
Abstract
Ubiquitous expression of mutant Cu/Zn-superoxide dismutase (SOD1) selectively affects motor neurons in the central nervous system (CNS), causing the adult-onset degenerative disease amyotrophic lateral sclerosis (ALS). The CNS-specific impact of ubiquitous mutant SOD1 expression is recapitulated in transgenic mouse models of the disease. Here we present outcomes for the metallo-complex CuII(atsm) tested for therapeutic efficacy in mice expressing SOD1G93A on a mixed genetic background. Oral administration of CuII(atsm) delayed the onset of neurological symptoms, improved locomotive capacity and extended overall survival. Although the ALS-like phenotype of SOD1G93A mice is instigated by expression of the mutant SOD1, we show the improved phenotype of the CuII(atsm)-treated animals involves an increase in mature mutant SOD1 protein in the disease-affected spinal cord, where concomitant increases in copper and SOD1 activity are also evident. In contrast to these effects in the spinal cord, treating with CuII(atsm) had no effect in liver on either mutant SOD1 protein levels or its activity, indicating a CNS-selective SOD1 response to the drug. These data provide support for CuII(atsm) as a treatment option for ALS as well as insight to the CNS-selective effects of mutant SOD1.
Similar content being viewed by others
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset disease in which motor neurons of the central nervous system (CNS) progressively deteriorate. Initial symptoms are relatively innocuous (e.g. weakness in a hand or slurred speech), but inevitably and relentlessly they escalate. People with ALS become paralysed, lose the ability to breathe, speak and swallow, and due to the absence of an effective treatment most will die within 5 years of diagnosis. The majority of ALS cases are sporadic but approximately 10% are familial and the heritable basis has been ascribed to mutations in over 20 different genes1.
Mutations in the copper-dependent antioxidant Cu/Zn-superoxide dismutase (SOD1) were the first described genetic cause of ALS2. Since the development of transgenic mice expressing human SOD1 containing ALS-causing substitution mutations3,4, these mouse models have provided a robust experimental approach to study ALS pathogenesis and progression, as well as opportunity to test new therapeutics in a system that entails basic yet clinically significant features (e.g. a mammalian blood-brain barrier and an adult-onset progressive phenotype). Moreover, mutant SOD1 expressing rodents also recapitulate a salient feature of clinical cases of ALS caused by SOD1 mutations; even though the mutant SOD1 is expressed ubiquitously and persistently from birth, the ALS-like phenotype only presents relatively late in the animals’ life and is the result of selective degeneration of motor neurons in the CNS3,4. Thus, mutant SOD1-expressing rodents provide opportunity to better understand why a ubiquitously expressed ALS-causing mutation selectively affects the CNS.
In the present study we used transgenic mice expressing human SOD1G93A on a mixed genetic background to assess the therapeutic effects of the metallo-compound CuII(atsm) and to partly investigate how the therapeutic activity of CuII(atsm) may be related to the CNS-selective effects of mutant SOD1 expression. CuII(atsm) – diacetylbis(N(4)-methylthiosemicarbazonato)-CuII – is a CuII complex of a bis(thiosemicarbazone) ligand5 which has been investigated as a potential therapeutic in animal models of ALS and Parkinson’s disease6,7,8,9,10 and as a PET imaging agent in the clinic for neurological11,12,13 and non-neurological conditions14. It is a low molecular weight compound (MW = 321) that is stable (KA = 1018) and able to cross the blood-brain barrier15. But despite the compound’s stability, an assessment of SOD1G37R mice revealed that approximately 50% of total SOD1 in the spinal cords of these mice exists in a Cu-deficient state, and diminution of this pool following oral administration of CuII(atsm) was shown to involve in vivo transfer of Cu from the compound to the Cu-deficient SOD1 in the affected spinal cord8. Transfer of Cu from CuII(atsm) to mutant SOD1 was ascribed to at least part of the compound’s therapeutic activity8 and this was supported by a subsequent study in which the compound was administered to alternate mutant SOD1 mouse models of ALS10.
Thus, biochemical and therapeutic outcomes for CuII(atsm) indicate the compound’s ability to improve Cu bioavailability to SOD1 may contribute, at least in part, to its therapeutic activity in mutant SOD1 mouse models of ALS. Recently, it was demonstrated that the bioavailability of endogenous Cu, but not Zn, is a limiting factor with respect to satiating the elevated requirement for Cu and Zn that is driven by SOD1 over-expression in SOD1G37R mice16. Significantly, despite ubiquitous expression of mutant SOD1 in these mice, the insufficient availability of endogenous Cu to SOD1 in these mice is only evident in the CNS16. In light of this, and given that the therapeutic activity of CuII(atsm) appears to involve the modulation of Cu bioavailability in vivo8,10, the present study was undertaken to assess whether CuII(atsm) may increase Cu bioavailability to SOD1 in peripheral tissues or only tissues from the CNS. To assess this in the context of the compound’s therapeutic activity, representative CNS (spinal cord) and non-CNS (liver) tissues were collected from SOD1G93A mice in which treating with CuII(atsm) translated into a robust therapeutic effect.
Results
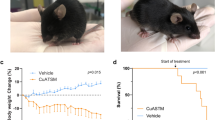
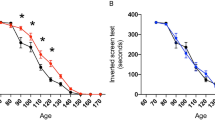
Litter- and gender-matched SOD1G93A mice on a mixed genetic background were treated daily with CuII(atsm) or sham control from the age of 50 days. Twice weekly assessment on the rotarod test revealed a sharp decline in locomotive function commencing when the mice were around 100 days old (Fig. 1A). This decline was delayed in mice that were treated with CuII(atsm), with the treatment effect attaining statistical significance at 113 days then persisting for the remainder of the study period. An alternate assessment of neurological function17 provided a comparable outcome; the neurological phenotype of the SOD1G93A mice noticeably and progressively worsened from around 100 days but treating with CuII(atsm) delayed the phenotype (Fig. 1B). The latter of these two methods for assessing phenotype progression revealed that the CuII(atsm) treatment delayed neurological symptom onset under the present experimental conditions by 9 days (Fig. 1C).

(A) Performance of sham- and CuII(atsm)-treated SOD1G93A mice on the rotarod test for locomotive function and (B) assessment for neurological symptoms via Neurological Score17. (C) Age of symptom onset defined as an individual mouse attaining a score of 1 in the Neurological Score system17. (D) Survival to phenotype end-point curves for sham- and CuII(atsm)-treated SOD1G93A mice and (E) box and whisker plots showing the overall treatment effect on survival. (F) Relationship between total daily dose of CuII(atsm) and percentage increase in mean survival. Data for the 100 mg dose are calculated from experiment presented in (D) (mean = 143 days, n = 24), data for the 200 mg dose calculated from experiments in Williams et al.10 (mean = 155 days, n = 20), and data for the 0 mg dose calculated across the two studies (mean = 131 days, n = 44). Solid lines in (A and B) are mean values. Grey dashed lines in (A,B and D) represent SEM. Data in (C,E and F) are presented as box (median ± 95% CI) and whisker (maximum and minimum) plots. P values in (A and B) represent statistical significance of the treatment effect (repeat measures ANOVA), whereas grey shaded boxes indicate periods for statistically significant differences between mean values for sham- and CuII(atsm)-treated mice (Sidak’s multiple comparisons test). P values in (C and E) indicate a statistically significant difference between mean values for sham- and CuII(atsm)-treated mice (unpaired t-test). P value in (D) represents statistically significant treatment effect (Cox proportional hazards model). Percentage values in F represent mean increase in survival for each CuII(atsm) dose. For A-E, n = 23 sham-treated mice and n = 24 CuII(atsm)-treated mice (treatments administered twice daily by gavage with CuII(atsm) administered per dose at 50 mg kg−1 mouse body weight). Vertical dashed lines in A and B represent the age at which a separate cohort of mice was killed for biochemical analyses.
The improved neurological phenotype of SOD1G93A mice in response to the CuII(atsm) treatment translated to an improvement in overall survival to phenotypic end-point (Fig. 1D). Treating with CuII(atsm) increased median survival by 8% from 130 to 141 days and mean survival by 11% from 129 to 143 days (Fig. 1E). The comparable effect that CuII(atsm) had on delaying neurological onset and extending survival to phenotypic end-point equated to no change in the duration of symptom progression: on average, the period of symptom progression from onset to end-point was 16 days in the sham-treated mice and 15 days in the CuII(atsm)-treated mice (P = 0.85, two-tailed t-test). These results are consistent with a previous study in which ALS mice expressing SOD1 with the G37R mutation4 were treated orally with CuII(atsm)8 and a more recent study in which CuII(atsm) was administered to SOD1G93A mice via a transdermal route10. Significantly, despite fundamental differences in the route of administration and performing the experiments across two different colonies of mice at two different institutes, doubling the daily dose effectively doubled the extension in survival elicited by administering CuII(atsm) to SOD1G93A mice (Fig. 1F).
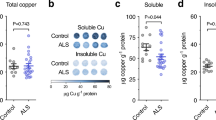
Assessing the influence of CuII(atsm) on levels of mutant SOD1 protein in spinal cord tissue from SOD1G93A mice at the mid-stages of symptom progression (indicated via vertical dashed lines in Fig. 1A and B) demonstrated that treating with CuII(atsm) increased levels of mutant SOD1 in the disease-affected CNS tissue (Fig. 2A). Catalytic activity of SOD1 is dependent upon the protein binding Cu18. Thus, we measured SOD1 activity in spinal cord extracts from sham- and CuII(atsm)-treated mice to assess whether the increase in mutant SOD1 protein in response to the CuII(atsm) translated to an increase in SOD1 activity. Reflecting overall differences in SOD1 protein levels between non-transgenic mice and the over-expressing SOD1G93A mice19, SOD1 activity was relatively low in extracts collected from non-transgenic mice and this was unchanged by the CuII(atsm) treatment (Fig. 2B). As a result of human SOD1 overexpression, and because the G93A mutation does not affect the enzyme’s dismutase activity3,20, SOD1 activity was relatively high in the spinal cords of the sham-treated SOD1G93A mice (Fig. 2B). This was further increased by the CuII(atsm) treatment (Fig. 2B). Moreover, analysing the Cu content of spinal cords supported outcomes from the SOD1G37R model8; elevated spinal cord Cu in CuII(atsm)-treated non-transgenic mice confirmed that oral administration of the compound affects Cu levels in the CNS, and the same dose administered to mice expressing mutant SOD1 elicits a greater response (Fig. 2C). In contrast to these effects in the spinal cord, administering CuII(atsm) to SOD1G93A mice had no influence on mutant SOD1 protein levels or activity in the liver (Fig. 2D,E), nor was there any statistically significant difference between non-transgenic and SOD1G93A mice with respect to liver Cu levels in response to the CuII(atsm) treatment (Fig. 2F, P = 0.99).

Relative abundance of mutant SOD1 protein in spinal cord (A) and liver (D) samples determined via western blot using an antibody that detects only human SOD1. Mutant SOD1 protein levels are expressed relative to the loading control GAPDH. Representative western blot images are shown. SOD1 activity in TBS-soluble extracts from mouse spinal cords (B) and livers (E) presented as pmol superoxide decay min−1 mg−1 tissue protein. The amount of Cu g−1 protein in spinal cord (C) and liver (F) tissue. Treatments were administered twice daily by gavage and commenced when the mice were 50 days old. CuII(atsm) administered per dose was 50 mg kg−1 mouse body weight. Mice were killed at 120 days old to collect tissues for analysis. Graphed data are box (median ± 95% CI) and whisker (maximum and minimum) plots and P value represents statistically significant treatment effect on mean values (unpaired t-test in (A and D) or one-way ANOVA with Tukey’s multiple comparisons test in (B,C,E and F)). NS = not statistically different. For all data shown, n = 6 mice per treatment group.
The increase in SOD1 activity in the spinal cords of CuII(atsm)-treated SOD1G93A mice (Fig. 2B) is supportive of reports which confirm the presence of a large pool of Cu-deficient SOD1 in the spinal cords of SOD1G93A and SOD1G37R mice8,10 and that in vivo transfer of Cu from CuII(atsm) to SOD1 can increase the concentration of Cu-containing SOD1, ergo its Cu-dependent dismutase activity8,10. The absence of any change to SOD1 activity in the livers of CuII(atsm)-treated SOD1G93A mice (Fig. 2E) by contrast, indicates that endogenous Cu bioavailability in the liver is able to meet the elevated requirement for Cu due to SOD1 over-expression and that SOD1 in the livers of the transgenic mice is therefore relatively Cu-replete (a possibility supported recently16), or that Cu delivered as CuII(atsm) does not become bioavailable to SOD1 in the liver. To partly interrogate these possibilities, we adopted a protocol in which Cu2+ ions are added to tissue extracts in order to assess whether SOD1 activity in the extracts is responsive to the available Cu21. Outcomes from this assay showed SOD1 activity in SOD1G93A mouse spinal cord extracts is increased by directly adding Cu2+ ions to the tissue extract (Fig. 3A) but activity in liver extracts from the same mice is not (Fig. 3B).

SOD1 activity in TBS-soluble extracts from SOD1G93A mouse spinal cords (A) and livers (B) presented as pmol superoxide decay min−1 mg−1 tissue protein. Tissue extracts were prepared from untreated SOD1G93A mice killed at 120 days old. All data are presented as box (median ± 95% CI) and whisker (maximum and minimum) plots and P values represent statistically significant differences between mean values for indicated groups (paired t-test). NS = not statistically different. For all data shown, n = 6 mice per treatment group.
Other important enzymes are dependent upon Cu for their catalytic activity, including cytochrome c oxidase, the terminal enzyme complex of the mitochondrial electron transfer chain. Consistent with a recent report10, cytochrome c oxidase activity is unaltered in the spinal cords of SOD1G93A mice and treating with CuII(atsm) has no detectable influence on its activity in these mice (Fig. 4A).

(A) Cytochrome c oxidase activity in non-transgenic and SOD1G93A mouse spinal cords presented as nmol cytochrome c oxidised min−1 mg−1 tissue protein. (B) Citrate synthase activity in non-transgenic and SOD1G93A mouse spinal cords presented as nmol DTNB reduced min−1 mg−1 tissue protein. Treatments were administered twice daily by gavage and commenced when the mice were 50 days old. CuII(atsm) administered per dose was 50 mg kg−1 mouse body weight. Mice were killed at 120 days old to collect tissues for analysis. Graphed data are box (median ± 95% CI) and whisker (maximum and minimum) plots. No statistically significant differences exist between any of the treatment groups (one-way ANOVA with Tukey’s multiple comparisons test). For all data shown, n = 6 mice per treatment group.
A multitude of dysfunctional pathways appear to contribute to symptom onset and progression in ALS. Considering that SOD1 activity is already higher in the spinal cords of the sham-treated mutant SOD1 mice due to over-expression of the transgene (Fig. 2B)3,4,8,16, and notwithstanding the presence of large pools of Cu-deficient and catalytically inactive SOD1, it is therefore unlikely that increasing SOD1 activity in the spinal cords of mutant SOD1 over-expressing mice per se is solely responsible for the CuII(atsm) induced improvement in the animals’ phenotype. Supporting this, our assessment of broad indications of spinal cord tissue health (oxidative damage, astrogliosis and motor neuron numbers) all demonstrated the beneficial effects of CuII(atsm) in the primary site of pathology in the SOD1G93A mice (Fig. 5).

(A) Quantitation of α-motor neurons per section in both ventral horn regions of spinal cord sections determined via cresyl violet staining. Only motor neurons with a diameter equivalent to 20 μm or greater were counted. (B) Abundance of oxidatively modified proteins determined using the OxyBlot assay in spinal cord tissue expressed relative to levels detected in sham-treated non-transgenic controls. Representative histology images for GFAP (C) and Iba-1 (D) immunoreactivity in spinal cord transverse sections. Data in (A and B) are presented as box (median ± 95% CI) and whisker (maximum and minimum) plots. P values represent statistically significant differences between mean values for indicated groups (one-way ANOVA with Tukey’s multiple comparisons test, n = 6 mice per treatment group). NS = not statistically different.
Discussion
Mutant SOD1 is a cause of familial ALS2 and transgenic mice expressing the mutant protein accurately recapitulate many features of the disease3,4. Significantly, this includes the onset of symptoms of motor neuron decline in adulthood, even though the causative mutation is expressed ubiquitously and persistently from birth. But to date, an unequivocal explanation for why ubiquitously expressed mutant SOD1 selectively affects the CNS in mice and humans has remained elusive.
In the present study, and in the context of the therapeutic agent CuII(atsm), we investigated the bioavailability of Cu as a potential contributing factor. The over-expression of mutant SOD1 in transgenic mice disrupts Cu homeostasis; some studies indicate increased levels of spinal cord Cu in multiple mutant SOD1 mouse models of ALS22 and the abundance of various Cu transporters and Cu chaperones is also altered22,23. Furthermore, the potential to improve the symptoms of ALS and protect motor neurons in the CNS via therapeutic strategies that modulate Cu bioavailability has already been demonstrated24; treating mutant SOD1 mice with Cu-chelating agents such as ammonium tetrathiomolybdate and D-penicillamine or with the Cu-delivery agent CuII(atsm) improves their neurological phenotype and extends survival7,8,9,10,22,25,26,27,28. Collectively these outcomes lend support to the notion that Cu bioavailability is an important factor in the ALS-like symptoms that develop in mutant SOD1 mice. Details of the deleterious mechanistic processes are yet to be elucidated, but the emerging consensus appears to be that disrupted Cu bioavailability, rather than Cu deficiency or Cu accumulation per se, is a primary feature of the neurodegenerative process.
This is consistent with some aspects of the potential SOD1 gain of function in ALS. SOD1 is a well-characterised metalloenzyme with a relative abundance of biochemical and biophysical information on its interaction with Cu and Zn. These interactions govern the protein’s maturation, stability and structure18,29,30,31,32, and Cu-associated perturbations to SOD1 maturation can promote aggregation via their differential effects on the seeding and growth of SOD1 fibrils33. This implicates Cu in the widely supported notion that SOD1 mis-folding and aggregation is a primary mechanism of toxicity for SOD1 in mutant SOD1 cases of ALS34,35. Further to this, altered interaction with Cu also provides a plausible mechanism by which SOD1 may contribute to motor neuron decline in sporadic cases of ALS that do not involve mutant SOD1; even in the absence of a disease-causing mutation, the bioavailability of Cu to SOD1 is an important determinant of the protein’s stability and structure36, and mis-folded and aggregated SOD1 is present in sporadic cases of ALS37. Moreover, the presence of Cu-deficient SOD1 in the disease-affected spinal cords of ALS model mice has been confirmed; direct assessment of metals bound to SOD1 via a quantitative mass spectrometry approach38 shows that almost half of the total SOD1 pool in the spinal cords of SOD1G37R mice is Cu-deficient and a similar pool of Cu-deficient SOD1 is present in the spinal cords of SOD1G93A mice10.
But although the role for disrupted Cu bioavailability in the pathogenesis of ALS is supported by several lines of evidence, a Cu-centric explanation for why the CNS is more susceptible to the effects of mutant SOD1 expression is less clear. In the present study we show that oral treatment with CuII(atsm) improves the neurological phenotype and survival of SOD1G93A mice (Fig. 1) and that the treatment increases the abundance of mutant SOD1 in the spinal cord (Fig. 2). These results are consistent with outcomes from previous studies10 and the increase in SOD1 activity in the spinal cord (Fig. 5A) is consistent with the demonstrated capacity for CuII(atsm) to make Cu bioavailable to SOD1 in vivo8. Overall, the in vivo effects for CuII(atsm) are consistent across multiple mutant SOD1 murine models of ALS and to date is reproduced via two distinct drug administration methods (summarised in Table 1). Across multiple studies it therefore appears that CuII(atsm) stabilises mutant SOD1 in vivo, in a seemingly non-toxic form, by satiating its requirement for Cu and converting Cu-deficient SOD1 to mature holo-SOD1.
But in contrast to these observations in the spinal cord, treating with CuII(atsm) had no influence on SOD1 levels or activity in the liver (Fig. 2D–F), indicating that SOD1 in the livers of SOD1G93A mice is relatively Cu-replete and/or that CuII(atsm) does not make Cu bioavailable to SOD1 in the liver. Our observation that supplementing tissue extracts with Cu increased SOD1 activity in the spinal cord but not the liver (Fig. 3) lends support to the former of these possibilities as does our recent assessment of SOD1 in SOD1G37R mice16. Due to ubiquitous expression of the transgene, SOD1 protein levels are elevated in various CNS and non-CNS tissues from SOD1G37R mice, and in the non-CNS tissues this increase in SOD1 protein is matched by a commensurate increase in SOD1 activity as well as a commensurate increase in Cu and Zn16. However, in the CNS tissues, although the increased level of SOD1 protein is matched by an increase in Zn, a comparable increase in Cu is not evident. As a result, the Cu-dependent activity of SOD1 in the CNS tissue is limited16.
It therefore appears that while the increased requirement for Cu due to SOD1 over-expression is met in non-CNS tissues, the natural bioavailability of Cu in CNS tissues is a limiting factor, leading to an accumulation of Cu-deficient SOD1 only in CNS tissue. This may, in part, be related to the endogenous pathways via which Cu is presented to SOD1. A key stage in SOD1 maturation involves the Cu chaperone for SOD1 (CCS) which acquires Cu for delivery to SOD1 and thereby facilitates SOD1 disulphide bond formation for structural stability. Endogenous mouse CCS appears relatively inefficient at facilitating human SOD1 maturation10. As such, under conditions whereby the natural bioavailability of CNS Cu becomes a rate limiting factor in human SOD1 over-expressing mice, the relative inefficiency of endogenous mouse CCS could become an additive exacerbating factor. Indeed, alleviating the limited availability of Cu to SOD1 via treating with CuII(atsm) induces a relatively modest increase in the Cu content of SOD1 in the spinal cords of SOD1G93A mice (and a relatively modest increase in mouse survival), but when the same treatment is applied to SOD1G93A mice that also express human CCS the effect on Cu delivery to mutant SOD1 (and on mouse survival) is dramatic10. Thus, when human CCS is expressed in SOD1G93A mice the inefficiency of Cu delivery to human SOD1 in the spinal cord is no longer an impediment, and increasing spinal cord Cu via CuII(atsm) therefore improves survival of the mutant SOD1 expressing mice to a remarkable extent.
Consistent with outcomes from previous studies which utilised alternate mutant SOD1 mouse models of ALS8,9, we show here that treating with CuII(atsm) potently decreases protein markers of oxidative stress in the SOD1G93A mice (Fig. 5B) and that markers of astrogliosis are also diminished (Fig. 5C,D). While the explicit source of oxidative stress leading to oxidative damage in the SOD1G93A mice is yet to be unequivocally demonstrated, disruptions to physiological electron flux through the mitochondrial respiratory chain is a widely mooted possibility39,40. Cytochrome c oxidase, complex IV of the respiratory chain, requires Cu for its catalytic activity. As such, and in the context of modulating Cu bioavailability as potential part of the mechanism of action for CuII(atsm) in the mutant SOD1 mice, this raises the possibility that an unmet requirement for Cu in cytochrome c oxidase could contribute to respiratory chain dysfunction, ergo oxidative stress, in the SOD1G93A mice. However, our analysis of cytochrome c oxidase activity in the spinal cords of these mice indicates no overt impediment to this Cu-dependent aspect of mitochondrial function (Fig. 4) and this is consistent with outcomes reported in a recent study for mice that only express mutant SOD110. However, decreased cytochrome c oxidase activity has been reported in mice expressing mutant SOD1. Co-expression of human Cu chaperone for SOD1 (CCS) with mutant SOD1 dramatically accelerates the ALS-like phenotype of the mutant SOD1 mice and induces a mitochondrial pathology10,41. Cytochrome c oxidase activity is decreased by 75% in the CCS x SOD1G93A mice yet is completely restored by treating with CuII(atsm)10. Thus, while treating with CuII(atsm) restores functionality to cytochrome c oxidase in SOD1G93A x CCS mice10, outcomes from the present study do not implicate Cu-dependent cytochrome c oxidase activity in the observed changes in oxidative stress in the CuII(atsm)-treated SOD1G93A mice.
The presence of a substantial pool of Cu-deficient SOD1 in the spinal cord but not liver could explain the apparent tissue-specific effect that CuII(atsm) has on overall levels of Cu in each tissue and this, in turn, could have implications for the clinical use of CuII(atsm) as a PET imaging agent. Treating with CuII(atsm) resulted in a greater elevation in Cu levels in the spinal cord of SOD1G93A mice when compared to non-transgenic mice yet there was no significant difference between SOD1G93A and non-transgenic mice with respect to the liver (Fig. 2C and F). CuII(atsm) labelled with positron emitting Cu isotopes shows greater retention of the signal in the motor cortex of ALS patients13 as well as disease-specific regions of the Parkinson’s disease-affected12 brain and the brains of people affected by MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes)11. The biochemical mechanisms that may govern selective retention of the tracer in the disease-affected regions have been investigated and include oxidative stress, hypoxia and mitochondrial respiratory chain dysfunction42,43. Central to these mechanisms is the presence of cellular proteins which will bind, and therefore retain, the Cu after is has dissociated from the atsmH2 scaffold44. Many proteins under physiological conditions will be able to bind Cu should cellular Cu levels rise relatively rapidly (e.g. metallothioneins), but it stands to reason that cells containing a higher concentration of Cu-deficient proteins will have a greater capacity to retain Cu under such conditions. A substantial pool of Cu-deficient SOD1 exists in CNS tissue from mutant SOD1 expressing mice8,10, and data presented here (Fig. 3) and previously16 indicate the accumulation of Cu-deficient SOD1 in these animals is most evident in CNS tissue.
Methods
CuII(atsm) treatment of SOD1G93A mice
All research involving live mice was approved by a University of Melbourne Animal Experimentation Ethics Committee (#1312908) and conformed with guidelines of the Australian National Health and Medical Research Council. Hemizygous mice expressing a transgene for human SOD1 containing the G93A substitution mutation (SOD1G93A) on the mixed B6/SJL background were from the Jackson Laboratories (strain B6SJL-Tg(SOD1*G93A)1GurJ) and generously provided by Prize4Life. Non-transgenic littermates were used as a control. Prior to treating, mice were allocated based on sex and litter to either the “survival” cohort or the “biochemical” cohort. Mice in the survival cohort were kept through to phenotypic end-point to collect data on the effects of treatment on survival and symptom progression, and mice in the biochemical cohort were killed at the age of 120 days to collect tissues for biochemical analyses. Within each cohort individual mice were allocated based on sex and litter to either the sham treatment or the CuII(atsm) treatment group. All treatments were thus spread evenly across sexes, litters and genotypes. Treatment commenced when the mice were 50 days old. Sham treatment involved gavage with standard suspension vehicle (SSV; 0.9% w/v NaCl, 0.5% w/v Na-carboxymethylcellulose, 0.5% v/v benzyl alcohol, 0.4% v/v Tween-80). CuII(atsm) treatment involved gavage using SSV supplemented with CuII(atsm). CuII(atsm) was synthesised as described previously5,45. Dose of CuII(atsm) administered to each animal was 50 mg kg−1 body weight. Treatments were administered twice daily, 7 days week−1 through to phenotypic end-point (survival cohort) or until the mice reached 120 days of age (biochemical cohort).
Phenotype assessment of mice
SOD1G93A mice were assessed for symptom progression using the rotarod assay for locomotive function and a Neurological Score system previously described17. Mice were habituated to the rotarod assay for 5 days prior to recording performance. During the recording period the rotation speed of the rotarod was accelerated from 4 rpm to 40 rpm over a 180 second period with the time taken to fail the task (latency to fall) recorded for each mouse. During assessment each mouse was subjected to two independent runs on the rotarod and only the higher latency to fall score used for subsequent analysis. Survival of SOD1G93A mice represents the age at which an individual mouse could no longer right itself within 15 seconds of being placed on its side. All phenotype assessments were performed by researchers blinded to mouse genotype and treatment.
Mouse tissue collection
SOD1G93A mice and non-transgenic littermates at 120 days old were anaesthetised by intraperitoneal injection of ketamine (120 mg kg−1) and xylazine (16 mg kg−1) in PBS, then perfused transcardially with PBS containing 0.25% (v/v) phosphatase inhibitor cocktail 2 (Sigma), 1% (v/v) Complete EDTA-free protease inhibitor (Roche), and 20 U mL−1 heparin (Sigma). Following perfusion, tissues were excised then (excepting regions of spinal cord used for histology as described below) snap frozen on dry ice and stored at −80 °C.
SDS-PAGE and western blotting
Tissue samples were homogenised using polypropylene pestles in TBS supplemented with 0.5% (v/v) phosphatase inhibitor cocktail 2 (Sigma), 2% (v/v) Complete EDTA-free protease inhibitor (Roche), and 5% (v/v) DNAse. Homogenates were then separated into TBS-soluble and TBS-insoluble fractions by centrifugation (18,000× g, for 30 minutes at 4 °C). TBS-soluble extracts were prepared in denaturing sample buffer containing 62.2 mM Tris, 5% (v/v) glycerol, 2% (w/v) SDS, and 0.0025% (w/v) bromophenol blue prior to loading onto 4–12% NuPAGE Novex Bis-Tris Midi gels (Life Technologies) and electrophoresis at 200 V for 40 minutes in MES SDS running buffer (Life Technologies). Resolved proteins were transferred onto PVDF membranes using iBlot gel transfer stacks (Life Technologies) as per manufacturer’s instructions. Membranes were blocked for 1 hour in PBS supplemented with 0.05% (v/v) Tween-20 (Chemsupply) and 4% (w/v) skim milk powder prior to incubation with primary antibodies in blocking buffer, overnight at 4 °C. Primary antibodies used were raised to detect human SOD1 (Abcam; 1:100,000) or GAPDH (Cell Signaling; 1:5,000). A horseradish peroxidase-conjugated secondary antibody for anti-rabbit IgG (Cell Signaling; 1:5,000) was then used, and subsequent immunoreactive protein bands visualised by adding Enhanced Chemiluminescence (ECL Advance, GE Healthcare) to membranes and detecting luminescence using a FujiFilm LAS-3000 imager. Quantitation of immunoreactivity was performed using ImageJ software on TIFF file images.
SOD1 activity
SOD1 activity in TBS-soluble tissue extracts (described above) was assessed via a pyrogallol assay based on published procedures46,47. Pyrogallol (Sigma) was added to a reaction buffer (50 mM Tris, 1 mM EGTA, pH 7.4) to a final concentration of 200 μM and allowed to equilibrate for 1 minute. TBS-soluble tissue extracts were added then reaction mixtures monitored at 325 nm. Reaction mixtures supplemented with 10 mM KCN were used to determine the KCN-sensitive activity attributable to SOD1. SOD1 activity was determined by calculating the rate of change through the linear phase of reaction. Reactions mixtures containing equivalent volumes of TBS homogenising buffer ±KCN were included as additional controls. For experiments in which tissue extracts were supplemented with Cu2+ (Fig. 3), TBS-soluble extracts were incubated with CuCl2 (final Cu2+ concentration = 10 μM) overnight at 4 °C before performing the activity assay.
Tissue copper analysis
Sections of frozen tissue were weighed then analysed for total copper levels following protocols described previously42. Briefly, tissue samples were homogenised in TBS as described above then aliquots assessed for total protein content. The remainder of the homogenate was dried down, digested using concentrated nitric acid, then analysed for copper content using an Agilent 7700 Series ICP-MS with a helium reaction cell.
Cytochrome c oxidase and citrate synthase activity
TBS-insoluble spinal cord material was solubilised by adding lauryl maltoside to a final concentration of 1.5% (v/v). Lauryl maltoside soluble extracts were recovered by centrifugation (21,000× g, 3 minutes, 4 °C) then normalised to a consistent protein concentration. Cytochrome c oxidase and citrate synthase activities were determined as described previously48.
Histology
All histology protocols were as previously described8. Briefly, lumbar regions of mouse spinal cord freshly dissected from mice at 120 days old were submersion fixed in 4% (v/v) paraformaldehyde, paraffin-embedded, then sectioned and mounted onto glass microscope slides. Sections were stained with cresyl violet for motor neuron counts or incubated with primary antibodies to GFAP (Dako) or Iba-1 (Wako) for assessing astrogliosis. Motor neuron values presented per mouse represent the average number of α-motor neurons from approximately 30 separate sections per mouse (spanning approximately 2 mm along the longitudinal plane of the spinal cord). For all motor neurons counted, the area was quantified using Image J software and only those motor neurons with an area equivalent to a 20 μm diameter or greater were considered as α-motor neurons. Data presented in Fig. 5A represent the average number of α-motor neurons in both ventral horn regions of the grey matter per section.
Oxidatively modified proteins
TBS-soluble spinal cord samples were analysed for oxidatively modified proteins using the OxyBlot Protein Oxidation Detection kit (Millipore) as described previously8.
Statistical analyses
Data sets were assessed for statistical significance of the CuII(atsm) treatment on age-related outcomes via the following tests: two-tailed repeat measures ANOVA (Fig. 1A,B); Cox proportional hazards model (Fig. 1D). Gender and litter were both excluded as potential confounding factors in the proportional hazards model. Data sets were assessed for the statistical significance of the CuII(atsm) treatment on static group means via the following tests: two-tailed t-test (Figs 1C,E and 2A,D); ordinary one-way ANOVA with Tukey’s multiple comparisons test (Figs 2B,C,E,F, 4A,B and 5A,B); two-tailed paired t-test (Fig. 3A,B). Experimental replicates are individual mice or tissues collected from individual mice. Phenotype assessment data shown in Fig. 1 involved n = 23 sham-treated mice (12 female, 11 male) and n = 24 CuII(atsm)-treated mice (13 female, 11 male). Biochemical data shown in Figs 2, 3, 4 and 5 involved tissues from n = 6 mice (3 female, 3 male) for each treatment group.
Additional Information
How to cite this article: Hilton, J. B. et al. CuII(atsm) improves the neurological phenotype and survival of SOD1G93A mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci. Rep. 7, 42292; doi: 10.1038/srep42292 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Renton, A. E., Chio, A. & Traynor, B. J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 (2014).
Rosen, D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
Gurney, M. E. et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994).
Wong, P. C. et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116 (1995).
Gingras, B. A., Suprunchuk, T. & Bayley, C. H. The preparation of some thiosemicarbazones and their copper complexes: Part III. Can. J. Chem. 40, 1053–1059 (1962).
Hung, L. W. et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 209, 837–854 (2012).
McAllum, E. J. et al. Therapeutic effects of CuII(atsm) in the SOD1G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 586–590 (2013).
Roberts, B. R. et al. Oral treatment with CuII(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 34, 8021–8031 (2014).
Soon, C. P. et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 286, 44035–44044 (2011).
Williams, J. R. et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 89, 1–9 (2016).
Ikawa, M. et al. PET imaging of redox and energy states in stroke-like episodes of MELAS. Mitochondrion 9, 144–148 (2009).
Ikawa, M. et al. Evaluation of striatal oxidative stress in patients with Parkinson’s disease using [62Cu]ATSM PET. Nucl. Med. Biol. 38, 945–951 (2011).
Ikawa, M. et al. Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology 84, 2033–2039 (2015).
Vavere, A. L. & Lewis, J. S. Cu-ATSM: a radiopharmaceutical for the PET imaging of hypoxia. Dalton Trans. 4893–4902 (2007).
Wada, K., Fujibayashi, Y., Tajima, N. & Yokoyama, A. Cu-ATSM, an intracellular-accessible superoxide dismutase (SOD)-like copper complex: evaluation in an ischemia-reperfusion injury model. Biol. Pharm. Bull. 17, 701–704 (1994).
Hilton, J. B., White, A. R. & Crouch, P. J. Endogenous Cu in the central nervous system fails to satiate the elevated requirement for Cu in a mutant SOD1 mouse model of ALS. Metallomics 8, 1002–1011 (2016).
Leitner, M., Menzies, S. & Lutz, C. Working with ALS mice: Guidelines for preclinical testing and colony management (Appendix A). The Jackson Laboratory (2009).
Forman, H. J. & Fridovich, I. On the stability of bovine superoxide dismutase. The effects of metals. J. Biol. Chem. 248, 2645–2649 (1973).
Gurney, M. E. et al. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann. Neurol. 39, 147–157 (1996).
Hayward, L. J. et al. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 277, 15923–15931 (2002).
Graffmo, K. S. et al. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 51–60 (2013).
Tokuda, E., Okawa, E., Watanabe, S., Ono, S. & Marklund, S. L. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol. Dis. 54, 308–319 (2013).
Tokuda, E., Okawa, E. & Ono, S. Dysregulation of intracellular copper trafficking pathway in a mouse model of mutant copper/zinc superoxide dismutase-linked familial amyotrophic lateral sclerosis. J. Neurochem. 111, 181–191 (2009).
Tokuda, E. & Furukawa, Y. Copper homeostasis as a therapeutic target in amyotrophic lateral sclerosis with SOD1 mutations. Int. J. Mol. Sci. 17 (2016).
Tokuda, E. et al. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp. Neurol. 213, 122–128 (2008).
Andreassen, O. A. et al. Effects of an inhibitor of poly(ADP-ribose) polymerase, desmethylselegiline, trientine, and lipoic acid in transgenic ALS mice. Exp. Neurol. 168, 419–424 (2001).
Hottinger, A. F., Fine, E. G., Gurney, M. E., Zurn, A. D. & Aebischer, P. The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur. J. Neurosci. 9, 1548–1551 (1997).
Nagano, S. et al. The efficacy of trientine or ascorbate alone compared to that of the combined treatment with these two agents in familial amyotrophic lateral sclerosis model mice. Exp. Neurol. 179, 176–180 (2003).
Ip, P., Mulligan, V. K. & Chakrabartty, A. ALS-causing SOD1 mutations promote production of copper-deficient misfolded species. J. Mol. Biol. 409, 839–852 (2011).
Lynch, S. M., Boswell, S. A. & Colon, W. Kinetic stability of Cu/Zn superoxide dismutase is dependent on its metal ligands: implications for ALS. Biochemistry 43, 16525–16531 (2004).
Rumfeldt, J. A., Lepock, J. R. & Meiering, E. M. Unfolding and folding kinetics of amyotrophic lateral sclerosis-associated mutant Cu,Zn superoxide dismutases. J. Mol. Biol. 385, 278–298 (2009).
Roberts, B. R. et al. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J. Mol. Biol. 373, 877–890 (2007).
Chattopadhyay, M. et al. The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu,Zn superoxide dismutase (SOD1) fibrillation. J. Biol. Chem. 290, 30624–30636 (2015).
Mulligan, V. K. & Chakrabartty, A. Protein misfolding in the late-onset neurodegenerative diseases: common themes and the unique case of amyotrophic lateral sclerosis. Proteins 81, 1285–1303 (2013).
Hilton, J. B., White, A. R. & Crouch, P. J. Metal-deficient SOD1 in amyotrophic lateral sclerosis. J. Mol. Med. (Berl.) 93, 481–487 (2015).
Furukawa, Y., Torres, A. S. & O’Halloran, T. V. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 23, 2872–2881 (2004).
Bosco, D. A. et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 (2010).
Rhoads, T. W. et al. Measuring copper and zinc superoxide dismutase from spinal cord tissue using electrospray mass spectrometry. Anal. Biochem. 415, 52–58 (2011).
Tan, W., Pasinelli, P. & Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1842, 1295–1301 (2014).
Vehvilainen, P., Koistinaho, J. & Gundars, G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8, 126 (2014).
Son, M. et al. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc. Natl Acad. Sci. USA 104, 6072–6077 (2007).
Donnelly, P. S. et al. An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copperII . Proc. Natl Acad. Sci. USA 109, 47–52 (2012).
Yoshii, Y. et al. Radiolabeled Cu-ATSM as a novel indicator of overreduced intracellular state due to mitochondrial dysfunction: studies with mitochondrial DNA-less rho(0) cells and cybrids carrying MELAS mitochondrial DNA mutation. Nucl. Med. Biol. 39, 177–185 (2012).
Holland, J. P., Lewis, J. S. & Dehdashti, F. Assessing tumor hypoxia by positron emission tomography with Cu-ATSM. Q. J. Nucl. Med. Mol. Imaging 53, 193–200 (2009).
Blower, P. J. et al. Structural trends in copper(II) bis(thiosemicarbazone) radiopharmaceuticals. Dalton Trans. 4416–4425 (2003).
Li, F. et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 89, 1308–1312 (2004).
Marklund, S. & Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 47, 469–474 (1974).
Trounce, I. A., Kim, Y. L., Jun, A. S. & Wallace, D. C. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Meth. Enzymol. 264, 484–509 (1996).
Acknowledgements
This work was funded by the National Health and Medical Research Council (Project Grant 1061550 to PJC and PSD; Career Development Fellowship 1084927 to PJC) the University of Melbourne (Research Grant Support Scheme to PJC), and the Motor Neurone Disease Research Institute of Australia (Zo-ee MND Research Grant to PJC and BRR). Part of this work was also supported by funding from the DoD Congressionally Directed Medical Research Program (AL140108 to JSB) and by Ice Bucket donations made to the ALSA (Research Award ALSA 320 to JSB). JBH was recipient of the Australian Postgraduate Award and the Nancy Frances Curry Scholarship. All SOD1G93A mice and non-transgenic littermates in this study were from The Jackson Laboratory and generously provided by Prize4Life. All ICP-MS analyses were performed by Ms Irene Volitakis at the Biometals Facility, Florey Institute of Neurosciences and Mental Health. Assoc. Prof. Peter Crack kindly provided the Iba-1 antibody.
Author information
Authors and Affiliations
Contributions
J.B.H. and P.J.C. conceived and designed the project; J.B.H., S.W.M., N.K.H.L. and P.J.C. performed experiments; G.B. and P.S.D. synthesised CuII(atsm); J.B.H., N.G.F. and P.J.C. performed data analysis; J.B.H., B.R.R., J.S.B., A.R.W. and P.J.C. wrote/edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
Collaborative Medicinal Development has licensed IP pertaining to CuII(atsm) from the University of Melbourne where the inventors include ARW and PSD.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hilton, J., Mercer, S., Lim, N. et al. CuII(atsm) improves the neurological phenotype and survival of SOD1G93A mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci Rep 7, 42292 (2017). https://doi.org/10.1038/srep42292
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42292
This article is cited by
-
Microglial ferroptotic stress causes non-cell autonomous neuronal death
Molecular Neurodegeneration (2024)
-
Evidence for disrupted copper availability in human spinal cord supports CuII(atsm) as a treatment option for sporadic cases of ALS
Scientific Reports (2024)
-
Copper Metabolism and Cuproptosis: Molecular Mechanisms and Therapeutic Perspectives in Neurodegenerative Diseases
Current Medical Science (2024)
-
Development of novel treatments for amyotrophic lateral sclerosis
Metabolic Brain Disease (2023)
-
Therapeutic Effects of Combination of Nebivolol and Donepezil: Targeting Multifactorial Mechanisms in ALS
Neurotherapeutics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
