1038/nrn.2015.2 (2016).
Zhang, D. et al. The neural crest: a versatile organ system. Birth defects research. Part C, Embryo today: reviews 102, 275–298, 10.1002/bdrc.21081 (2014).
Long, C. et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science, 10.1126/science.aad5725 (2015).
Lambeth, L. S., Cummins, D. M., Doran, T. J., Sinclair, A. H. & Smith, C. A. Overexpression of aromatase alone is sufficient for ovarian development in genetically male chicken embryos. PloS one 8, e68362, 10.1371/journal.pone.0068362 (2013).
Kim, J. H. et al. Condensin I associates with structural and gene regulatory regions in vertebrate chromosomes. Nature communications 4, 2537, 10.1038/ncomms3537 (2013).
Guschin, D. Y. et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649, 247–256, 10.1007/978-1-60761-753-2_15 (2010).
Sato, Y., Yasuda, K. & Takahashi, Y. Morphological boundary forms by a novel inductive event mediated by Lunatic fringe and Notch during somitic segmentation. Development 129, 3633–3644 (2002).
Yokota, Y., Saito, D., Tadokoro, R. & Takahashi, Y. Genomically integrated transgenes are stably and conditionally expressed in neural crest cell-specific lineages. Developmental biology 353, 382–395, 10.1016/j.ydbio.2011.02.001 (2011).
Oriol Arqués, I. C., Stephan, T., Isabel, P. & Héctor, G. Palmer. Standardized Relative Quantification of Immunofluorescence Tissue Staining. Nature Protocol Exchange, 10.1038/protex.2012.008 (2012).
Acknowledgements
We thank Damien Hudson (MCRI) and Craig Smith (MCRI) for the DT40 and DF-1 cells respectively, Marguerite Evans-Galea (MCRI) for the mouse brain and Yoshiko Takahashi (Kyoto, Japan) for Tol2 and transposase plasmids. We thank Matthew Burton and Paul Lau (Flow Cytometry-MCRI), Paul Kalitsis (Chromosomal Research-MCRI), Hwee Ong (Genetic Health Research-MCRI) and Alex Combes (Kidney Development, Disease and Regeneration) for their technical advice and support. The mCherry monoclonal antibody (3A11) was developed and supplied by the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. This research was supported by NHMRC grant 1069757 and MCRI. HSCR-related constructs were contributed to by Foundation for Children 2014-211. We acknowledge the Victorian Government’s Operational Infrastructure Support Program to MCRI. K.D.A.-B. holds an IPRS and APA(Int) PhD Scholarship through the Department of Paediatrics, University of Melbourne.
Author information
Authors and Affiliations
Contributions
K.D.A.-B. designed and performed the in vitro experiments. D.Z. and K.D.A.-B. performed and harvested the in vivo electroporation experiments. K.D.A.-B. and D.F.N. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Abu-Bonsrah, K., Zhang, D. & Newgreen, D. CRISPR/Cas9 Targets Chicken Embryonic Somatic Cells In Vitro and In Vivo and generates Phenotypic Abnormalities. Sci Rep 6, 34524 (2016). https://doi.org/10.1038/srep34524
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34524
This article is cited by
-
Study of the regulatory elements of the Ovalbumin gene promoter using CRISPR technology in chicken cells
Journal of Biological Engineering (2023)
-
CRISPR/Cas9 gene editing in a chicken model: current approaches and applications
Journal of Applied Genetics (2020)
-
Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR–Cas9 and ssODN in human cancer cells
Cell & Bioscience (2018)
-
Avian transcriptomics: opportunities and challenges
Journal of Ornithology (2018)
-
Gene editing in birds takes flight
Mammalian Genome (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
