
Abstract
Genomic prediction shows promise for personalised medicine in which diagnosis and treatment are tailored to individuals based on their genetic profiles for complex diseases. We present a theoretical framework to demonstrate that prediction accuracy can be improved by targeting more informative individuals in the data set used to generate the predictors (“discovery sample”) to include those with genetically close relationships with the subjects put forward for risk prediction. Increase of prediction accuracy from closer relationships is achieved under an additive model and does not rely on any family or interaction effects. Using theory, simulations and real data analyses, we show that the predictive accuracy or the area under the receiver operating characteristic curve (AUC) increased exponentially with decreasing effective size (Ne), i.e. when individuals are closely related. For example, with the sample size of discovery set N = 3000, heritability h2 = 0.5 and population prevalence K = 0.1, AUC value approached to 0.9 and the top percentile of the estimated genetic profile scores had 23 times higher proportion of cases than the general population. This suggests that there is considerable room to increase prediction accuracy by using a design that does not exclude closer relationships.
Similar content being viewed by others
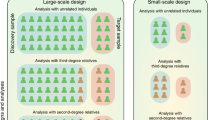


Introduction
The genomics era has demonstrated the polygenic nature of complex genetic traits, and genomic prediction shows much promise for personalised medicine in which diagnosis and treatment are tailored to individuals based on the profiles recorded in their genome. This creates the opportunity for ‘stratified medicine’1 in which individuals are classified into higher and lower risk groups and intervention or treatment of relevant sub-categories is based on profiles that incorporate information from both genomic and environmental risk factors. The utility of this approach, of course, will depend on the reliability of these risk predictions.
A key feature of risk predictors is that their use does not necessarily require an understanding of the aetiology of disease1. Usefulness of such prediction is demonstrated by success in genetic selection programs in animals and plants. Risk prediction in human medicine can also have an important impact even in absence of a full understanding of the underlying biology of diseases and disorders. Aggregate effects from causal variants tagged by single nucleotide polymorphisms (SNPs) across the genome can quantify and assess individual risk for a particular disease or disorder, deemed “genomic prediction”.
Genomic prediction has recently been tested and shown to be promising for diseases of which genetic variance is largely explained by a number of major genes2,3,4. However, for polygenic diseases and disorders caused by numerous genes with small effect, which is the case for most complex traits, the accuracy of genomic prediction has been considered too low to be useful in actual clinical applications5,6,7,8,9. Most of these studies employed population-based prediction based on unrelated individuals. Several studies have reported a considerable increase in prediction accuracy when the training data set included individuals that were closely related to the target sample, from data on humans10,11,12,13 as well as from other species14,15,16. Some have argued that the use of close relatives may inflate estimated genetic variance due to common environmental effects, or gene-environment or gene-gene interaction17,18,19, and therefore such effects may also bias genomic risk prediction. However, theoretical work from previous studies20,21,22 has shown that genomic predictions are more accurate in populations of smaller effective size, i.e. where individuals tend to be more closely related. In such cases there are effectively fewer chromosome segments to estimate across the genome, which allows a higher prediction accuracy from the same size of data20,21,22. This suggests that subjects that are closely related could be a valuable resource for genomic risk prediction. For predicting human diseases, the area under the receiver operating characteristic curve (AUC) or odds ratio (OR) of case-control status contrasting the higher or lower risk group is a typical measure of prediction accuracy. However, we have no adequate insight in predicting the improvement in AUC or OR when using more related subjects, and how this accuracy may vary between individuals or between populations.
In this study, we revisit the theory on genomic prediction accuracy as presented previously20,21,22, and derive an improved method linking effective population size (Ne) and effective number of chromosome segments (Me) to prediction accuracy, assuming that trained individuals and predicted subjects are from the same homogenous population. We use simulated as well as real data to demonstrate that prediction accuracy can be increased when predicting from more related subjects. We extend this work to a case-control data set, which is a typical design for human diseases, so that the outcomes of this study are applicable to a clinical program for human diseases.
Results
Effective number of chromosome segments and prediction accuracy
A key parameter for determining the accuracy of the genomic prediction is Me, the effective number of chromosome segments segregating in a population. Intuitively, this makes sense, as fewer independent chromosomal segments require fewer independent parameters to be estimated from the same data. We show that Me depends on effective population size Ne, the number of chromosomes and the length of genomic region (eqs (10) and (11) in Methods), and that allowing close genetic relationships to exist between the discovery sample (used to generate predictors) and the target sample (where prediction is applied) is equivalent to reducing Me. We present an improved derivation of the expected Me by taking into account that there is a covariance between the relationship between individuals at different chromosomes (Supplementary Tables 1–3). We validated the theory for estimation of Me using the stochastic coalescence gene-dropping method (see simulation I in Methods). The expected Me (from eqs (10) or (11), eq. (11) accounts for historical mutations whereas eq. (10) does not, although the difference is small) were compared to the estimated Me from the variation in genomic relationships between discovery and target samples, i.e. using the elements in the off-diagonal block of the matrix relating to target × discovery sample, using eq. (12) (Supplementary Figures 1A–3A). Furthermore, the expected prediction accuracy from theory (eq. (1)) and the observed accuracy from the simulated genotypes and phenotypes were compared (Supplementary Figures 1B–3B). The observed accuracy was obtained from the correlation between true and estimated genetic profile scores in the target data set (see Methods).
The empirically estimated Me from the genomic relationships (using eq. (12)) agreed with the expected Me from eqs (10) or (11) whether using a small or large sample size (Supplementary Figures 1A and 2A). From the estimated Me, the expected prediction accuracy could be obtained from eq. (1). The expected prediction accuracy was within the confidence interval of the actual observed prediction accuracy over 100 replicates (Supplementary Figures 1B and 2B). With a larger number of chromosomes the estimated Me from the genomic relationship matrix (GRM) was close to the expected Me from eqs (10) and (11) that accounts for the correlation between chromosomes, and the expected prediction accuracy from eq. (1) coincided with the confidence interval of the observed prediction accuracy over 100 replicates (Supplementary Figure 3).
Theoretical prediction accuracy in relation to Ne as a key design parameter
Accuracy of the genomic prediction is determined by the genetic variance, the number of phenotypic observations and Me. We theoretically quantified prediction accuracy. Using the theory (eqs (2), (10) and (11)), the prediction accuracy for a quantitative trait was quantified in relation to Ne, using h2 = 0.5, 30 chromosomes each with a genomic length of L = 1 Morgan (30 Morgan in total) and N = 3000 (number of records for the discovery sample) that mimics a typical GWAS. It is noted that 23 chromosomes each with L = 1.3 Morgan (30 Morgan in total) gave similar values (result not shown). Figure 1 shows that when Ne was smaller, the correlation between the estimated genetic profile scores and phenotypes for the target samples was increased, approaching the square root of the heritability. With Ne = 10,000, this correlation was only 0.18, but the accuracy became larger rapidly with smaller Ne. For example, the correlation was 0.65 with Ne = 100.

The number of records (N) in the discovery data set is 3000, the true heritability is 0.5 and the number of chromosome is 30 each with a genomic length of 1 Morgan.
The prediction accuracy was also derived for case-control data using the same parameters as above for an underlying quantitative trait. A disease or disorder with population prevalence of K = 0.1 and a proportion of cases in the sample of P = 0.5 was used. With these parameters, we obtained the expected values for AUC (eq. (3)), the odds ratio of case-control status contrasting the top and bottom 20% of the genetic profile scores (eq. (4)) and that contrasting the top 1% of the genetic profile scores and the general population (eq. (5)). The expected values were verified by comparing them with the observed values from simulation II, showing that the expectation and observation agreed well (Supplementary Figures 4, 5 and 6). Furthermore, we tested the prediction accuracy with a rare disease or disorder with population prevalence of K = 0.01, which also showed a good agreement between the expectation and observation (Supplementary Figures 7, 8 and 9).
When using Ne = 10,000, the value for AUC was just 0.60, rising to a value of 0.85 with Ne = 100 (Fig. 2). The odds ratio of the case-control status, contrasting the top and bottom 20% according to estimated genetic profile scores, ranged from 2.7 with Ne = 10,000 to 131.9 with Ne = 100 (Fig. 3). The odds ratio of the case-control status contrasting the top 1% of estimated genetic profile scores and normal population was 2.3 with Ne = 10,000, and 23.0 with Ne = 100 (Fig. 4). With a larger N or higher h2, the prediction accuracy was further dramatically increased (Supplementary Table 4).

The number of records (N) is 3000, the true heritability is 0.5, the number of chromosome is 30 each with a genomic length of 1 Morgan, the population prevalence is K = 0.1 and the proportion of cases in the sample is P = 0.5.

The number of records (N) is 3000, the true heritability is 0.5, the number of chromosome is 30 each with a genomic length of 1 Morgan, the population prevalence is K = 0.1 and the proportion of cases in the sample is P = 0.5.

The number of records (N) is 3000, the true heritability is 0.5, the number of chromosome is 30 each with a genomic length of 1 Morgan, the population prevalence is K = 0.1 and the proportion of cases in the sample is P = 0.5.
Real data application
We applied the approach to a real data set, the Framingham heart study (see Methods). In 100 cross-validation replicates, the real data was randomly divided into two sets - one for discovery and the other for target, where sampling was either family wise to create a larger Ne or within family to create a low Ne. For the family-wise sampling, 80% of the available families were selected as the discovery data set, with the remaining 20% of families used as the target data set. For the within-family sampling, each member in every family was assigned an 80% chance to be a discovery sample and the rest was in the target sample (see Methods). The discovery set had an average of 3394 individuals and the target set had an average of 849 individuals over 100 cross-validation replicates (Table 1). The estimated Me from the genomic relationship between the discovery and target samples was 4,434 and 31,080 (from eq. (12)) when generating a smaller and a larger Ne, respectively. The distribution of variance of relationships, calculated for each target individual when paired with discovery individuals is shown in Supplementary Figure 10 for designs with smaller and larger Nevalues. Table 1 shows that the average correlation between the estimated genetic profile scores and the phenotypes (height) in the target set was 0.549 (SD 0.021) and 0.091 (SD 0.043) when using a design with small and large Ne, respectively, clearly indicating the advantage of using a design with a smaller Ne.
Interestingly, the results were consistent with the estimated heritability from family-based studies (i.e. h2 = 0.823,24,25) or population-based studies (i.e. h2 = 0.4526,27), which is numerically illustrated in Supplementary Table 5. When using h2 = 0.8 and Me = 4,434, the expected accuracy of genomic prediction was 0.551 (from eq. 2), which was close to the observed accuracy of 0.549 (Table 1). By contrast, using h2 = 0.45 and Me = 31,080 would give an expected accuracy of genomic prediction of 0.145, approximately similar to the observed accuracy of 0.091 (Table 1).
Mimicking case-control data, the top 10% of the phenotypes were selected and treated as cases (i.e. K = 0.1), and 11.1% of the remaining 90% of phenotypes were chosen to be controls. Therefore, the case-control ratio was 1:1 (i.e. P = 0.5). The two sampling strategies used in cross-validation for the quantitative traits, were also used for the case-control data generating higher and lower variance of relationships between discovery and target sets (smaller and larger Ne, respectively). The discovery set had an average of 680 individuals and the target set had an average of 170 individuals over 100 cross-validation replicates (Table 1). The estimated Me from the genomic relationship between the discovery and target samples was 3,247 and 29,479 from eq. (12) for smaller and larger Ne, respectively. In Table 1, the average AUC for the two scenarios was 0.687 (SD 0.037) and 0.535 (SD 0.038), indicating that the AUC was considerably higher with a smaller Ne than with larger Ne. The observed AUC values were very similar to the expected values, based on eq. (3), for the small Ne design (0.682 with Me = 3247 and h2 = 0.823,24,25) and the large Ne design (0.537 with Me = 29,479 and h2 = 0.4526,27), respectively (Supplementary Table 5).
The odds ratio of case-control status comparing each 20 percentile to the bottom 20% of the ranked genetic profile scores demonstrates that the contrasting power was substantially higher with a smaller Ne than with a larger Ne. (Fig. 5). The observed odds ratio of case-control status contrasting the top and bottom 20% of the genetic profile scores was similar to the expected odds ratio from eq. (5) for the small Ne design with Me = 3,247 and h2 = 0.823,24,25 and the large Ne design with Me = 29,479 and h2 = 0.4526,27, respectively (Supplementary Table 5).

The odds ratio of the case-control status contrasting the top and bottom 20% of the ranked genetic profile scores estimated from a design with a smaller or larger Ne, in the Framingham data.
We additionally analysed BMI phenotypes, which gave a similar result in that the prediction accuracy was considerably higher with a smaller Ne than with larger Ne, and that the observed and expected values agreed with each other (Supplementary Table 6).
As a second real data set, we used genetic data from the European ancestry participants of the Kaiser Permanente genetic epidemiology research on adult health and aging (GERA) cohort. When using the GERA dataset that does not have a clear family structure, the prediction accuracy for hypertension phenotypes is significantly higher for 25% of the target sample with the highest variance of pair-wise relationships with the discovery sample (Fig. 6 and Supplementary Figure 11). The prediction accuracy was 0.118 (0.114–0.123) for the top 25% and 0.106 (0.104–0.107) for the entire target sample. Moreover, the prediction accuracy was significantly decreased to 0.097 (0.095–0.099) (Fig. 7) when higher relationships were removed from the sample (>relatedness of 0.025), therefore increasing Me (Supplementary Figure 12). These results demonstrate that a higher variance of pair-wise relationships, hence smaller Me, results in a higher prediction accuracy even when using data from an extensive population-based sample. We also confirmed these results by using the real genotype data (GERA) and simulating phenotypes with the total variance fully or partly explained by the SNPs in order to support the results from the real data analysis (Figs 6 and 7) by showing that higher prediction accuracy for the top 25% group and the lower accuracy after removing one from a pair of individuals with higher relationships was not due to non-genetic confounding factors such as artefact batch effects (Supplementary Figures 13–16).

GERA data with hypertension phenotypes are used. The error bar is 95% confidence interval of the observed prediction accuracy over 100 replicates.

GERA data with hypertension phenotypes are used. The same number of discovery and target sample is used for both tests. The error bar is 95% confidence interval of the observed prediction accuracy over 100 replicates.
We also analysed dyslipidemia phenotypes and found a consistent result showing that the prediction accuracy was significantly increased for 25% of the target sample with the highest variance of pair-wise relationships with the discovery sample (Supplementary Figure 17), and that it was decreased (although non-significant) when higher relationships were removed from the sample (Supplementary Figure 18).
Discussion
In this study, we have shown, by theory, simulations and analyses of real data that genomic prediction based on a discovery set that includes individuals with close relationships to the predicted subjects leads to higher prediction accuracy, assuming that reference sample and predicted subjects are from the same homogenous population. The accuracy can be predicted from the variation in relationships of the target individual with the individuals in the discovery data set. The variation in relationship can be linked back to the number of effective chromosome segments to be estimated, which in turn is a result of a certain effective population size, i.e. the size of a homogeneous unstructured population where the amount of chromosome segment sharing is similar, leading to the same accuracy of prediction. We showed that there is merit in designing the discovery population such that variation of genetic relationships is maximized.
Current studies for polygenic diseases or disorders have reported that the accuracy of genomic prediction is not useful for actual clinical practice5,6,7,8,9 due to low prediction accuracies. However, it is common practice to deliberately exclude close relatives and use samples from the population that are genetically distant resulting in Ne values of more than a few thousand and a resulting Me across the genome in the tens of thousands, even when predictions are just within populations of pure European descent. The effective number of chromosome segments is a key parameter on which prediction accuracy depends20,21,22. A desirable design for genomic prediction should have a discovery set that is well related to the target set of individuals, resulting in a smaller Me. Note that this is similar to predicting in a population with a smaller Ne, in other words, including closer relatives in the discovery set has the same effect as predicting in a population with a smaller Ne. It was shown that the prediction accuracy (AUC and ORs) increased with a design of a smaller Ne, compared to that with a larger Ne, using theory, simulations and real data analyses. Note that the term ‘effective population size’ is used here not as a property of the population at large, but rather the term refers to the effective information that arises from the relatedness between the discovery set and the subject(s) to be predicted.
The utility of genomic prediction was illustrated with an example where the top percentile of the estimated genetic profile scores had a substantially higher proportion of cases than a random population sample (23-fold) especially when using a design that includes closer relatives, which effectively leads to a smaller Ne, and even when using a moderate sample size in the discovery set (N = 3,000) and a heritability of 0.5 (Fig. 4). This could be increased to 32-fold with a larger sample size (N = 24,000), or 176-fold with a higher heritability (h2 = 0.8) (Supplementary Table 4). This demonstrates that relatives (i.e. smaller Ne) are a valuable resource for genomic prediction that can be used in stratified medicine. This is an important implication because currently the information on relatives is often discarded in predicting human complex traits and diseases based on genome-wide SNP data.
Even for a data set of unrelated individuals based on a random population sample, such as the case in the GERA data set, when using the discovery individuals that are more related to the target individuals, the genomic prediction accuracy increased (Figs 6 and 7 and Supplementary Figures 13 and 14), because of the larger variance of pair-wise relationships to the target sample (implying lower Me and Ne). This may have important implications when only considering population-based samples in genomic risk prediction for human complex traits and diseases where higher relationships are subject to be excluded.
One challenge with this approach is that a large number of records or samples needs to be collected within a local community or from extended families. However, increasingly databases are built with phenotypic and genotypic information from closer relatives28. In practice, a composite discovery population combining population- and family-based samples may be an alternative and desirable design, as demonstrated here for the Framingham study as well as in other studies29,30. In fact, personalised medicine based on family-based databases are in line with the very concept of family medicine31,32,33,34,35.
In many previous studies, it was observed that family-based estimates are considerably higher than population-based estimates of heritability23,25,36. There are plausible explanations for this phenomenon, including inflation due to family effects, gene-gene (G × G) or gene-environment interactions (G × E)17,18,19,37, or imperfect linkage disequilibrium (LD)11 and this has led to many studies discarding information from more closely related individuals. However, the theory and simulation in this study have clearly shown that even in absence of these inflatory effects there was a substantial increase in prediction accuracy (Figs 1, 2, 3 and 4), suggesting that information from close relatives should not be discarded. The results from real data showed that designing the discovery data set to include individuals that are closely related to those in the target sample could give substantially higher prediction accuracy for the target sample (Table 1 and Fig. 5). In the real data, this is unlikely to be driven by population stratification, as 10 PCs were included in the analysis model (see Methods). However, it is not impossible that non-additive genetic effects could contribute to the increase in prediction accuracy (Table 1 and Supplementary Table 5), but one could argue that this is not unwarranted when predicting individual risk. A further study about the possible role of non-additive genetic factors, and whether they can be estimated separately, may be needed.
In the near future, hundreds of thousands or more records on genotype and phenotype of people will be available for a reference sample to predict genetic risk for a target individual, e.g. all newborn babies could be genotyped and there are improved data bases for recording phenotypes, consisting of one large reference panel that can be generally applied to a nation-wide genomic prediction. Using eqs (1) and (12), we show that either adding more relatives or more genetically distant individuals increased the prediction accuracy substantially (Supplementary Table 4 and Supplementary Figure 19). The number of relatives required to get the same high accuracy is much lower than that of distant individuals, implying that the information from relatives is of much higher value in genomic prediction.
Practically, the genotypic and phenotypic information of a subject’s relatives (including parents, siblings, cousins and more distant relatives) can be used effectively as a part of the unified reference panel that also include a large number of individuals that are not related to the predicted subject to improve the accuracy further as illustrated in Supplementary Figure 19 (B). An optimal design should consist of both close relatives and unrelated individuals, e.g. a composite design, to maximise the prediction accuracy. That is, the composite design takes advantage of effective information from smaller number of relatives while it also use information from a greater number of unrelated individuals38.
When using case-control data by selecting 10% of the highest phenotypes, the estimated Me was slightly reduced (Table 1). Specifically, Me was diminished from 4,434 to 3,247 with a smaller Ne and from 31,080 to 29,479 with a larger Ne. This would be expected because selection on the heritable traits might lower Ne39, therefore Me was therefore decreased.
In this study, we present a theoretical framework, simulation and real data analyses to demonstrate that prediction accuracy can be improved by targeting more informative individuals in a discovery set with closer relationships with the subjects, making prediction more similar to those in populations with small effective size (Ne). This work is also extended to case-control data analyses so that the outcomes of this study are applicable to a clinical program for human diseases. It is argued that there is considerable room to increase prediction accuracy for polygenic phenotypes so that genomic prediction can be useful for clinical applications in the near future.
Methods
Accuracy of genomic prediction
Genomic prediction uses phenotypes alongside genome-wide SNPs or sequence data to estimate the effects of observed variants that are projected onto independent subjects and to estimate the subjects’ individual genetic profile scores (i.e. breeding values in the context of animal and plant breeding). The accuracy of the genomic prediction depends on the captured genetic variance as a proportion of the total variance, the number of phenotypic observations and the number of independent genomic regions expressed as the effective number of chromosome segments20,21,22,40, that is

where  is the correlation coefficient between the true and estimated genetic profile scores, h2 is the heritability of the trait, Me is the effective number of chromosome segments, N is the number of phenotypic observations and b is the proportion of genetic variance captured by observed variants (e.g. SNPs) that can be written as20,21,22
is the correlation coefficient between the true and estimated genetic profile scores, h2 is the heritability of the trait, Me is the effective number of chromosome segments, N is the number of phenotypic observations and b is the proportion of genetic variance captured by observed variants (e.g. SNPs) that can be written as20,21,22

where M is the number of observed variants. Owing to dense SNP genotypes or sequence data being available, b is often close to unity. Therefore, with dense markers, the genomic prediction accuracy can be simplified as

The correlation coefficient between phenotypes and estimated genetic profile scores in a target data set is

When using case-control studies for human diseases, the correlation coefficient between true and estimated genetic profile scores can be written as41,42

where u is a genetic profile score on the 0, 1 disease scale41,43, K is the population prevalence for the disease, P is the proportion of cases in the total sample N of cases and controls, and z is the density at the threshold on the normal distribution in the liability threshold model. The AUC as a measure of the accuracy of genomic prediction in a target data set for case-control studies is44,45

where i is the mean liability for cases, i2 is the mean liability for controls, t is the threshold on the normal distribution that truncates the proportion of disease prevalence K and Φ is the cumulative density function of the normal distribution. Another approach to assess the predictive utility of a continuous risk score of diseases, which is common in epidemiology studies, is to stratify individuals into percentiles according to ranked values of the genetic profile scores and estimate the odds ratio of case-control status by contrasting the top percentile with the bottom percentile5, that is

where the probability of being a case in the top/bottom percentile is

and

where itop and ibottom are the mean genetic profile scores for the top and bottom percentiles, respectively, ttop and tbottom are the thresholds on the normal distribution that truncates the proportion of the top and bottom percentiles (for detailed derivation, see Supplementary Note).
In more general terms, one can obtain the odds ratio of case-control status by contrasting the top percentile against the general population, that is

Effective number of chromosome segments
The effective number of chromosome segments (Me) is a key parameter for determining the accuracy of the genomic prediction as fewer segments require fewer independent parameters to be estimated from the same data, i.e. a higher accuracy. Me depends on the effective population size (Ne) and the length of genomic region (L)20,21,22. There are several studies that derive Me based on population parameters but there are some inconsistencies between these20,21,22. We revisit the theory and provide another derivation of Me as a function of Ne and L, using the theory of a squared correlation matrix between all SNPs46.
Considering a genomic region spanning 1 Morgan with M SNPs that are equally distributed over the region, one can construct an M × M squared correlation matrix S in which the elements are the squared correlation coefficients between each pair of SNPs (r2)46. The squared correlation coefficients can be approximated as r2 = 1/(1 + 4Ne × c) where Ne is the effective population size and c is the distance in Morgan between each pair of SNPs under a standard neutral model without mutation47. Unless the off-diagonal elements in S are all zero, the effective number of SNPs (or chromosome segments) is less than M. In order to obtain the effective number of SNPs, each SNP can be weighted and the weights can be obtained as46

and

where w is an M vector of SNP weights derived from the correlation structure among the SNPs and e is a vector of length M with all elements equal to one. In fact, the effective number of chromosome segments is calculated from the sum of the SNP weights as

The underlying linear system of order M can be written as

where  is a correlation coefficient between the ith and jth SNPs in the matrix S. This linear system can be simplified as
is a correlation coefficient between the ith and jth SNPs in the matrix S. This linear system can be simplified as

where  .
.
As the covariance between t and w is usually small, it can be approximated as

From eq. (6), the term  can be replaced with Me, resulting in
can be replaced with Me, resulting in

This agrees with Goddard (2009)20 who derived this same expression from the covariance statistic between two linked variants while we derived it from the SNP squared correlation matrix theory46.
It is noted that the pattern of the same values is repeated in the matrix S because of the SNPs being equally distributed. For example, the values for  are the same for all combinations for which |i−j| is the same, e.g.
are the same for all combinations for which |i−j| is the same, e.g.  . Therefore, the sum in eq. (7) can be written as
. Therefore, the sum in eq. (7) can be written as

where the first part of each term refers to the estimated r2 based on the distance, and the second part refers to the frequency of SNP pairs with such an r2 value. When scaling the equation by M2, this can be rewritten as

For the right hand side with M approaching infinity, the expression can be approximately transformed to a function of x with infinity data points ranging from 0 to 1, which can be written as

The sum of the function f(x) in the variable x ranging from 0 to 1 is defined by an integration. Therefore the denominator in eq. (7),  can be obtained as
can be obtained as

It is straightforward to extend this formula to a genomic region with length L rather than 1 Morgan (see Supplementary Note). For an L Morgan region, this is

Therefore,

When accounting for mutation48, therefore using the correlation coefficients between SNPs from the formula 1/(2 + 4Ne × c), eq. (8) is slightly modified to

The equivalence between eqs (6) and (7), and the approach linking eqs (7) and (8) (or (7) and (9)) were validated with actual analyses of the squared correlation matrix (Supplementary Tables 1 and 2).
Equations (8) and (9) are analytically confirmed and agreed well with eq. (7) (Supplementary Tables 1 and 2) and improved from the previous derivations20,21,22 (Supplementary Table 3). Difference became more remarkable when using more chromosomes. It is noted that a genomic length of 30 Morgan is more realistic (Supplementary Table 3). Previous studies20,21,22 ignored the correlation between chromosomes, however this is not negligible. Following Goddard (2009)20 but based on the individual level (rather than the gametic level), the probability of a random pair of individuals having the same parents (i.e. full sibs) in the last generation is (2/Ne)2 and that of having one parent in common (i.e. half sibs) is 4/Ne − (2/Ne)2. This generates a variance of the relationships among the pairs, which can be analytically approximated as 1/(4Ne). For the previous generations, the variance is 1/(16Ne), 1/(32Ne), 1/(64Ne) and so on. Summing all these variances gives 1/(3Ne), therefore, the covariance of the pairwise relationships between two chromosomes is 1/(3Ne).
Assuming there are multiple chromosomes having an equal genomic length, the chromosome covariance matrix (with order equal to the number of chromosomes) can be written as

where the diagonal (1/Me) can be obtained from eqs (8) or (9) and the off-diagonal (1/(3Ne)) is the covariance of the pairwise relationships between two chromosomes. As in eq. (7), the inverse of the overall Meacross the multiple chromosomes can be obtained such that all elements in the covariance matrix should be averaged. Hence, eqs (8) and (9) for multiple chromosomes can be expressed as

and when considering historical mutations,

where Nchr is the number of chromosomes each with length L.
Me from the genomic relationship matrix
Me can be empirically obtained when a GRM is given21, which can be interpreted as an observed Me from the genotype data on which the GRM is based, which can be written as21

where Aij is the genomic relationship between individual i from the target and j from the discovery sample. More details are in the Supplementary Note.
Estimated genetic profile scores
The MTG2 software9,49 was applied to a discovery data set to estimate SNP effects jointly. The estimated SNP effects were projected onto the target samples, resulting in a genomic best linear unbiased prediction (GBLUP)50 of the genetic profile score for each target individual in the target data set. Dudbridge (2013)42 showed that the standard genetic profile score method8,51 and GBLUP have the same power and accuracy using theory that assumed all causal variants are unlinked and observed. However, in real situations where there are complex LD structures among SNPs, GBLUP is a preferred method, therefore, we used GBLUP in this study.
Simulation I
Equations (10) and (11) were validated with a stochastic coalescence simulation and genomic prediction approach. A stochastic gene-dropping method52,53 was used to simulate 20,000 SNPs across a single chromosome of L = 1 Morgan with Ne = 500, 1000, 2000 and 4000 for 500, 1000, 2000 and 4000 generations, respectively. Recombinations occurred across the genomic region according to the genetic distance between SNPs that were equally distributed across the region. The mutation rate was 1e-08 per site per generation54. Random mating and random selection were used according to the standard genetic drift model55. In the final generation, as a discovery data set, we generated 2000 or 5000 individuals having genotype data for 10,000 causal SNPs, a subset of the 20,000 SNPs, of which the minor allele frequency was more than 1%. For the discovery data set, phenotypes were simulated such that the variance explained by the SNPs was 1% of the total phenotypic variance where SNP and residual effects were drawn from normal distributions. For the target data set, another set of 1000 or 2500 individuals was chosen to estimate the observed accuracy of genomic prediction, i.e. the correlation between true and estimated genetic profile scores. We also conducted simulations of 5 chromosomes each being L = 1 Morgan long, with a total number of 50, 000 SNPs, resulting in variance explained by the SNPs being 5% of the total phenotypic variance.
Using the genotype data of the discovery data set, a GRM was constructed and Me was estimated from eq. (12) as the observed Me from the simulated data. We used eqs (10) and (11) to get the expected Me given Ne and L. The observed and expected Me values were compared. In addition, the expected accuracy of the genomic prediction was obtained from eq. (1) using the observed Me, which was compared to the correlation (as the observed accuracy) between true and estimated genetic profile scores (GBLUP) in the target data set.
Simulation II
In order to confirm the theory in deriving AUC and odds ratios (eqs (3), (4) and (5)), we simulated disease data (binary responses) using a liability threshold model with a population prevalence of 10% (K = 0.1). In the discovery data set, cases were over-sampled such that the ratio of cases and controls was equal (P = 0.5), mimicking a typical case-control design. The total number of samples in the discovery set was N = 3000. We simulated Me independent SNPs, the effects of which were normally distributed, and a residual effect such that the heritability on the liability scale was h2 = 0.5. We used 5 different values for Me = 254, 1188, 4506, 10891 and 21248, reflecting the expected values for Ne = 100, 500, 2000, 5000 and 10000, respectively when using a genomic length of 30 Morgan (30 chromosomes each with 1 Morgan long) and the coalescence formula 1/(2 + 4Ne × c) (eq. (9)). SNP effects were estimated in the discovery data set and these estimates were used to predict genetic profile scores in an independent population sample of N = 30000 as the target data set. For the target sample, we used a large population sample to reduce empirical sampling error and mimic a realistic situation, e.g. screening newborn babies. We obtained AUC from the genetic profile scores predicting the disease status in the target data set. Additionally, we obtained the odds ratio contrasting the top and bottom 20% of the normal population sample according to the genetic profile scores. We also obtained the odds ratio contrasting the top 1% according to the genetic profile scores and the normal population. These observed AUC and odds ratios from the simulated data were compared to the expected values from the theory (eqs (3), (4) and (5)).
Real data
Framingham heart study
Publicly available data from the Framingham heart study (phs000007.v26.p10.c1)56 were used. Stringent quality control (QC) was applied to the available genotypes, including SNP call rate > 0.95, individual call rate > 0.95, HWE p-value > 0.0001, MAF > 0.01 and individual population outliers < 6 SD from the first and second principal components (PC). After QC, 6920 individuals and 389,265 SNPs remained. Among them, 4243 individuals from 628 families were phenotyped for height and body mass index (BMI). The mean number of members per family was 6.76 (SD 12.77). Phenotypes were adjusted for birth year, sex, and the first 10 PCs. We calculated the ancestry PCs from the POPRES reference sample57,58,59 because direct PC analysis on the sample could be confounded with family structure58,60.
In order to contrast designs with smaller and larger Ne(and Me) values, two kinds of cross-validation schemes were implemented. For a design with larger Ne, 80% of the 628 families were selected as the discovery data set, with the remaining 20% of families used as the target data set. Therefore, the discovery and target sample shared distant common ancestors, hence a larger Ne and Me. In contrast, for a design with smaller Ne, each member in every family was assigned an 80% chance to be a discovery sample and the rest was in the target sample. Therefore, the discovery and target sample shared recent common ancestors, hence a smaller Ne and Me.
Using the real genotype data, the genomic relationships between the discovery and target sample were constructed, and Me was estimated from eq. (12). The correlation between the phenotypes (that were not used in the analyses) and estimated genetic profile scores in the target data set was estimated.
Genetic epidemiology research on adult health and aging cohort
As a second real data set, we used genetic data from the European ancestry participants of the Kaiser Permanente genetic epidemiology research on adult health and aging (GERA) cohort (phs000674.v1.p1)61, an extensive population sample. The same QC process was applied to the available genotypes. After QC, 62,318 individuals each with 575,760 SNP genotypes remained. We used the trait “hypertension” and “dyslipidemia” for the prediction analyses. Phenotypes were adjusted for birth year, sex, and the first 10 PCs that were inferred from the POPRES reference sample57,58,59.
Unlike the Framingham data, GERA does not have an explicit family structure, i.e. the proportion of pair-wise relationship more than 0.2 was only 0.0002%. Therefore, the family-wise cross-validation schemes used in the Framingham data could not be used. Instead, we randomly selected 46,000 individuals, and randomly assigned 80% and 20% to a discovery data set (N = 36,800) and a target data set (N = 9,200), respectively, in 100 cross-validation replicates. We calculated the variance of pair-wise relationships with the individuals in the discovery data set for each individual in the target data set, and identified the top 25% of the target individuals with the highest variance of the relationships. Then, we tested if the prediction accuracy for the top group (N = 2,300) was higher than that for the entire target sample, to show if a larger variance, hence smaller Me, resulted in a higher prediction accuracy even when using a population-based sample without a substantial family structure. In addition, we obtained the prediction accuracy from a subset of the sample that excluded higher relationships (>0.025). We first applied the relatedness cut-off to all individuals, and then selected 46,000 individuals that were subsequently divided into the discovery (N = 36,800) and target data sets (N = 9,200). It is noted that for each target individual, the variance of pair-wise relationships (eq. (12)) with the discovery individuals was reduced due to the relatedness cut-off. In any case, we used the same number of discovery samples (N = 36,800) in order to have the same power and for fair comparisons.
Data access
We used publicly available data. Accession codes are in the following. The Framingham heart study (phs000007.v26.p10.c1). The European ancestry participants of the Kaiser Permanente genetic epidemiology research on adult health and aging (GERA) cohort (phs000674.v1.p1).
Software
Theory, simulation models and GBLUP used in this study have been fully implemented in publicly available software, MTG2. The source code, executive binary file, manual and examples are readily available to use, and can be downloaded from https://sites.google.com/site/honglee0707/mtg2.
Additional Information
How to cite this article: Lee, S. H. et al. Using information of relatives in genomic prediction to apply effective stratified medicine. Sci. Rep. 7, 42091; doi: 10.1038/srep42091 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Kapur, S., Phillips, A. G. & Insel, T. R. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol Psychiatry 17, 1174–1179 (2012).
Wei, Z. et al. From Disease Association to Risk Assessment: An Optimistic View from Genome-Wide Association Studies on Type 1 Diabetes. PLOS Genet 5, e1000678 (2009).
Abraham, G. et al. Accurate and Robust Genomic Prediction of Celiac Disease Using Statistical Learning. PLoS Genet 10, e1004137 (2014).
Wei, Z. et al. Large Sample Size, Wide Variant Spectrum, and Advanced Machine-Learning Technique Boost Risk Prediction for Inflammatory Bowel Disease. American Journal of Human Genetics 92, 1008–1012 (2013).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Zhou, X., Carbonetto, P. & Stephens, M. Polygenic Modeling with Bayesian Sparse Linear Mixed Models. PLOS Genet 9, e1003264 (2013).
Moser, G. et al. Simultaneous discovery, estimation and prediction analysis of complex traits using a Bayesian mixture model. PLoS Genet 11, e1004969 (2015).
Purcell, S. M. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009).
Maier, R. et al. Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder and major depression disorder. Am J Hum Genet 96, 283–294 (2015).
Tucker, G. et al. Two-Variance-Component Model Improves Genetic Prediction in Family Datasets. The American Journal of Human Genetics 97, 677–690 (2015).
de los Campos, G., Vazquez, A. I., Fernando, R., Klimentidis, Y. C. & Sorensen, D. Prediction of Complex Human Traits Using the Genomic Best Linear Unbiased Predictor. PLoS Genet 9, e1003608 (2013).
Makowsky, R. et al. Beyond Missing Heritability: Prediction of Complex Traits. PLoS Genet 7, e1002051 (2011).
Aulchenko, Y. S. et al. Predicting human height by Victorian and genomic methods. Eur J Hum Genet 17, 1070–5 (2009).
Lee, S., van der Werf, J., Hayes, B., Goddard, M. & Visscher, P. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLOS Genet 4, e1000231 (2008).
Clark, S. A., Hickey, J. M., Daetwyler, H. D. & van der Werf, J. H. The importance of information on relatives for the prediction of genomic breeding values and the implications for the makeup of reference data sets in livestock breeding schemes. Genet Sel Evol 44, 4 (2012).
Legarra, A., Robert-Granie, C., Manfredi, E. & Elsen, J. M. Performance of genomic selection in mice. Genetics 180, 611–8 (2008).
Manolio, T. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Gibson, G. Rare and Common Variants: Twenty arguments. Nature reviews. Genetics 13, 135–145 (2011).
Zuk, O., Hechter, E., Sunyaev, S. R. & Lander, E. S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci USA 109, 1193–8 (2012).
Goddard, M. E. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136, 245–257 (2009).
Goddard, M. E., Hayes, B. J. & Meuwissen, T. H. E. Using the genomic relationship matrix to predict the accuracy of genomic selection. Journal of Animal Breeding and Genetics 128, 409–421 (2011).
Meuwissen, T., Hayes, B. & Goddard, M. Accelerating Improvement of Livestock with Genomic Selection. Annual Review of Animal Biosciences 1, 221–237 (2013).
Silventoinen, K. et al. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res 6, 399–408 (2003).
Macgregor, S., Cornes, B. K., Martin, N. G. & Visscher, P. M. Bias, precision and heritability of self-reported and clinically measured height in Australian twins. Hum Genet 120, 571–80 (2006).
Visscher, P. M. et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2, e41 (2006).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42, 565–569 (2010).
Yang, J. et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet 43, 519–525 (2011).
Ott, J., Kamatani, Y. & Lathrop, M. Family-based designs for genome-wide association studies. Nat Rev Genet 12, 465–474 (2011).
MacInnis, R. J. et al. A risk prediction algorithm based on family history and common genetic variants: application to prostate cancer with potential clinical impact. Genetic Epidemiology 35, 549–556 (2011).
Do, C. B., Hinds, D. A., Francke, U. & Eriksson, N. Comparison of Family History and SNPs for Predicting Risk of Complex Disease. PLoS Genet 8, e1002973 (2012).
Rich, E. C. et al. Reconsidering the Family History in Primary Care. Journal of General Internal Medicine 19, 273–280 (2004).
Future of Family Medicine Project Leadership Committee. The Future of Family Medicine: A Collaborative Project of the Family Medicine Community. The Annals of Family Medicine 2, S3–S32 (2004).
Khoury, M. J., Feero, W. G. & Valdez, R. Family History and Personal Genomics As Tools for Improving Health in an Era of Evidence-Based Medicine. American Journal of Preventive Medicine 39, 184–188 (2010).
Yoon, P. W. et al. Can family history be used as a tool for public health and preventive medicine? Genet Med 4, 304–310 (2002).
Feero, W. G., Guttmacher, A. E. & Collins, F. S. Genomic Medicine—An Updated Primer. New England Journal of Medicine 362, 2001–2011 (2010).
van Dongen, J., Slagboom, P. E., Draisma, H. H. M., Martin, N. G. & Boomsma, D. I. The continuing value of twin studies in the omics era. Nat Rev Genet 13, 640–653 (2012).
Clarke, A. J. & Cooper, D. N. GWAS: heritability missing in action? European Journal of Human Genetics 18, 859–861 (2010).
Pszczola, M., Strabel, T., Mulder, H. A. & Calus, M. P. Reliability of direct genomic values for animals with different relationships within and to the reference population. J Dairy Sci 95, 389–400 (2012).
Robertson, A. Inbreeding in artificial selection programmes. Genetics Research 2, 189–194 (1961).
Daetwyler, H. D., Villanueva, B. & Woolliams, J. A. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLOS ONE 3, e3395 (2008).
Lee, S. H. & Wray, N. R. Novel genetic analysis for case-control genome-wide association studies: quantification of power and genomic prediction accuracy. PLOS ONE 8, e71494 (2013).
Dudbridge, F. Power and Predictive Accuracy of Polygenic Risk Scores. PLOS Genet 9, e1003348 (2013).
Lee, S. H., Wray, N. R., Goddard, M. E. & Visscher, P. M. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet 88, 294–305 (2011).
Wray, N. R., Yang, J., Goddard, M. E. & Visscher, P. M. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet 6, e1000864 (2010).
Lee, S. H., Goddard, M. E., Wray, N. R. & Visscher, P. M. A better coefficient of determination for genetic profile analysis. Genet Epidemiol 36, 214–224 (2012).
Speed, D., Hemani, G., Johnson, M. R. & Balding, D. J. Improved Heritability Estimation from Genome-wide SNPs. Am J Hum Genet 91, 1011–1021 (2012).
Sved, J. A. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol 2, 125–141 (1971).
Tenesa, A. et al. Recent human effective population size estimated from linkage disequilibrium. Genome Research 17, 520–526 (2007).
Lee, S. H. & van der Werf, J. H. J. MTG2: An efficient algorithm for multivariate linear mixed model analysis based on genomic information. Bioinformatics 32, 1420–1422 (2016).
Meuwissen, T. H. E., Hayes, B. J. & Goddard, M. E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 (2001).
Wray, N. R. et al. Research Review: Polygenic methods and their application to psychiatric traits. Journal of Child Psychology and Psychiatry 55, 1068–1087 (2014).
MacCluer, J. W., VandeBerg, J. L., Read, B. & Ryder, O. A. Pedigree analysis by computer simulation. Zoo Biology 5, 147–160 (1986).
Lee, S. & van der Werf, J. The efficiency of designs for fine-mapping of quantitative trait loci using combined linkage disequilibrium and linkage. Genetics Selection Evolution 36, 145–161 (2004).
Roach, J. C. et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science 328, 636–639 (2010).
Masel, J. Genetic drift. Current Biology 21, R837–R838 (2011).
Splansky, G. L. et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 165, 1328–35 (2007).
Nelson, M. R. et al. The Population Reference Sample, POPRES: A Resource for Population, Disease, and Pharmacological Genetics Research. Am J Hum Genet 83, 347–358 (2008).
Chen, C.-Y. et al. Improved ancestry inference using weights from external reference panels. Bioinformatics 29, 1399–1406 (2013).
Chen, G.-B., Lee, S. H., Zhu, Z.-X., Benyamin, B. & Robinson, M. R. EigenGWAS: finding loci under selection through genome-wide association studies of eigenvectors in structured populations. Heredity, doi: 10.1038/hdy.2016.25 (2016).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–909 (2006).
Banda, Y. et al. Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics 200, 1285–1295 (2015).
Acknowledgements
This research is supported by the Australian National Health and Medical Research Council (1080157, 1087889, 1078901), the Australian Research Council (DP160102126, FT160100229) and the Australian Sheep Industry Cooperative Research Centre. We thank Prof. Peter M. Visscher for helpful discussion in general and his valuable contribution in deriving the theory of odds ratio of contrasting case-control status in percentile analyses. The Framingham Heart Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195). This manuscript was not prepared in collaboration with investigators of the Framingham Heart Study and does not necessarily reflect the opinions or views of the Framingham Heart Study, Boston University, or NHLBI. Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02‐HL‐64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. GERA data came from a grant, the Resource for Genetic Epidemiology Research in Adult Health and Aging (RC2 AG033067; Schaefer and Risch, PIs) awarded to the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and the UCSF Institute for Human Genetics. The RPGEH was supported by grants from the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation, Kaiser Permanente Northern California, and the Kaiser Permanente National and Northern California Community Benefit Programs. The RPGEH and the Resource for Genetic Epidemiology Research in Adult Health and Aging are described in the following publication, Schaefer C., et al., The Kaiser Permanente Research Program on Genes, Environment and Health: Development of a Research Resource in a Multi-Ethnic Health Plan with Electronic Medical Records, In preparation, 2013.
Author information
Authors and Affiliations
Contributions
S.H.L. and J.H.J.W. conceived the idea. J.H.J.W. and M.E.G. provided key elements in deriving theory. S.H.L. and J.H.J.W. derived formulas. S.H.L. and W.M.S.P.W. performed the analyses. S.H.L., J.H.J.W. and N.R.W. drafted the manuscript. M.E.G. and N.R.W. provided critical feedback. All authors contributed to editing and approval of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lee, S., Weerasinghe, W., Wray, N. et al. Using information of relatives in genomic prediction to apply effective stratified medicine. Sci Rep 7, 42091 (2017). https://doi.org/10.1038/srep42091
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42091
This article is cited by
-
Genome-wide fine-mapping identifies pleiotropic and functional variants that predict many traits across global cattle populations
Nature Communications (2021)
-
An integrative analysis of genomic and exposomic data for complex traits and phenotypic prediction
Scientific Reports (2021)
-
Maximizing efficiency of genomic selection in CIMMYT’s tropical maize breeding program
Theoretical and Applied Genetics (2021)
-
A deterministic equation to predict the accuracy of multi-population genomic prediction with multiple genomic relationship matrices
Genetics Selection Evolution (2020)
-
Efficient polygenic risk scores for biobank scale data by exploiting phenotypes from inferred relatives
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
