
Abstract
Glucocorticoids are commonly used to prevent chemotherapy-induced nausea and vomiting despite a lack of understanding of their direct effect on cancer progression. Recent studies suggest that glucocorticoids inhibit cancer cell migration. However, this action has not been investigated in estrogen receptor (ER)-negative breast tumour cells, although activation of the glucocorticoid receptor (GR) is associated with a worse prognosis in ER-negative breast cancers. In this study we have explored the effect of glucocorticoids on the migration of the ER-negative MDA-MB-231 human breast tumour cell line and the highly metastatic MDA-MB-231-HM.LNm5 cell line that was generated through in vivo cycling. We show for the first time that glucocorticoids inhibit 2- and 3-dimensional migration of MDA-MB-231 cells. Selection of cells for high metastatic potential resulted in a less migratory cell phenotype that was resistant to regulation by glucocorticoids and showed decreased GR receptor expression. The emergence of glucocorticoid resistance during metastatic selection may partly explain the apparent disparity between the clinical and in vitro evidence regarding the actions of glucocorticoids in cancer. These findings highlight the highly plastic nature of tumour cells, and underscore the need to more fully understand the direct effect of glucocorticoid treatment on different stages of metastatic progression.
Similar content being viewed by others


Introduction
Glucocorticoids are used extensively and best known as anti-inflammatory and immunosuppressive agents. However, over 30 years ago, the glucocorticoid dexamethasone was reported to be effective in preventing chemotherapy-induced nausea and vomiting1,2. Since that time, glucocorticoids have also been shown to prevent postoperative3,4 and radiotherapy-induced5 nausea and vomiting. Thus, glucocorticoids constitute one of the major classes of drugs that are considered first-line anti-emetic agents, together with 5HT3 receptor antagonists, such as ondansetron, tachykinin NK1 receptor antagonists, such as aprepitant, and dopamine D2 receptor antagonists, such as metoclopramide6. Despite the common use of glucocorticoids as anti-emetics in cancer patients receiving chemotherapy, the mechanism of action of this effect remains unclear. Several mechanisms have been proposed7 including: decreasing the inflammatory response from chemotherapy or radiation8,9,10; direct actions on the solitary tract nucleus in the central nervous system which receives emetogenic inputs from the afferent sympathetic and parasympathetic nervous systems11; effects on key mediators/receptors in emetic pathways such as 5HT and NK receptors12,13,14; and effects on the hypothalamic-pituitary-adrenal (HPA)-axis15. Rapid glucocorticoid actions related to increased levels of endocannabinoid CB1 ligands may also play a role in the antiemetic effect of glucocorticoids16,17. Furthermore, there is a surprising lack of characterisation of the direct effect of glucocorticoids on the development and progression of different cancers.
At a molecular level, glucocorticoids act through the glucocorticoid receptor (GRα, herein referred to as GR) and predominantly work by modulating gene transcription, although non-genomic mechanisms are increasingly being reported18. In the absence of ligand, the GR is principally located in the cytoplasm in an inactive multi-protein complex19. Upon ligand binding, the GR undergoes a conformational change whereby the complexed proteins dissociate and nuclear localisation signals are exposed, promoting rapid nuclear translocation. Once in the nucleus, the ligand-bound GR is able to: initiate transactivation of genes containing glucocorticoid response elements (GREs); repress transcription of genes containing a negative GRE (nGRE); and trans-repress the activity of other transcription factors such as activator protein-1 (AP-1) and nuclear factor kappa B (NF-κB)20,21. The number of genes modulated by glucocorticoid administration varies depending on cell type and context20,21. However, in some circumstances over 10% of the entire genome can be regulated following a single dose of glucocorticoid22.
Breast cancer is the second most common cancer worldwide, and the most common cancer in females23. Breast cancer mortality is primarily due to metastatic spread of the tumour to secondary sites, such as bone and lung. Functional GR is expressed in almost all cell types, including breast tumour cells and the surrounding stromal tissue24,25, highlighting the imperative to understand what direct effects glucocorticoid use may have on breast tumour cell biology. Unlike in lymphoid cells where glucocorticoids cause cell death, glucocorticoids inhibit apoptosis of epithelial cell types26 including breast tumour cells27. This promotion of cell survival has been proposed to interfere with the cytotoxic effects of chemotherapy28,29, although there is a lack of clinical studies to support this idea30. Glucocorticoids may also decrease cell invasion by reducing tissue permeability31. Importantly, activation of the GR has been shown to be associated with a worse prognosis in estrogen receptor (ER)-negative breast cancer32,33. This is particularly significant, since ER-negative tumours are often intrinsically more metastatic than ER-positive tumours34. Furthermore, few treatment or prevention strategies are available for patients with ER-negative breast cancer35. Recent studies have shown that glucocorticoids can inhibit migration of neuronal precursor cells36, A549 lung adenocarcinoma cells37, MCF10A breast epithelial cells38, and estrogen-receptor positive T47D breast cancer cells39. However, the effect of glucocorticoids on estrogen-receptor negative breast tumour cell migration is yet to be established.
In this study, we have explored the effect of glucocorticoids on the migration of the ER-negative MDA-MB-231 human breast tumour cell line. Cell migration is a critical component of metastasis, allowing cells to intravasate and travel via the circulation to seed in secondary organs, forming metastases. We have therefore also examined the changes in migratory phenotype and glucocorticoid sensitivity that occur following selection of cells with high metastatic potential, using the MDA-MB-231-HM.LNm5 cell line. This cell line was generated by in vivo passaging of tumour cells which has been demonstrated to be a useful method to select for highly metastatic cell lines40.
Results
Glucocorticoids inhibit the migration of the ER-negative MDA-MB-231 human breast tumour cells
Fetal calf serum at a sub-maximal concentration of 1% (Supplementary Figure S1) was used as a stimulus for MDA-MB-231 cell migration in the scrape wound healing assay (Fig. 1A). Dexamethasone concentration-dependently inhibited FCS-induced MDA-MB-231 cell migration with complete inhibition observed at 10 nM Dex (Fig. 1B). Dexamethasone at 100 nM had no effect on cell proliferation as measured by direct cell enumeration and by the resazurin assay (Supplementary Figure S2). Other glucocorticoids, including methylprednisolone (100 nM), budesonide (100 nM) and hydrocortisone (1 μM), similarly inhibited MDA-MB-231 cell migration (Fig. 1C). Furthermore, dexamethasone fully inhibited the migration of the non-invasive ER-positive MCF-7 breast tumour cell line (Fig. 1D).

Serum-starved MDA-MB-231 cells (A,B,C) and MCF-7 cells (D) were scrape-wounded and then pre-incubated with glucocorticoids (dexamethasone, Dex; hydrocortisone, HC; methylprednisolone, MP; budesonide, Bud) for 30 min prior to the addition of FCS (1% v/v) to promote cell migration. Wound infiltration was measured by the change in grey-value using ImageJ/FIJI software (A). The extent of wound closure 15 h after FCS addition is expressed as a percentage of basal and presented as mean ± SEM of n = 4 independent experiments. Chemotactic and chemokinetic migration of serum-stared MDA-MB-231 cells was observed using uncoated (E) and Matrigel™-coated (F) transwell inserts in a modified Boyden chamber assay. Dexamethasone was added to both compartments 30 min before FCS (1% v/v) that was added to both compartments to stimulate chemokinetic migration, or bottom compartment only to stimulate chemotactic migration. After 20 h, migrated cells were stained using Diff-Quick™ and visualised by light microscopy at a magnification of 400×. Data are expressed as the number of migrated cells per high powered field (HPF, 400×) and presented as mean ± SEM of n = 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 from repeated measures one-way ANOVA with Dunnett’s post-hoc test (B,C) or two-way ANOVA with Bonferroni post-hoc test (D,E,F).
Glucocorticoids inhibit 3-dimensional MDA-MB-231 human breast tumour cell migration and invasion
Dexamethasone inhibited both chemotactic and chemokinetic migration (induced by 1% FCS) in the modified Boyden chamber assay (Fig. 1E). The chemotactic and chemokinetic migratory effects of FCS were enhanced when the Transwell inserts were coated with Matrigel™ (Fig. 1F). However, this enhanced migration remained sensitive to attenuation by dexamethasone (Fig. 1F). Dexamethasone also slowed the basal and FCS-stimulated matrix invasion of MDA-MB-231 cells in a 3-dimensional cell-spheroid based assay (Fig. 2).

3-dimensional MDA-MB-231 cell spheroids were generated in low-adhesion round bottom 96 well plates then embedded into type-I fibrillar collagen derived from rat-tails (450 μg/mL). Collagen-embedded spheroids were then treated with dexamethasone (100 nM) or vehicle control (0.01% DMSO) for 30 min prior to the addition of fetal calf serum (FCS, 5% v/v). Static images were taken every 24 hours for 96 h total using an Olympus IX51 microscope (A). The cell-containing area was traced using ImageJ software to determine the extent of invasion (B). Data are expressed as the area of invasion in pixels/1000 and presented as mean ± SEM from n = 8 independent experiments. *P < 0.05, ***P < 0.001 from two-way ANOVA with Bonferroni post-hoc test.
Glucocorticoid inhibition of MDA-MB-231 cell migration is sensitive to the GR antagonist RU486 and the protein synthesis inhibitor cycloheximide
The GR transactivation antagonist and transrepression partial agonist RU486 (1 μM) had no effect on FCS-induced cell migration, but completely prevented the inhibition caused by dexamethasone (Fig. 3A). This concentration of RU486 was sufficient to fully inhibit GRE transactivation by dexamethasone in a reporter assay system (Fig. 3B).

Glucocorticoid receptor antagonist RU486 prevents dexamethasone (10 nM) inhibition of wound closure (A) at a concentration that fully inhibits pGRE-SEAP reporter construct activation (B). The extent of wound closure 15 h after FCS addition is expressed as a percentage of basal (A) and the extent of pGRE-SEAP activation is expressed as relative light units (RLU) from a chemiluminescence SEAP detection kit (B). The protein synthesis inhibitor cycloheximide also attenuates dexamethasone inhibition of cell migration (C). To account for differences in extent of migration from different FCS concentrations, results are expressed as percentage inhibition of wound closure (C). Dexamethasone-conditioned medium (4 h exposure) in which the glucocorticoid activity is prevented by RU486 does not influence cell migration in scrape wound healing assay (D). All data are presented as mean ± SEM of n = 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 from two-way ANOVA with Bonferroni post-hoc test.
In order to determine whether de novo protein synthesis was required for the inhibition of cell migration, cells were incubated with cycloheximide for 1 h prior to dexamethasone addition. Due to the capacity of cycloheximide to influence cell migration per se, higher concentrations of FCS were employed to generate a positive migration stimulus (Supplementary Figure S3). The extent of dexamethasone-induced inhibition of cell migration was decreased in the presence of cycloheximide over all the FCS concentrations used (Fig. 3C).
Glucocorticoid inhibition of MDA-MB-231 cell migration is not mediated by a secreted product
Given cycloheximide attenuated the glucocorticoid inhibition of migration, the synthesis of new protein products is required to manifest the anti-migratory effect. We therefore next investigated whether a newly formed anti-migratory protein may be released from glucocorticoid-treated cells and act in an autocrine or paracrine manner to inhibit cell migration. As expected, conditioned medium from dexamethasone-treated cells inhibited FCS-induced migration of naïve cells (Fig. 3D). However, when RU486 was added to block the activity of the dexamethasone in the conditioned-medium, the FCS-induced migration was not influenced by the conditioned medium (Fig. 3D). These results suggest no stable anti-migratory mediator is released by MDA-MB-231 cells in response to 4 hours of dexamethasone treatment.
MDA-MB-231HM.LNm5 breast cancer cells are spontaneously metastatic to lymph node, liver and lung in immuno-compromised mice
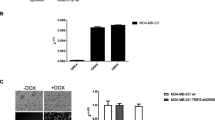
The metastatic capacity of MDA-MB-231HM.LNm5 cells was compared with that of parental MDA-MB-231 cells in immuno-compromised NSG mice using in vivo and ex vivo imaging. MDA-MB-231HM.LNm5 cells spontaneously metastasised to liver, lung and axillary lymph nodes in all three mice inoculated (Supplementary Figure S4, Table S1). In addition, spleen metastases were also observed in one mouse while large paraspinal metastases between the kidneys were evident in two animals. Notably, the extent of metastasis to liver, lung, and lymph node appeared independent of local tumour recurrence following surgical resection of the primary tumour. Distant metastasis was not evident in mice inoculated with parental MDA-MB-231 cells (data not shown).
MDA-MB-231-HM.LNm5 cells show decreased cell migration and decreased glucocorticoid sensitivity compared to parental MDA-MB-231 cells
The highly metastatic MDA-MB-231-HM.LNm5 cell line showed reduced basal migration in the scrape wound healing assay compared to the parental line (Fig. 4A). FCS (1%) enhanced wound closure in both the highly metastatic MDA-MB-231-HM.LNm5 cell line and in the parental cell line. Remarkably, dexamethasone influenced neither basal migration, nor FCS-induced migration in the highly metastatic variant (Fig. 4A). Similar observations were made in the Boyden chamber assay, where the highly metastatic variant showed reduced FCS-induced migration and no effect of dexamethasone treatment (Fig. 4B). In the 3D spheroid invasion assay, the highly metastatic variant cell line showed similar levels of collagen invasion to the parental cell line (Fig. 4C). However, no effect of dexamethasone on MDA-MB-231-HM.LNm5 collagen invasion was observed (Fig. 4C).

Migration of parental MDA-MB-231 and highly metastatic MDA-MB-231-HM.LNm5 cell line derived from lymph node metastasis of MDA-MB-231 cells were compared in scrape wound healing assay (A), modified Boyden chamber assay (B) and spheroid matrix invasion assay (C). Dexamethasone (100 nM) was added for 30 min prior to stimulation with 1% v/v FCS (A,B) or 5% v/v FCS (C). Data are presented as mean ± SEM for n = 3 (A), n = 3–7 (B), n = 4 (C) independent experiments. *P < 0.05, ***P < 0.001 from two-way ANOVA with Bonferroni post-hoc test.
MDA-MB-231-HM.LNm5 cells show decreased glucocorticoid receptor expression and decreased glucocorticoid-responsive gene expression compared to parental MDA-MB-231 cells
Upon examination by qPCR, GR expression was significantly lower in MDA-MB-231-HM.LNm5 cells compared to parental cells (Fig. 5). There was also reduced expression of the dexamethasone-responsive genes encoding glucocorticoid-induced leucine zipper (GILZ, gene name TSC22D3) and epithelial sodium channel 1 alpha (ENaCα, gene name SCNN1A) (Fig. 5).

qPCR was performed on cDNA derived from highly metastatic MDA-MB-231-HM.LNm5 cells and parental MDA-MB-231 cells treated with dexamethasone for 24 h. Target gene expression of glucocorticoid receptor (GRα; gene name: NR3C1), epithelial sodium channel alpha subunit (ENaCα; gene name: SCNN1A), glucocorticoid inducible leucine zipper (GILZ; gene name: TSC22D3) was normalised to 18 S rRNA housekeeping gene and is expressed as fold-change from levels in parental MDA control cells. Data presented as mean ± SEM from n = 4 independent experiments. *P < 0.05, **P < 0.01 from two-way ANOVA with Bonferroni post-hoc test.
Gene Ontology enrichment analysis identifies the most highly enriched gene ontologies relate to cell motility and migration in dexamethasone-treated MDA-MB-231 cells compared to MDA-MB-231-HM.LNm5 cells
Transcriptomic analysis of the parental and MDA-MB-231-HM.LNm5 cells detected 10352 genes above threshold levels of 10 cpm. A hierarchical list of differentially expressed genes between the Dex-treated MDA-MB-231 and MDA-MB-231-HM.LNm5 cell lines were subject to Gene Ontology (GO) enrichment analysis and 9703 genes were found to be associated with a GO term. Dex-treated parental MDA-MB-231 cells were found to have 199 enriched ontologies (p < 0.001) compared to Dex-treated MDA-MB-231-HM.LNm5 cells (Supplementary Figure S5A). Of these, the 8 most enriched ontologies (p < 10−10) all relate to locomotion and migration (Table 1, Supplementary Figure S5B).
MDA-MB-231-HM.LNm5 cell transcriptome shows decreased dexamethasone-induced changes in migratory genes compared to parental MDA-MB-231 cell transcriptome
Further transcriptomic analysis was performed on dexamethasone-induced changes in migratory genes by examining the magnitude of change of Dex over control for genes directly annotated with GO terms positive regulation of cell migration” (GO:0030335) and “negative regulation of cell migration” (GO:0030336).
Parental and MDA-MB-231-HM.LNm5 cells revealed the detectable expression of 233 genes annotated with the term “positive regulation of cell migration” (GO:0030335). Of these 233 genes, 78 genes were regulated by dexamethasone in either cell line (Fig. 6A). Of the 36 genes annotated with the term “positive regulation of cell migration” that were repressed to less than 0.66 fold by dexamethasone in the parental cell line (Fig. 6A, cluster 1, 3, 4), 15 were repressed to a lesser extent (Fig. 6A, cluster 4) and 3 were stimulated (Fig. 6A, cluster 1) in the MDA-MB-231-HM.LNm5 cells. Of the 24 genes annotated with the term “positive regulation of cell migration” which were stimulated by ≥1.5 fold by dexamethasone in the parental cell line (Fig. 6A, cluster 5 & 6), 9 were repressed to a lesser extent (Fig. 6A, cluster 6) and 5 were repressed (Fig. 6A, cluster 5) in the MDA-MB-231-HM.LNm5 cells. 9 genes (Fig. 6A, cluster 7) were only induced in the MDA-MB-231-HM.LNm5 cell line and not in the parental cell line.

RNA-Seq analysis was performed on dexamethasone-treated and control MDA-MB-231-HM.LNm5 and parental MDA-MB-231 cell lines. Dex-induced changes in expression of genes annotated with (A) GO:0030335 ‘positive regulation of cell migration’ or (B) GO:0030336 ‘negative regulation of cell migration’ in the amiGO Gene Ontology (GO) database were calculated as log2(fold change, FC, of Dex/Control) for each cell line. Genes with low read count (<10 cpm in all samples) or low Dex regulation (<1.5 fold) were excluded from the analysis. The remaining genes were hierarchically clustered using an uncentered correlation as distance measure and an average linkage using Cluster 3.0 software and are presented as heat maps generated by java TreeView. Clusters of genes showing a similar pattern of Dex-regulation have been numbered 1–7 for GO:0030335 and 1–6 for GO:0030336.
128 genes annotated with the term “negative regulation of cell migration” (GO:0030336) were detected in the RNA-Seq analysis above threshold levels as specified in the methods section. Of these, 33 genes were regulated by dexamethasone in either the parental or MDA-MB-231-HM.LNm5 cell line (Fig. 6B). 2 genes were selectively induced in the parental line (Fig. 6B, cluster 1) and 5 genes were selectively induced in the MDA-MB-231-HM.LNm5 cell line (Fig. 6B, cluster 3). In contrast, 3 genes were selectively repressed in the parental line (Fig. 6B, cluster 4) and 7 genes were selectively repressed in the MDA-MB-231-HM.LNm5 cell line (Fig. 6B, cluster 6).
Discussion and Conclusions
This work has shown for the first time that glucocorticoids inhibit 2- and 3-dimensional migration of the ER-negative MDA-MB-231 human breast cancer cell line. This study has added to the growing body of evidence that glucocorticoids inhibit cell migration36,37,38,39. We found similar inhibitory effects in ER-positive MCF-7 and ER-negative MDA-MB-231 breast tumour cells suggesting that the presence of the ER is not permissive for the anti-migratory effects of dexamethasone, despite a known connection between the ER and cellular migration41 and the fact that ER-negative tumours are often intrinsically more metastatic than ER-positive tumours34.
Given cellular migration is a critical component of metastasis, we also examined the changes in migratory phenotype and glucocorticoid sensitivity that occur during selection of cells with high metastatic potential. The highly metastatic MDA-MB-231-HM.LNm5 cell line showed decreased basal and FCS-induced migration and, surprisingly, glucocorticoid treatment of this cell line caused no inhibition of migration. We therefore conclude that selection for high metastatic potential has resulted in a less migratory cell phenotype and a decreased responsiveness to the anti-migratory effect of glucocorticoids.
The observation that the highly metastatic cell line has decreased migratory phenotype is unexpected although not entirely unprecedented, as inverse relationships between migratory behaviour and metastasis have been previously observed in melanoma42. This finding highlights the complex nature of metastasis whereby cells do not merely acquire a pro-migratory and pro-invasive phenotype but also become highly proliferative, evade apoptotic mechanisms, become insensitive to anti-growth signals, promote sustained angiogenesis and develop a limitless replicating potential43. Our results might suggest that the metastatic selection was driven by a change in one or more of these other mechanisms, and the resulting effect on migration was secondary to these changes. Alternatively the decrease in migratory phenotype may have promoted the metastatic phenotype by allowing cells more time to adhere and grow at metastatic sites, or may have facilitated evasion of immune recognition mechanisms.
Interestingly, the sensitivity of MDA-MB-231 cells to glucocorticoid treatment was altered by the selection for metastatic potential. MDA-MB-231-HM.LNm5 cells showed no change in cell migration or invasion upon treatment with dexamethasone. We also found that the MDA-MB-231-HM.LNm5 cell line expresses much lower levels of GRalpha mRNA and this correlated with reduced dexamethasone-induction of glucocorticoid-responsive genes. The emergent resistance to the actions of glucocorticoids through metastatic selection is to our knowledge a novel finding. Importantly, it was not a result of selection pressure following drug administration, since the in vivo cycling method40,44 used to generate the highly metastatic cell line did not expose the cells to high levels of glucocorticoid, but rather the resistance occurs against a physiological background concentration of corticosterone. Our data also suggest that the decrease in GRalpha and concomitant decrease in glucocorticoid sensitivity may be permissive to the establishment of the more metastatic phenotype. For example, glucocorticoids are well known to be anti-angiogenic45, through both direct effects on endothelial cells46, as well as through suppression of angiogenic factor release from cells neighbouring the vasculature, including tumour cells47. It is therefore intriguing to speculate that a reduction in glucocorticoid sensitivity has promoted the release of pro-angiogenic mediators from the MDA-MB-231-HM.LNm5 cell line, causing an increase in metastatic potential through an increase in angiogenesis.
Several steps in this study have been taken to ensure that migratory responses rather than cellular proliferation were assessed. Several distinct assays of migration assessed: movement of tumour cells as 2-dimensional cellular monolayers; individual cell migration through transwell pores in the presence and absence of extracellular matrix; as well as invasion in 3-dimensions through extracellular matrix. These separate approaches allowed us to increase our confidence in our conclusions. Each experiment was carefully designed to use minimal FCS concentrations to stimulate migration and we have kept the duration of experiments as short as possible in order to minimise the contribution of proliferation. Despite these precautions, it was still necessary to directly assess the influence of dexamethasone exposure on MDA-MB-231 cell proliferation. As expected, FCS stimulation caused an increase in proliferation as measured by both cell enumeration and Resazurin assay. However, dexamethasone exposure had no effect on basal or FCS-stimulated cell number in either assay. We therefore interpret the glucocorticoid influences on cell migration as direct rather than secondary to changes in cell number.
The inhibition of migration by glucocorticoids in the parental MDA-MB-231 cell line was completely prevented by the GR antagonist RU486, suggesting that this effect is mediated through canonical GR transactivation signalling mechanisms48. The glucocorticoid effect required production of new protein products, as evidenced by reduced inhibition in the presence of cycloheximide. This observation ruled out non-genomic mechanisms of glucocorticoid actions, such as modulation of PI3K and JNK signalling pathways18,20. However, there was no evidence of mediation by a secreted product. Further work will be required to determine the precise mechanism through which GR transactivation leads to inhibition of migration. However, this could be difficult to establish due to the multitude of inter-dependent GR mechanisms20,21,49. Interestingly, we found distinct transcriptomic patterns between the highly metastatic and parental cell lines following dexamethasone treatment in genes controlling both positive and negative regulation of migration. This suggests that selection for metastatic potential has resulted in altered glucocorticoid signalling mechanisms and an impaired ability to activate pathways leading to inhibition of migration. Interestingly, different glucocorticoids have recently been shown to differentially activate the transcription of different genes products50 foreshadowing future drug optimisation strategies for improved efficacy49 in attenuating cell migration. Importantly, since the discovery of the anti-emetic effect of glucocorticoids in the early 1980 s, there has been little or no optimisation for this indication. It remains likely that certain glucocorticoid agonists have better efficacy/safety profiles than those currently used (dexamethasone or methylprednisolone).
In this study, we have presented evidence that the sensitivity of MDA-MB-231 breast tumour cells to dexamethasone is dependent on the stage of metastatic progression. We do not yet know what molecular alterations have occurred during the phenotype change from parental MDA-MB-231 cell line to the highly metastatic MDA-MB-231-HM.LNm5 cell line. Nor do we know at what stage of metastatic phenotype selection the resistance to glucocorticoid inhibition emerged. In future work it will also be important to establish whether other similar sublines express similar characteristics following metastatic selection since the current study is limited to one cell line. Importantly, the emergence of glucocorticoid resistance during metastatic selection may go some way to explaining the apparent disparity between clinical evidence that activation of the GR is associated with a worse prognosis in estrogen receptor (ER)-negative breast cancer32,33, and in vitro studies showing inhibitory actions of glucocorticoids in commonly used cell lines. These findings highlight the highly plastic nature of tumour cells during metastasis, and underscore the need to more fully understand the direct effect of adjuvant glucocorticoid treatment on cancer cell biology at different stages of progression.
Methods
Cell Culture
MDA-MB-231 and MCF-7 breast tumour cells (ATCC, Manassas, VA, USA) were cultured in phenol-red free RPMI 1640 and DMEM respectively supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FCS), 2 mM L-glutamine, 1 mM nonessential amino acids, 1 mM sodium pyruvate, 15 mM HEPES solution, 100 U/mL penicillin and 100 μg/mL streptomycin as previously described51,52. The highly metastatic MDA-MB-231-HM.LNm5 cell line was generated through a two-step process whereby a cell population initially selected by 6 cycles of transplanting spontaneous MDA-MB-231 pulmonary metastases to the mammary fat pad44 (designated MDA-MB-231-HM and kindly provided by ZM Shao and ZL Ou (Breast Cancer Institute, Fudan University, Shanghai, China) were inoculated into the mammary fat pad of a female BALB/c-SCID mouse, following which the axillary lymph node metastases were isolated. All experiments involving mice had approval from the Peter MacCallum Animal Experimentation Ethics Committee and were carried out in accordance with the Australian code for the care and use of animals for scientific purposes (8th edition, 2013). MDA-MB-231-HM.LNm5 cells were cultured in RPMI 1640 supplemented as above.
Assessment of metastatic potential
The metastatic capacity of MDA-MB-231HM.LNm5 cells was compared with that of parental MDA-MB-231 cells in immuno-compromised NOD scid gamma (NSG) mice. To facilitate optical imaging of tumours in vivo, both parental MDA-MB-231 and MDA-MB-231HM cells were transduced with retrovirus encoding tdTomato fluorescent protein, selected with Blasticidin S and bulk sorted for tdTomato expression by flow cytometry (FACSAria, Beckton Dickinson) as previously described53. Both populations were also transduced with retrovirus encoding Firefly luciferase and selected using puromycin53. Cells (n = 3 for each line) were inoculated into the 4th inguinal mammary gland of NSG mice and tumours were allowed to form (one tumour per mouse). Primary mammary tumours were surgically resected 22 days after inoculation. In vivo bioluminescence imaging was performed 13 days after primary tumour resection to detect luciferase-positive proximal metastatic lesions. Mice were culled 21 days following resection and distant metastases were evaluated by ex vivo imaging of tdTomato fluorescence in whole organs (lung, liver, spine, lymph node and spleen) using fluorescent stereomicroscopy.
Scrape wound healing assay
MDA-MB-231 cells were subcultured in 24-well plates at a density of 105 cells/cm2 and grown in serum-containing media until confluency was reached before being starved for 24 hours in serum-free media containing 0.25% BSA. A sterile 200 μL pipette tip was used to scrape away a central portion of cells and media was replaced to remove dislodged cells and debris. Cells were treated with glucocorticoids (1–100 nM dexamethasone, 1 μM hydrocortisone, 100 nM methylprednisolone, 100 nM budesonide) for 30 minutes prior to stimulation with FCS (0.1–5%). The concentrations of glucocorticoids used are able to fully saturate the glucocorticoid receptor and show maximal effects in a wide variety of cell types including airway smooth muscle cells54, airway epithelial cells50, breast cancer cells55, peripheral blood mononuclear cells56. In selected experiments, GR antagonist 1 μM RU486 was added 30 min prior to glucocorticoids. Protein synthesis inhibitor cycloheximide (1 μM) was added 1 h prior to glucocorticoids. Conditioned media was generated by exposing confluent, serum-starved MDA-MB-231 cells to dexamethasone or vehicle for 4 hours before media was transferred to a denuded 24-well plate and treatments added as above. Cells were imaged every 30 minutes for 15 hours using the Leica DMI6000B live cell imaging microscope, being maintained at 37 °C in an atmosphere of room air containing 5% CO2. The optical density (grey value) of the denuded region was determined by manual delineation of the wound area using ImageJ (version 1.46). This value was subtracted from the grey values of the same area in subsequent frames. In individual experiments, the wound closure at 15 hours in untreated (control) culture was used to normalise all other values.
Boyden chamber assay
Chemotactic and chemokinetic migration was observed using polycarbonate cell culture inserts containing 8 μm pores (Costar #3422). Some inserts were coated with 7 μL Matrigel™ (BD Biosciences, North Ryde, NSW, Australia), diluted 1:3 with serum-free RPMI to assess cell invasion. Inserts were centrifuged for 10 minutes at 270 g to generate an even coating. MDA-MB-231 cells grown to confluence in 25 cm2 flasks were serum-starved for 24 h then dislodged using trypsin-EDTA. 50,000 cells in 150 μl serum-free RPMI were seeded into the upper compartment of cell culture inserts and left to adhere for 6 h with 500 μl serum-free RPMI in the bottom compartment. 100 nM dexamethasone was added to both compartments 30 min before FCS (1% v/v) was added to stimulate chemokinetic migration (FCS added to both compartments) or chemotactic migration (FCS added to bottom compartment only). Inserts were incubated for 20 hours at 37 °C in 5% CO2 at which time non-migrated cells were scraped from the upper side of the polycarbonate membrane and migrated cells on the underside of the membrane were stained using Diff-Quick™ (Thermo Fisher, Scoresby, VIC, Australia). Briefly, inserts were washed twice with PBS, fixed with DiffQuick™ fix for 2 min then stained in DiffQuick™ red stain for 10 seconds, followed by DiffQuick™ blue for 6 seconds, and then washed twice in PBS. Membranes were excised from inserts using a scalpel, placed onto microscope slides to air dry and coverslipped using DEPEX mountant. Cells were visualised by light microscopy at a magnification of 400×. The number of cells was counted, with five central, adjacent fields of view averaged for each insert.
3D spheroid matrix invasion assay
Round bottom 96-well plates (Corning, Tewksbury, MA, USA) were coated with 0.5% poly(2-hydroxyethyl methacrylate) (poly-HEMA) to minimise cell adhesion. Cell spheroids were generated in 96 h from 5,000 cells which were seeded into each poly-HEMA coated well in a non-gel forming concentration (2.5%) of Cultrex® extracellular matrix (R&D Systems, Minneapolis, USA) in RPMI medium.
Spheroid matrix invasion was assessed by embedding cell spheroids in type-I fibrillar collagen derived from rat-tails (450 μg/mL). The spheroid-containing collagen was incubated at 37 °C for 1 hour to allow the collagen to set before 100 μL serum-free RPMI was added along with dexamethasone (100 nM) or vehicle (0.01% DMSO) for 30 minutes prior to the addition of FCS (5%). Static images of the spheroids were taken every 24 hours using an Olympus IX51 microscope. The cell-containing area was traced using ImageJ software to determine the extent of invasion.
pGRE-SEAP reporter assay
MDA-MB-231 cells were transiently transfected with a pGRE-SEAP reporter construct (Clontech, Mountain View, CA, USA) as previously described57. Briefly, cells were transfected for 6 hours with a pGRE-SEAP reporter construct (150 ng/well) that was complexed with Lipofectamine 2000 reagent (1 μL/well) in 100 μL/well of OptiMEM for 20 min at room temperature prior to addition to cells. The day after transfection, cells were treated with RU486 (1 μM) for 30 min prior to glucocorticoid stimulation. After 24 h, supernatants were collected and the amount of secreted human placental alkaline phosphatase (SEAP) was determined using a chemiluminescence kit (Roche Applied Science, NSW, Australia).
RNA extraction and RT-qPCR
MDA-MB-231 and MDA-MB-231-HM.LNm5 cells were subcultured into 24-well plates at 105 cells/cm2 and grown for 48 hours before being serum-starved for 24 h. Cells were treated with dexamethasone (100 nM) for 24 hours before total RNA was harvested using TRIzol® (Life technologies). Reverse transcription was carried out using the High Capacity cDNA Reverse Transcription Kit (Invitrogen, Mulgrave, VIC, Australia). Reactions of 5 μL total volume were performed using 2 μl total RNA (containing 100 ng RNA), 2.5 μL 2× RT Buffer, 0.25 μL 20× RT Enzyme Mix and 0.25 μL DEPC-treated water. Thermal conditions for the reaction were 37 °C, 60 minutes; 95 °C, 5 minutes which was performed on a Mastercycler® Pro (Eppendorf, Hamburg, Germany). The resulting cDNA was diluted with 145 μL DEPC-treated water and stored at −20 °C. Real-time PCR was performed in triplicate for each gene of interest in a 384-well plate using ABI Prism 7900HT sequence detection system (Applied Biosystems). Each 5 μL reaction consisted of 1.5 μL diluted cDNA, 2.5 μL iTaq™ universal SYBR ® Green Supermix (Bio Rad, Gladesville, NSW, Australia) and 0.5 μL of each of the relevant forward and reverse primers (Table 2). The conditions for PCR amplification were: 50 °C for 2 min then 95 °C for 10 min followed by 40 cycles of 95 °C for 0.15 min, and 60 °C for 1 min. The threshold cycle determined for each gene was normalized against that obtained for 18S ribosomal RNA, which was measured as internal control.
Cell enumeration
MDA-MB-231 cells were subcultured in 24-well plates at a density of 50,000 cells/cm2 and grown in serum-containing media for 24 hours before being starved for 24 hours in serum-free media containing 0.25% BSA. Cells were treated with 100 nM dexamethasone for 30 min prior to stimulation with FCS 5% for 48 hours. Cells were then exposed to 0.12% (w/v) trypsin for 5 minutes before being neutralised in a 0.067% Trypan blue/FCS solution. The number of cells per well was then manually counted using a haemocytometer in duplicate.
Resazurin reduction assay
Reduction of the redox dye resazurin to resorufin was used as a measure of cellular proliferation58. MDA-MB-231 cells were seeded in 96 well flat-bottomed plates at 36,000 cells/cm2 and allowed to adhere/proliferate for 24 hours before being starved for 24 hours in serum-free RPMI. Cells were pre-incubated with 100 nM dexamethasone for 30 min before stimulation with 1–5% (v/v) FCS for 48 h. Medium was aspirated and cells were incubated with resazurin reagent containing 1.5% (w/v) Resazurin, 0.25 (w/v) methylene blue, 2.9% (w/v) potassium hexacyanoferrate (III) and 4.22% (w/v) potassium hexacyanoferrate (II) trihydrate for 2 h at 37˚C in 5% CO2. Resorufin formation was measured fluorometrically at excitation of 570 nm and emission of 620 nm using a FlexStationIII (Molecular Devices, Sunnyvale, CA, USA).
Transcriptomic Analysis
MDA-MB-231 and MDA-MB-231-HM.LNm5 cells were serum-starved for 24 h then treated with dexamethasone (100 nM) or vehicle control for 24 hours after which total RNA was extracted using TRIzol® (as above). RNA-Seq libraries were prepared from 500 ng total RNA using NEBNext Ultra RNA library prep kit for Illumina (#E7530) with NEBNext Poly(A) mRNA Magnetic Isolation Module (#E7490). Prior to library preparation, RNA was confirmed to be of high quality (RIN > 8) by Agilent Bioanalyzer 2100 analysis. Paired end 2 × 50 bp rapid sequencing was performed on an Illumina HiSeq 2500 in the Melbourne Translational Genomic Platform, University of Melbourne. Raw data was filtered by removing reads with adaptor sequences, reads where the percentage of unknown bases is greater than 10%, and reads considered low quality (where bases with quality value less than or equal to 5 constitute greater than 50% of base reads) to obtain clean reads. All subsequent analyses are based on clean reads only. Clean reads were mapped to the human reference genome GRCh38-hg38 assembly using Rsubread package59. Reads that were aligned to annotated coding regions of the genome were counted using featureCounts60 and gene expression values were calculated for each gene as counts per million (cpm). Genes that did not reach a threshold level of 10 cpm in at least one condition were excluded from further analysis. Gene Ontology (GO) enrichment analysis was performed using GOrilla61,62 (http://cbl-gorilla.cs.technion.ac.il) on a hierarchical list of differentially expressed genes between the Dex-treated MDA-MB-231 and MDA-MB-231-HM.LNm5 cell lines. Further GO annotation analysis was carried out on genes annotated with GO:0030335 ‘positive regulation of migration’ and GO:0030336 ‘negative regulation of migration’ according to the AmiGO 2 database (version 2.3.2). For this analysis, Dex-induced changes were calculated as log2(fold change, FC) of Dex over Control for each cell line. We then performed hierarchical clustering using an uncentred correlation as distance measure and an average linkage using Cluster 3.0 software (http://bonsai.ims.utokyo.ac.jp/~mdehoon/software/cluster) and visualized clustered Heat maps using java TreeView (http://jtreeview.sourceforge.net).
Statistical analysis
Results are expressed at mean ± SEM from n independent experiments (performed on separate days on cells from a different passage) and analysed as grouped data. Scrape wound healing is expressed as a percentage of the total control migration observed over 15 hours. Modified Boyden chamber data are expressed as the number of migrated cells per high-powered field (average of 5 fields). pGRE-SEAP activity data are expressed as relative light units. Quantitative PCR data are expressed as the fold change in expression from control. Cell viability data are expressed as a percentage of the number of control cells counted. One-way or Two-way ANOVA with repeated measures was performed using GraphPad Prism 5.0 on non-normalised data (GraphPad, San Diego, CA, USA). Post hoc Dunnett’s and Bonferroni tests were then performed to ascertain significance. P < 0.05 was considered to be statistically significant.
Additional Information
How to cite this article: Fietz, E. R. et al. Glucocorticoid resistance of migration and gene expression in a daughter MDA-MB-231 breast tumour cell line selected for high metastatic potential. Sci. Rep. 7, 43774; doi: 10.1038/srep43774 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Aapro, M. S. & Alberts, D. S. Dexamethasone as an antiemetic in patients treated with cisplatin. N Engl J Med 305, 520 (1981).
Markman, M., Sheidler, V., Ettinger, D. S., Quaskey, S. A. & Mellits, E. D. Antiemetic efficacy of dexamethasone. Randomized, double-blind, crossover study with prochlorperazine in patients receiving cancer chemotherapy. N Engl J Med 311, 549–552 (1984).
Wang, J. J. et al. The prophylactic effect of dexamethasone on postoperative nausea and vomiting in women undergoing thyroidectomy: a comparison of droperidol with saline. Anesth Analg 89, 200–203 (1999).
Wang, J. J. et al. Dexamethasone reduces nausea and vomiting after laparoscopic cholecystectomy. Br J Anaesth 83, 772–775 (1999).
Kirkbride, P. et al. Dexamethasone for the prophylaxis of radiation-induced emesis: a National Cancer Institute of Canada Clinical Trials Group phase III study. J Clin Oncol 18, 1960–1966 (2000).
Navari, R. M. & Aapro, M. Antiemetic Prophylaxis for Chemotherapy-Induced Nausea and Vomiting. N Engl J Med 374, 1356–1367 (2016).
Chu, C. C. et al. The cellular mechanisms of the antiemetic action of dexamethasone and related glucocorticoids against vomiting. Eur J Pharmacol 722, 48–54 (2014).
Hesketh, P. J. Chemotherapy-induced nausea and vomiting. N Engl J Med 358, 2482–2494 (2008).
Rudd, J. A., Bunce, K. T. & Naylor, R. J. The interaction of dexamethasone with ondansetron on drug-induced emesis in the ferret. Neuropharmacology 35, 91–97 (1996).
Tanihata, S., Oda, S., Nakai, S. & Uchiyama, T. Antiemetic effect of dexamethasone on cisplatin-induced early and delayed emesis in the pigeon. Eur J Pharmacol 484, 311–321 (2004).
Ho, C. M., Ho, S. T., Wang, J. J., Tsai, S. K. & Chai, C. Y. Dexamethasone has a central antiemetic mechanism in decerebrated cats. Anesth Analg 99, 734–739 (2004).
Ihara, H. & Nakanishi, S. Selective inhibition of expression of the substance P receptor mRNA in pancreatic acinar AR42J cells by glucocorticoids. J Biol Chem 265, 22441–22445 (1990).
Mantovani, G., Maccio, A., Massa, E., Lai, P. & Esu, S. Cisplatin induces serotonin release from human peripheral blood mononuclear cells of cancer patients and methylprednisolone inhibits this effect. Oncol Rep 4, 1051–1053 (1997).
Suzuki, T., Sugimoto, M., Koyama, H., Mashimo, T. & Uchida, I. Inhibitory effect of glucocorticoids on human-cloned 5-hydroxytryptamine3A receptor expressed in xenopus oocytes. Anesthesiology 101, 660–665 (2004).
Hursti, T. J. et al. Endogenous cortisol exerts antiemetic effect similar to that of exogenous corticosteroids. Br J Cancer 68, 112–114 (1993).
Hill, M. N. & Tasker, J. G. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience 204, 5–16 (2012).
Malcher-Lopes, R., Franco, A. & Tasker, J. G. Glucocorticoids shift arachidonic acid metabolism toward endocannabinoid synthesis: a non-genomic anti-inflammatory switch. Eur J Pharmacol 583, 322–339 (2008).
Jiang, C. L., Liu, L., Li, Z. & Buttgereit, F. The novel strategy of glucocorticoid drug development via targeting nongenomic mechanisms. Steroids 102, 27–31 (2015).
Pratt, W. B. & Toft, D. O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev 18, 306–360 (1997).
Keenan, C. R., Radojicic, D., Li, M., Radwan, A. & Stewart, A. G. Heterogeneity in mechanisms influencing glucocorticoid sensitivity: The need for a systems biology approach to treatment of glucocorticoid-resistant inflammation. Pharmacol Ther 150, 81–93 (2015).
Oakley, R. H. & Cidlowski, J. A. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132, 1033–1044 (2013).
Whirledge, S., Dixon, D. & Cidlowski, J. A. Glucocorticoids regulate gene expression and repress cellular proliferation in human uterine leiomyoma cells. Horm Cancer 3, 79–92 (2012).
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer(2014).
Buxant, F., Engohan-Aloghe, C. & Noel, J. C. Estrogen receptor, progesterone receptor, and glucocorticoid receptor expression in normal breast tissue, breast in situ carcinoma, and invasive breast cancer. Appl Immunohistochem Mol Morphol 18, 254–257 (2010).
Smith, R. A., Lea, R. A., Weinstein, S. R. & Griffiths, L. R. Progesterone, glucocorticoid, but not estrogen receptor mRNA is altered in breast cancer stroma. Cancer Lett 255, 77–84 (2007).
Keenan, C. R., Salem, S., Fietz, E. R., Gualano, R. C. & Stewart, A. G. Glucocorticoid-resistant asthma and novel anti-inflammatory drugs. Drug Discov Today 17, 1031–1038 (2012).
Moran, T. J., Gray, S., Mikosz, C. A. & Conzen, S. D. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res 60, 867–872 (2000).
Wu, W., Pew, T., Zou, M., Pang, D. & Conzen, S. D. Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J Biol Chem 280, 4117–4124 (2005).
Herr, I. et al. Glucocorticoid cotreatment induces apoptosis resistance toward cancer therapy in carcinomas. Cancer Res 63, 3112–3120 (2003).
Grunberg, S. M. Antiemetic activity of corticosteroids in patients receiving cancer chemotherapy: dosing, efficacy, and tolerability analysis. Ann Oncol 18, 233–240 (2007).
Nauck, M. et al. Induction of vascular endothelial growth factor by platelet-activating factor and platelet-derived growth factor is downregulated by corticosteroids. Am J Respir Cell Mol Biol 16, 398–406 (1997).
Pan, D., Kocherginsky, M. & Conzen, S. D. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res 71, 6360–6370 (2011).
Skor, M. N. et al. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res 19, 6163–6172 (2013).
Alanko, A., Heinonen, E., Scheinin, T., Tolppanen, E. M. & Vihko, R. Significance of estrogen and progesterone receptors, disease-free interval, and site of first metastasis on survival of breast cancer patients. Cancer 56, 1696–1700 (1985).
Putti, T. C. et al. Estrogen receptor-negative breast carcinomas: a review of morphology and immunophenotypical analysis. Mod Pathol 18, 26–35 (2005).
Fukumoto, K. et al. Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol Psychiatry 14, 1119–1131 (2009).
Mayanagi, T., Morita, T., Hayashi, K., Fukumoto, K. & Sobue, K. Glucocorticoid receptor-mediated expression of caldesmon regulates cell migration via the reorganization of the actin cytoskeleton. J Biol Chem 283, 31183–31196 (2008).
Lauriola, M. et al. Diurnal suppression of EGFR signalling by glucocorticoids and implications for tumour progression and treatment. Nat Commun 5, 5073 (2014).
Meng, X. G. & Yue, S. W. Dexamethasone disrupts cytoskeleton organization and migration of T47D Human breast cancer cells by modulating the AKT/mTOR/RhoA pathway. Asian Pac J Cancer Prev 15, 10245–10250 (2014).
Banyard, J. et al. Identification of genes regulating migration and invasion using a new model of metastatic prostate cancer. BMC Cancer 14, 387 (2014).
Piccolella, M., Crippa, V., Messi, E., Tetel, M. J. & Poletti, A. Modulators of estrogen receptor inhibit proliferation and migration of prostate cancer cells. Pharmacol Res 79, 13–20 (2014).
Gallagher, S. J. et al. Beta-catenin inhibits melanocyte migration but induces melanoma metastasis. Oncogene 32, 2230–2238 (2013).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Chang, X. Z. et al. Identification of the functional role of AF1Q in the progression of breast cancer. Breast Cancer Res Treat 111, 65–78 (2008).
Hadoke, P. W., Iqbal, J. & Walker, B. R. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol 156, 689–712 (2009).
Logie, J. J. et al. Glucocorticoid-mediated inhibition of angiogenic changes in human endothelial cells is not caused by reductions in cell proliferation or migration. PLoS One 5, e14476 (2010).
Yano, A., Fujii, Y., Iwai, A., Kageyama, Y. & Kihara, K. Glucocorticoids suppress tumor angiogenesis and in vivo growth of prostate cancer cells. Clin Cancer Res 12, 3003–3009 (2006).
Ronacher, K. et al. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol 299, 219–231 (2009).
Keenan, C. R., Lew, M. J. & Stewart, A. G. Biased signalling from the glucocorticoid receptor: Renewed opportunity for tailoring glucocorticoid activity. Biochem Pharmacol 112, 6–12 (2016).
Joshi, T., Johnson, M., Newton, R. & Giembycz, M. An analysis of glucocorticoid receptor-mediated gene expression in BEAS-2B human airway epithelial cells identifies distinct, ligand-directed, transcription profiles with implications for asthma therapeutics. Br J Pharmacol 172, 1360–1378 (2015).
Khau, T. et al. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J 25, 483–496 (2011).
Al-Zaubai, N. et al. Resolvin D2 supports MCF-7 cell proliferation via activation of estrogen receptor. J Pharmacol Exp Ther 351, 172–180 (2014).
Johnstone, C. N. et al. Parvin-beta inhibits breast cancer tumorigenicity and promotes CDK9-mediated peroxisome proliferator-activated receptor gamma 1 phosphorylation. Mol Cell Biol 28, 687–704 (2008).
Stewart, A. G., Fernandes, D. & Tomlinson, P. R. The effect of glucocorticoids on proliferation of human cultured airway smooth muscle. Br J Pharmacol 116, 3219–3226 (1995).
Lippman, M., Bolan, G. & Huff, K. The effects of glucocorticoids and progesterone on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res 36, 4602–4609 (1976).
Smit, P. et al. Differential regulation of synthetic glucocorticoids on gene expression levels of glucocorticoid-induced leucine zipper and interleukin-2. J Clin Endocrinol Metab 90, 2994–3000 (2005).
Keenan, C. R. et al. Bronchial epithelial cells are rendered insensitive to glucocorticoid transactivation by transforming growth factor-beta1. Respir Res 15, 55 (2014).
Czekanska, E. M. Assessment of cell proliferation with resazurin-based fluorescent dye. Methods Mol Biol 740, 27–32 (2011).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Eden, E., Lipson, D., Yogev, S. & Yakhini, Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol 3, e39 (2007).
Eden, E., Navon, R., Steinfeld, I., Lipson, D. & Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 (2009).
Acknowledgements
The authors acknowledge the expert assistance from the Centre for Translational Pathology, The University of Melbourne, for library preparation and RNA-Seq in this study. This study was funded by NHMRC (Australia) project grants #1059655 and #1023185.
Author information
Authors and Affiliations
Contributions
A.G.S., E.F., C.K. designed experiments; E.F., Y.T., C.N.J., T.H. performed experiments; E.F., C.K., Y.T., G.L.C. analysed the results; C.K., G.L.C. performed the statistical analysis; E.F., C.K., A.G.S. interpretation of data; C.N.J. contributed new reagents or analytical tools; C.K., E.F. wrote the manuscript; A.G.S. conceived the study; All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fietz, E., Keenan, C., López-Campos, G. et al. Glucocorticoid resistance of migration and gene expression in a daughter MDA-MB-231 breast tumour cell line selected for high metastatic potential. Sci Rep 7, 43774 (2017). https://doi.org/10.1038/srep43774
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43774
This article is cited by
-
A semiconductor 96-microplate platform for electrical-imaging based high-throughput phenotypic screening
Nature Communications (2023)
-
Mechanisms behind context-dependent role of glucocorticoids in breast cancer progression
Cancer and Metastasis Reviews (2022)
-
Glucocorticoid receptors are required effectors of TGFβ1-induced p38 MAPK signaling to advanced cancer phenotypes in triple-negative breast cancer
Breast Cancer Research (2020)
-
Progesterone suppresses the invasion and migration of breast cancer cells irrespective of their progesterone receptor status - a short report
Cellular Oncology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
