

Abstract
In pioneering work by Cederbaum et al an excitation mechanism was proposed that occurs only in loosely bound matter (Cederbaum et al 1997 Phys. Rev. Lett. 79 4778): it turned out, that (in particular) in cases where a local Auger decay is energetically forbidden, an excited atom or molecule is able to decay in a scheme which was termed 'interatomic Coulombic decay' (or 'intermolecular Coulombic decay') (ICD). As ICD occurs, the excitation energy is released by transferring it to an atomic or molecular neighbor of the initially excited particle. As a consequence the neighboring atom or molecule is ionized as it receives the energy. A few years later the existence of ICD was confirmed experimentally (Marburger et al 2003 Phys. Rev. Lett. 90 203401; Jahnke et al 2004 Phys. Rev. Lett. 93 163401; Öhrwall et al 2004 Phys. Rev. Lett. 93 173401) by different techniques. Since this time it has been found that ICD is not (as initially suspected) an exotic feature of van der Waals or hydrogen bonded systems, but that ICD is a very general and common feature occurring after a manifold of excitation schemes and in numerous weakly bound systems, as revealed by more than 200 publications. It was even demonstrated, that ICD can become more efficient than a local Auger decay in some system. This review will concentrate on recent experimental investigations on ICD. It will briefly introduce the phenomenon and give a short summary of the 'early years' of ICD (a detailed view on this episode of investigations can be found in the review article by U Hergenhahn with the same title (Hergenhahn 2011 J. Electron Spectrosc. Relat. Phenom. 184 78)). More recent articles will be presented that investigate the relevance of ICD in biological systems and possible radiation damage of such systems due to ICD. The occurrence of ICD and ICD-like processes after different excitation schemes and in different systems is covered in the middle section: in that context the helium dimer (He2) is a particularly interesting (and exotic) system in which ICD was detected. It was employed in several publications to elucidate the strong impact of nuclear motion on ICD and its longrange-character. The review will present these findings and their initial theoretical predictions and give insight into most recent time-resolved measurements of ICD.
Export citation and abstract BibTeX RIS
1. Introduction
Electron correlation is a phenomenon that is responsible for a manifold of effects found in atomic and molecular physics. Already the most basic processes involving more than one electron, as for example the single photon double ionization of a He atom, are only possible because electron correlation is present [6]. While this process can be understood using an intuitive picture of one electron knocking out the second one from the ionized atom, a vast majority of electronic processes is lacking such pictures. A whole genre of such processes is formed, for example, by 'non-local electronic effects'. Already known for many years is, for example, Penning ionization. In Penning ionization [7] an excited atom or molecule de-excites by ionizing a second atom or molecule (typically) in a collision: as the two particles approach each other, the excitation energy is transferred via a charge exchange leading to the aforementioned ionization. The non-local character of this de-excitation scheme is due to the fact the the two colliding particles are not chemically bound, but usually treated as separate entities. Equally well known, but usually restricted to biological systems, is the so called 'Förster resonance energy transfer' (FRET) [8], or in more general descriptions just 'resonance energy transfer' (RET) [9]). In FRET energy is transferred between loosely bound complexes of molecules after a resonant excitation of one of the molecules. Again, the participating molecules do not share electrons in a sense of being covalently bound yielding the non-local attribute of FRET. It is obvious, that electron correlation is vital for this process: the energy is transferred by a non-radiative dipole-dipole coupling of the involved electrons. This was confirmed, as—for example—the dependence of the efficiency of FRET on the intermolecular distance R between the donor and the acceptor molecule drops with  [8, 10–12].
[8, 10–12].
Interatomic (or intermolecular) Coulombic decay (ICD) is a phenomenon that occurs in the same setting as these two examples of non-local processes. Even though being initially predicted in small compounds of HF and water molecules [1], a most clean prototype system, where ICD was observed, is the neon dimer ( ) [13–15]. ICD is an interatomic decay process, as depicted in figure 1 (from [3]). Upon 2s-ionization of one of the atoms of the dimer, a 2p-electron fills the vacancy. The de-excitation energy is transferred to the atomic neighbor, i.e. the second atom of the dimer, causing an ionization of a 2p-electron from that neon atom. From the side of theory the leading contribution to ICD can be treated just as a single site Auger decay [1]. Thus, the decay rate is proportional to
) [13–15]. ICD is an interatomic decay process, as depicted in figure 1 (from [3]). Upon 2s-ionization of one of the atoms of the dimer, a 2p-electron fills the vacancy. The de-excitation energy is transferred to the atomic neighbor, i.e. the second atom of the dimer, causing an ionization of a 2p-electron from that neon atom. From the side of theory the leading contribution to ICD can be treated just as a single site Auger decay [1]. Thus, the decay rate is proportional to  , where the two electron–electron Coulomb matrix elements
, where the two electron–electron Coulomb matrix elements
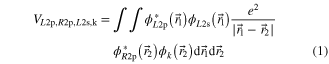
and
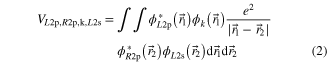
are termed 'direct' and 'exchange' contribution, respectively [16–18]. The indices of the wavefunctions in equations (1) and (2) are chosen such that 'R' denotes an orbital from the 'right' neon atom of the dimer and 'L' one from its left atom. '2s' and '2p' describe the involved shells and 'k' refers to the continuum photoelectron. The two contributions  and
and  arise from the fact that the electrons that are involved in the decay are indistinguishable. Therefore an essential difference to a single site Auger decay occurs, if the participating electrons are located at two different atoms, as in the case of ICD in a neon dimer. The direct integral (equation (1)) describes the case in which a 2p-electron of the same atom drops into the previously created 2s hole and a 2p-electron from the other atom of the dimer is emitted (see figure 1(b)). The exchange integral (equation (2)) describes a process of an electron transfer: the 2s-hole at the left neon atom is filled by a 2p-electron from the right atom, leading to the emission of another 2p-electron from the left atom (see figure 2). The contributions from these two integrals to the decay rates depend very differently on the internuclear distance R of the involved atoms [17, 19]. This fact has been used, for example, to experimentally distinguish the two terms in the decay of different shakeup states of neon dimers [18]. It was found that for the case of inner-valence ionization of a neon dimer (and other neon/rare gas clusters) ICD occurs almost completely due to the direct contribution [19].
arise from the fact that the electrons that are involved in the decay are indistinguishable. Therefore an essential difference to a single site Auger decay occurs, if the participating electrons are located at two different atoms, as in the case of ICD in a neon dimer. The direct integral (equation (1)) describes the case in which a 2p-electron of the same atom drops into the previously created 2s hole and a 2p-electron from the other atom of the dimer is emitted (see figure 1(b)). The exchange integral (equation (2)) describes a process of an electron transfer: the 2s-hole at the left neon atom is filled by a 2p-electron from the right atom, leading to the emission of another 2p-electron from the left atom (see figure 2). The contributions from these two integrals to the decay rates depend very differently on the internuclear distance R of the involved atoms [17, 19]. This fact has been used, for example, to experimentally distinguish the two terms in the decay of different shakeup states of neon dimers [18]. It was found that for the case of inner-valence ionization of a neon dimer (and other neon/rare gas clusters) ICD occurs almost completely due to the direct contribution [19].
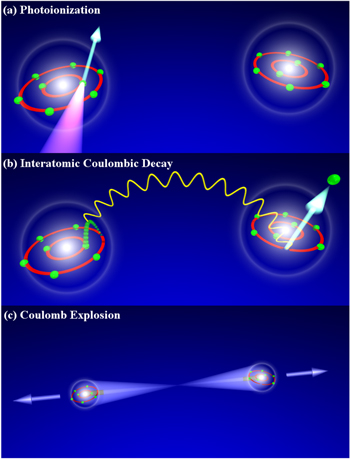
Figure 1. Interatomic Coulombic decay in a neon dimer: (a) A 2s-inner-valence-electron is removed from one atom of a neon dimer. (b) As the vacancy is filled by a 2p-electron the energy gained is transferred to the other atom of the dimer in a dipole/dipole-interaction. (c) As two singly charged neon ions are facing each other after the decay, the dimer fragments in a Coulomb explosion. The process sketched in (b) corresponds to the 'direct' contribution to ICD (see text). Reprinted with permission from [3]. Copyright 2004 by the American Physical Society.
Download figure:
Standard image High-resolution image
Figure 2. Top: the exchange contribution to interatomic Coulombic decay. Bottom: involved energy levels for the case of ICD of a neon dimer, showing the exchange contribution (left) and the aforementioned direct contribution (right).
Download figure:
Standard image High-resolution imageConcerning the aforementioned processes of FRET and Penning ionization the theoretical description is similar to that of ICD: the direct term of ICD is basically equivalent to the theoretical description of FRET, as it incorporates the dipole-dipole interaction and therefore scales with  . However, a major difference arises from the fact that for ICD the resonance conditions of the process are always fulfilled other than in the case of FRET. While the initial description of Penning ionization is in line with that of ICD [20, 21], the leading contribution to Penning ionization is usually described according to the exchange term of equation (2). This is mainly due to the fact that Penning ionization (other than ICD) is typically describing a collision process involving a metastable excited state. As the decay of a metastable state is incompatible to the dipole selection rule and as the collision times are typically very short, the direct term (equation (1)) is neglected [22] and, just as the exchange term of ICD, Penning ionization is mediated by orbital overlap with an exponential increase of efficiency with decreasing internuclear distance R. In many cases Penning ionization is even described as a molecular Auger decay of intermediate molecular states that are created during the collision [23].
. However, a major difference arises from the fact that for ICD the resonance conditions of the process are always fulfilled other than in the case of FRET. While the initial description of Penning ionization is in line with that of ICD [20, 21], the leading contribution to Penning ionization is usually described according to the exchange term of equation (2). This is mainly due to the fact that Penning ionization (other than ICD) is typically describing a collision process involving a metastable excited state. As the decay of a metastable state is incompatible to the dipole selection rule and as the collision times are typically very short, the direct term (equation (1)) is neglected [22] and, just as the exchange term of ICD, Penning ionization is mediated by orbital overlap with an exponential increase of efficiency with decreasing internuclear distance R. In many cases Penning ionization is even described as a molecular Auger decay of intermediate molecular states that are created during the collision [23].
Since the first predictions of ICD it has been shown that its main scheme of a non-local interatomic (or intermolecular) energy transfer is a very common phenomenon. The review article by U Hergenhahn [5] summarizes many of the obtained findings in detail and the following section will try to give a short overview of the different excitation schemes and systems in which ICD-like processes were found. While an exact delimitation of the phenomenon of ICD against other non-local ionization and energy transfer processes makes sense and is of scientific importance in some cases (see e.g. [24], where a clean distinction between ICD and FRET could be made by regarding the resonance conditions of the energy transfer process (more details are given in section 2.2)), a paramount contribution of the research on ICD to AMO physics lies (to the opinion of the author) in revealing that a manifold of mechanisms—even though this might not be surprising from examining their theoretical description—actually happen in nature. From the experimental AMO physics point of view it is often a major task to prepare a target, that is going to be examined, in a way, that it is separated most from its environment. That way a maximum level of control is achieved. A similar practice is often chosen in theoretical studies, as well, in order to being able to handle a system computationally. This approach, however, inhibits a whole genre of processes and effects that occur only due to a loosely bound chemical surrounding (i.e. in a setting which is actually very common in nature), as revealed by the research on ICD (which was triggered by the pioneering theoretical work of the Cederbaum group). 'ICD is everywhere' is a statement that was given in [25] and since then cited many times. It summarizes the findings of the last ten years of research on the topic: in many cases, even in those where more than two loosely bound atoms or molecules need to be involved, non-local ICD or ICD-like processes are able to outpace other local de-excitation pathways.
1.1. Early (experimental) work
After the prediction of ICD in 1997 [1] it took several years before first evidence for ICD was found in an electron spectroscopy experiment [2]. Shortly after these findings, two further studies reported on an experimental observation of ICD. Öhrwall et al concluded from the measured width of the 2s-photoline that a very fast decay mechanism must be present [4]. An unambiguous proof for the existence of ICD was given by a coincidence measurement [3]. By measuring the momenta of the electrons and ions emitted after photoionization of a 2s-electron of a neon dimer, a unique fingerprint for ICD (in small, atomic systems) was detected: as the potential energy curve of the 2s-ionized dimer is almost flat, the sum Esum of the kinetic energy of the ions (i.e. the 'kinetic energy release' (KER)) and the emitted ICD electron is a constant with a value of:
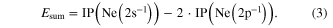
This value was observed in the experiment, as shown in figure 3: in a representation of the KER versus the kinetic energy of the electrons, events, where the sum of these two quantities is constant, occur on a diagonal line with a slope of  .
.
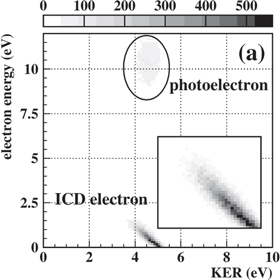
Figure 3. Unambiguous proof for ICD in a neon dimer. Reprinted with permission from [3]. Copyright 2004 by the American Physical Society. As the sum of the kinetic energy release and the ICD electron kinetic energy is a constant, events of ICD occur in this representation as a diagonal feature.
Download figure:
Standard image High-resolution imageAfter the experimental verification of the existence of ICD, the ubiquitous nature of ICD became more and more apparent. ICD was found not only following inner-valence ionization [26], but as well after resonant excitation [27, 28], as satellite states decay [18, 29–32] and subsequent to Auger cascades and therefore even as a terminal step after inner shell ionization [33–45]. Some experiments were able to show, that ICD occurs in solutions [46, 47] and water clusters [48, 49], as well. For a detailed review on these topics, please refer to [5].
As the present review will mainly cover the topic from an experimentalist's point of view, the two theoretical review articles available should at least be mentioned briefly: apart from the early review on theoretical work on ICD [17], a more recent one can be found in [50].
2. 'ICD is everywhere'
The title of this section is a very strong statement which was actually given in [25]. Obviously being only valid 'in quotation marks', it basically reflects the fact that during this time several groups realized that the concept of ICD is a very general phenomenon in loosely bound matter. Apart from the different excitation schemes mentioned in the previous section it was found that ICD happens in a manifold of different systems that are bound by the van der Waals force or hydrogen bonds. At that time it was suggested that ICD should have strong implications, as well, on biological systems and especially on radiation damage of such systems. These assumptions were nourished as several publications pointed out that DNA single and double strand breaks might occur efficiently after low energy electrons attaching to it (see e.g. [51, 52]). As ICD electrons are in most cases of low kinetic energy and as ICD was found to occur as a terminal decay step after Auger cascades [33–45] a corresponding conclusion was made. Experimental efforts yielded the demonstration of the existence of ICD in water, as a first step towards the realm of biology [48, 49]. Recently ICD was identified in theoretical investigations to be responsible for a DNA repair mechanism [24]. This section reports on the latter finding, more detailed investigations of ICD in water and on ICD occurring after further excitation schemes, that were not covered, so far, as ion and electron impact. A short summary of the work performed on ICD in quantum dots and on ICD at interfaces will be given, demonstrating the wealth of phenomena connected to ICD nowadays.
2.1. On the efficiency of ICD in water
Already shortly after confirming the occurrence of ICD in loosely bound matter, a manifold of articles were published stating that a connection between ICD and radiation damage might exist (see e.g. [34]). As molecular compounds of biological interest are (to some extend still) at the frontier of experimental and theoretical AMO-physics-investigations, many groups focused their research on ICD in water as a first step towards finding biological implications. Along with the initial predictions [1, 53, 54] Müller et al performed calculations on the kinetic energy spectrum of ICD electrons originating from small (n = 1..4) water clusters [55]. These calculations confirmed the expected behavior of mainly low energy ICD electrons being emitted upon ICD after inner-valence ionization. Due to the multitude of accessible states a broad distribution of electron kinetic energies below 10 eV peaking at 0 eV were expected. Two experiments confirmed these predictions in 2010 [48, 49]. It turned out, however, that the situation is more complicated already in small compounds of water molecules, as proton migration is a very common process after electronic excitation of such clusters. While in [49] it was concluded for water dimers, that due to the absence of the unsymmetrical breakup channel (H2OH OH+) ICD outpaces proton migration, Svoboda et al [56] mentioned that ICD might be quenched as the decaying state's energy is decreased due to nuclear dynamics down to a level that inhibits ICD. In that case the dimer would dissociate without a second charge being created, i.e explaining the observed lacking of H2OH
OH+) ICD outpaces proton migration, Svoboda et al [56] mentioned that ICD might be quenched as the decaying state's energy is decreased due to nuclear dynamics down to a level that inhibits ICD. In that case the dimer would dissociate without a second charge being created, i.e explaining the observed lacking of H2OH OH+. A tentative estimate of the efficiency of ICD after inner-valence ionization of 20 to 40% was given. Following pioneering experiments [57], Förstel et al were able to show, that ICD after 2s-ionization of neon clusters has an efficiency of unity (0.99 ± 0.11) [58] by employing an electron/electron-coincidence approach using a magnetic bottle spectrometer [59]. A corresponding measurement revealed that this is not the case for inner-valence ionized states of water clusters [60]. Here, ICD occurs with a probability of significantly less than 100%.
OH+. A tentative estimate of the efficiency of ICD after inner-valence ionization of 20 to 40% was given. Following pioneering experiments [57], Förstel et al were able to show, that ICD after 2s-ionization of neon clusters has an efficiency of unity (0.99 ± 0.11) [58] by employing an electron/electron-coincidence approach using a magnetic bottle spectrometer [59]. A corresponding measurement revealed that this is not the case for inner-valence ionized states of water clusters [60]. Here, ICD occurs with a probability of significantly less than 100%.
2.2. ICD in biological systems
In a recent publication by Harbach et al theoretical investigations come to the conclusion, that ICD is indeed relevant for a real biological system [24]. Certain damages to DNA that are caused by UV-irradiation can be repaired by photolyases. These DNA repair enzymes require visible light in order to donate an electron that will trigger the DNA repair. It was found that ICD is the mechanism underlying the generation of that (quasi) free electron. A common feature of different photolyases is its composition of two cofactors: a reduced flavin adenine dinucleotide (FADH) that acts as an electron donor and an antenna chromophore. Harbach et al demonstrate, that after the initial resonant excitation of the antenna chromphore (in their case a 8-hydroxy-5-deazaflavin (8-HDF)) the excitation energy is transferred to the neighboring FADH-molecule (see figure 4). It is emphasized, that on the level of detail of the theoretical calculations, ICD can be distinguished from a Förster-type energy transfer, as the latter yields a reduced efficiency of the energy transfer process: while a 'classical' Förster excitation energy transfer is restricted to bound acceptor-states, the accepting state in ICD can be electronically unbound. Therefore the freed electron can always be emitted with the right kinetic energy, thus always ensuring perfect resonance conditions.

Figure 4. Repair of a photolesion: a HDF-molecule absorbs a photon. The excitation energy is transferred from the HDF-antenna molecule to the FADH-molecule that acts as an electron donor, triggering the DNA repair. Reprinted with permission from [24]. Copyright 2013 American Chemical Society.
Download figure:
Standard image High-resolution imageAn extension of the work reported in [24] covering the complete photolesion repair mechanism from a theoretical point view is given in [61].
2.3. Resonant Auger decay-induced ICD
After the existence of ICD after resonant excitation (termed 'RICD') was demonstrated in publications by Barth et al [62] and Aoto et al [28] and its occurrence after Auger cascades was found [39–41, 43], the group of Cederbaum suggested that even resonant Auger decay might be a profitable excitation scheme after which ICD might happen, as well (figure 5(a)). After resonant excitation of a core electron an excited atom or molecule may deexcite in an Auger decay. Two different pathways are possible. Firstly, the excited electron drops back into the core hole. This is known as 'participator Auger decay'. In a second scenario a different electron from e.g. an inner-valence shell can fill the core vacancy leaving the initially excited electron in a highly excited state. Gokhberg et al proposed that therefore in many cases an excited state remains after the Auger decay that will further decay by ICD [63]. Additionally, depending on the exact state that is populated in the resonant excitation the electron energy of the ICD electron emitted in the terminal step can be tuned. As the excitation happens resonantly it was pointed out, that a specific site inside a large system consisting of many atoms can be addressed (figure 5(b)). As a testbench for the theoretical work ArKr dimers were chosen. The calculations revealed that ICD after resonant Auger decay is an efficient channel and that by choosing core excited states that are slightly different in energy (i.e. 246.51 eV corresponding to the Ar( ) parent state or 246.93 eV for the Ar(
) parent state or 246.93 eV for the Ar( ) parent state) ICD electron spectra that differ by up to 7 eV are created. In line with these findings a scheme for a possible radiation therapy employing ICD was proposed: the field of x-ray resonant theranostics [64, 65] explores the possibility to employ marker molecules that consist of at least one high-Z element to tag malignant cells. By resonantly exciting the high-Z atom of the marker molecule energy is deposited selectively at the site of the cell to treat. The results of Cederbaum et al demonstrated that ICD will directly create a genotoxic low energy electron in the closest vicinity of the selected site.
) parent state) ICD electron spectra that differ by up to 7 eV are created. In line with these findings a scheme for a possible radiation therapy employing ICD was proposed: the field of x-ray resonant theranostics [64, 65] explores the possibility to employ marker molecules that consist of at least one high-Z element to tag malignant cells. By resonantly exciting the high-Z atom of the marker molecule energy is deposited selectively at the site of the cell to treat. The results of Cederbaum et al demonstrated that ICD will directly create a genotoxic low energy electron in the closest vicinity of the selected site.
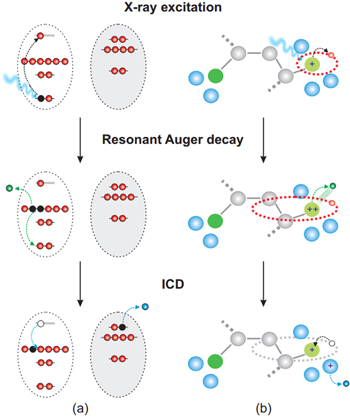
Figure 5. (a) ICD after resonant Auger decay. (b) The parent state of ICD is exclusively created at a certain atom of an embedded system, as this atom is excited resonantly. Reprinted with permisson from [63]. Copyright 2013, rights managed by Nature Publishing Group.
Download figure:
Standard image High-resolution imageSeveral publications confirmed the existence of ICD after resonant Auger decay (RA-ICD). Kimura et al observed it in Ar2 after resonant  excitation [66] and were furthermore able to demonstrate the expected tunability of the ICD electron spectrum [67] by detuning the intial photon energy as proposed in [63]. The latter work was performed on Ar2, ArKr and ArXe dimers. Independently O'Keeffe et al examined the process in ArNe and Ar2 at excitation energies corresponding to the resonant excitation of Ar(
excitation [66] and were furthermore able to demonstrate the expected tunability of the ICD electron spectrum [67] by detuning the intial photon energy as proposed in [63]. The latter work was performed on Ar2, ArKr and ArXe dimers. Independently O'Keeffe et al examined the process in ArNe and Ar2 at excitation energies corresponding to the resonant excitation of Ar( ), Ar(
), Ar( ) and Ar(
) and Ar( ) [68]. Their work demonstrates nicely, as well, the potential to control the emission energy of the ICD electron. By using a molecular target information on the efficiency of RA-ICD was gathered in [69]. After core-exciting N2 and CO dimers to a
) [68]. Their work demonstrates nicely, as well, the potential to control the emission energy of the ICD electron. By using a molecular target information on the efficiency of RA-ICD was gathered in [69]. After core-exciting N2 and CO dimers to a  -state, excited molecular states are created after spectator Auger decay. These states, however, are typically dissociative with rather steep potential energy curves. The lifetime of these states prior to dissociation is in the range of 10 fs yielding a molecular clock: if dissociation happens prior to ICD it will quench its occurrence. In turn, as ICD was observed it had to take place before dissociation happened, i.e. within approximately 10 fs.
-state, excited molecular states are created after spectator Auger decay. These states, however, are typically dissociative with rather steep potential energy curves. The lifetime of these states prior to dissociation is in the range of 10 fs yielding a molecular clock: if dissociation happens prior to ICD it will quench its occurrence. In turn, as ICD was observed it had to take place before dissociation happened, i.e. within approximately 10 fs.
2.4. ICD after ion and electron impact
Most experimental investigations on ICD were carried out employing photons in order to create the excited state, that is able to decay via ICD. Only a few cases can be found in literature, so far, where—for example—ion beams were used to trigger ICD. Pioneering experiments by Titze et al investigated the breakup of He2 into He+/He+ after bombardment with alpha-particles of an energy of 150 keV u−1 [70]. Among other channels, clearly the fingerprint of ICD [3, 31] was observed in the ion/ion/electron-coincidence data as shown in figure 6. Here, the following reaction was observed:
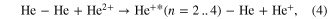
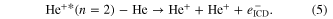
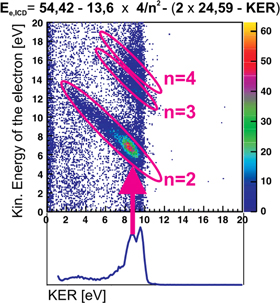
Figure 6. ICD after ion impact: as the sum kinetic energy of the ions and the ICD electron is constant, events of ICD occur along diagonals of 45o in a representation of KER versus electron energy. Reprinted with permission from [70]. Copyright 2011 by the American Physical Society. The different diagonals belong to an excitation of the He/He+ dimer ion into a state with a principal quantum number of n due to the ion impact.
Download figure:
Standard image High-resolution imageThe most detailed study of ICD after ion collisions can be found in [71] on neon and argon dimers. Here, a wide range of ionic projectiles was used (namely: 11.37 MeV u−1 S14+, 0.125 MeV u−1 He+, 0.1625 MeV u−1 He+, and 0.150 MeV u−1 He2+) resulting in final charge states q of up to q = 4 of the dimers.
In further experiments involving ion collisions by Kim et al the implications ICD might have on living tissue that is irradiated with ionizing beams were demonstrated. The model system under investigation, however, was not living tissue, but again a neon dimer [72]. Due to the simplicity of the irradiated target, it was possible to determine the amount of low energy electrons that is created solely because of ICD. The results are shown in figure 7. Plotted is the electron kinetic energy spectrum after irradiating neon monomers and neon dimers with He+ projectiles of an energy of 0.65 MeV. The projectile energy was chosen such that the energy is most efficiently deposited to the target, i.e. on the maximum of the Bragg peak. The result is striking: a surplus of 14 times more low energy electrons is found for the case of ICD as compared to the case where ICD is not possible, i.e. a case where neon monomers were ionized.
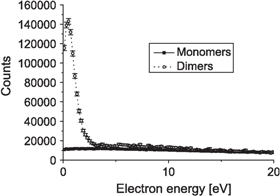
Figure 7. Kinetic energy of the electrons emitted from neon monomers (solid squares) and neon dimers (open circles) after collision with a 0.65 MeV He+ projectile. Reprinted with permission from [72].
Download figure:
Standard image High-resolution image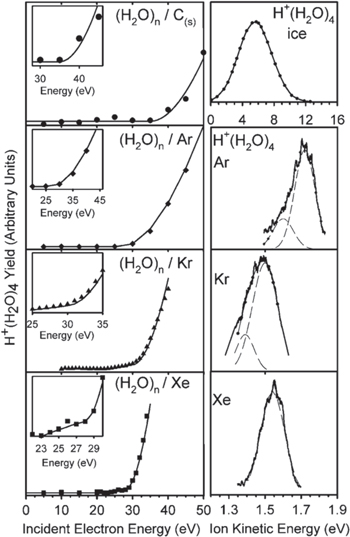
Figure 8. Evidence for ICD at heterogenous interfaces. (Left) Incident electron kinetic energy versus cluster ion yield for monolayer water films of water on graphite, Ar, Kr and Xe. (Right) Kinetic energy of the emitted H+(H2O)4 ions for the different substrates. Reprinted with permission from [76]. Copyright 2011 by the American Physical Society.
Download figure:
Standard image High-resolution image
Figure 9. Temporal evolution of the electron density for the three setups shown in the upper right corner. On the right the corresponding reaction probability (RP) in dependence of the incident energy  is depicted. Reprinted with permission from [81]. Copyright 2013 by the American Physical Society.
is depicted. Reprinted with permission from [81]. Copyright 2013 by the American Physical Society.
Download figure:
Standard image High-resolution imageSurprisingly, only little work can be found in literature on ICD after electron impact ionization, so far. Very recently Yan et al reported on a corresponding observation [73, 74]. A pulsed high energy electron beam ( ) was used to create multiply charged Ar dimers [73] and trimers [74]. In the case of electrons impinging on Ar2 two prominent features were observed in the KER distribution [73]: firstly, a structure was found, that belongs to a decay by electron transfer mediated decay (ETMD) (see figure 10 for a sketch of the process), occurring after an initial triple ionization of the dimer:
) was used to create multiply charged Ar dimers [73] and trimers [74]. In the case of electrons impinging on Ar2 two prominent features were observed in the KER distribution [73]: firstly, a structure was found, that belongs to a decay by electron transfer mediated decay (ETMD) (see figure 10 for a sketch of the process), occurring after an initial triple ionization of the dimer:
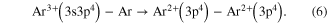
Secondly, at lower KERs and for a different sharing of the charges, a peak, that was assigned to ICD after initial triple ionization of the dimer, was measured:
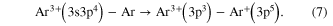
The assignment of these processes is in line with the findings reported in [75], where the same excited initial state was created by Auger decay or direct triple ionization using a synchrotron photon source.
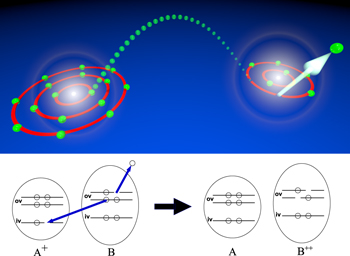
Figure 10. Sketch of ETMD(2). Top: an inner-valence vacancy in a heteronuclear dimer is filled by an electron from a neighboring atom. The excess energy is used to emitted a further electron from that atom. Bottom: representation depicting the inner-valence (iv) and outer-valence (ov) energy levels involved in ETMD(2). After ETMD(2) the atom A is neutralized, while atom B is doubly charged (right).
Download figure:
Standard image High-resolution imageBy measuring the KER after Coulomb explosion various decay mechanisms involving rich nuclear dynamics were found for Ar3 [74]. Particularly, the pathway of the breakup of Ar and Ar
and Ar is disentangled by comparison to previous work employing photons [38]. Both channels
is disentangled by comparison to previous work employing photons [38]. Both channels
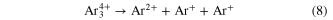
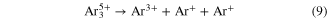
are found to be due to ICD with simultaneous single ionization of one of the argon atoms of the trimer by electron impact of the outgoing electrons.
2.5. ICD at weakly coupled heterogenous interfaces
In 2011 Grieves and coworkers examined ICD at weakly coupled condensed-phase targets [76]. The targets consisted of  monolayer (ML) of water deposited at a temperature of 30 K on clean graphite or preadsorbed multilayers (10–20 ML) of Ar, Kr or Xe. By measuring the yield and the kinetic energy of H+(H2O)n cluster after irradiating these targets with low energy electrons the mechanism responsible for the cluster desorption was retrieved. As a first result an increase of the cluster ion yield of more than one order of magnitude was observed as the water was adsorbed on rare gases. Furthermore, the onset of the cluster ion production was attributed to the energy threshold of corresponding inner-valence shells that can be subject to ICD. Depending on the rare gas species the ion yield increased as the incident electron kinetic energy reached the Ar(
monolayer (ML) of water deposited at a temperature of 30 K on clean graphite or preadsorbed multilayers (10–20 ML) of Ar, Kr or Xe. By measuring the yield and the kinetic energy of H+(H2O)n cluster after irradiating these targets with low energy electrons the mechanism responsible for the cluster desorption was retrieved. As a first result an increase of the cluster ion yield of more than one order of magnitude was observed as the water was adsorbed on rare gases. Furthermore, the onset of the cluster ion production was attributed to the energy threshold of corresponding inner-valence shells that can be subject to ICD. Depending on the rare gas species the ion yield increased as the incident electron kinetic energy reached the Ar( (29.3 eV), Kr(
(29.3 eV), Kr( (27.5 eV) and Xe(
(27.5 eV) and Xe( -level (23.3 eV) and, additionally, (as ICD may also occur after
-level (23.3 eV) and, additionally, (as ICD may also occur after  -ionization of water) the
-ionization of water) the  -threshold (
-threshold ( eV). The corresponding results are shown in figure 8 on the left panels. On the right panels the measured kinetic energy of H+(H2O)4 clusters is shown for the different substrates. Depending on the relative spatial proximities of the involved holes, the kinetic energy distribution shows distinct structures. Especially, in cases where energetically more than one hole-state is accessible by ICD, the observed peak consists of two contributions. This is the case for Ar and Kr, where a
eV). The corresponding results are shown in figure 8 on the left panels. On the right panels the measured kinetic energy of H+(H2O)4 clusters is shown for the different substrates. Depending on the relative spatial proximities of the involved holes, the kinetic energy distribution shows distinct structures. Especially, in cases where energetically more than one hole-state is accessible by ICD, the observed peak consists of two contributions. This is the case for Ar and Kr, where a  or
or  hole can be generated in water. In contrast, as the decay of the
hole can be generated in water. In contrast, as the decay of the  -state of Xe releases only enough energy to create a
-state of Xe releases only enough energy to create a  vacancy, the measured kinetic energy distribution consists of a single, narrower peak. From this threshold behavior and the measured kinetic energy of the ions it was concluded, that the H+(H2O)n-cluster ions are generated via Coulomb explosion due to ICD with a proton transfer occurring during or immediately after ICD.
vacancy, the measured kinetic energy distribution consists of a single, narrower peak. From this threshold behavior and the measured kinetic energy of the ions it was concluded, that the H+(H2O)n-cluster ions are generated via Coulomb explosion due to ICD with a proton transfer occurring during or immediately after ICD.
2.6. Quantum dots
So far, only theoretically explored is the emission of ICD electrons from quantum dots. In quantum dots, de-excitation by emission of a photon, as well, as intradot Auger decay are common processes with an atomic analogue. As quantum dots are often referred to as 'artificial atoms', it seems a logic extension, that ICD occurs, just as all other relaxation phenomena observed in atoms, in these entities, as well. In a first report by Cherkes et al ICD is proposed as a valid electron relaxation mechanism in quantum dot dimers [77]. It is shown, that in such systems an excitation energy can be transferred by ICD over a distance of 10 nm on a timescale of picoseconds.
Extending the approach, Bande et al demonstrated by applying time-dependent methods to study the electron dynamics of the decay, that resonant ICD is not restricted to atoms, molecules or specific quantum dot systems. It is a possible process in systems consisting of a sufficient amount of electrons with an arbitrary binding potential [78] with singlet states decaying more efficiently than triplet states [79] at short inter-dot distances. At larger distances between the quantum dots the decay width is dominated by the expected  -behavior. The numerical methods applied furthermore allowed for a description of the complete process of excitation of a quantum dot system employing a laser and its decay [80].
-behavior. The numerical methods applied furthermore allowed for a description of the complete process of excitation of a quantum dot system employing a laser and its decay [80].
In [81] the long-range electron interaction in double quantum dots is theoretically examined as it drives an energy transfer that is initiated by an electron capture. Again, the process described here has an analogue predicted earlier for loosely bound matter called 'interatomic Coulombic electron capture' (ICEC) [82, 83]. As an electron is captured into an atom, its excess energy needs to be emitted, typically in a process, which is the inverse of photoemission or photodetachment. In ICEC the excess energy is transferred—just as in ICD—to a neighboring atom, which, in turn, is ionized. Figure 9 shows the results obtained for ICEC in quantum dots by solving the time-dependent Schrödinger equation using a multiconfiguration time-dependent Hartree approach. As only a single quantum dot is present, the incident electron cannot be captured and is reflected as seen in figure 9(a). After adding a second quantum dot with a single energy level, electron capture is possible with an emission of an electron from the right quantum dot. In accordance with energy conservation, the momentum of the emitted electron is increased as compared to that of the impinging electron, as affirmed by the change of the slope of the wavepacket trajectory in figure 9(b). In figure 9(c) an enhanced reaction probability is observed, as the left quantum dot incorporates a second energy level. This can be explained by the resonance character of the double quantum dot  -state, i.e as such two electron states efficiently decay via ICD. If an electron approaches with a kinetic energy in the range of the two-electron resonance, it is captured with much higher probability than in the case (b), thus increasing the efficiency of ICEC.
-state, i.e as such two electron states efficiently decay via ICD. If an electron approaches with a kinetic energy in the range of the two-electron resonance, it is captured with much higher probability than in the case (b), thus increasing the efficiency of ICEC.
The results provided in [77, 78, 81] suggest, that specifically designed, ICD-capable quantum dot systems can be employed in technical applications, as for example photodetectors for detection of very weak IR-radiation with a specific wavelength.
3. Further ICD-like processes
The preceding section on ICD of quantum dots already briefly introduced the process of ICEC. Apart from ICD occurring in several different systems and after different excitation schemes, it was found throughout the last years, that further non-local ionization/excitation mechanisms are possible, as soon as an excited species is located in a chemical environment. The following section will cover some of these 'ICD-like' processes.
3.1. Electron transfer mediated decay
Already in 2001 a further decay route in loosely bound matter was predicted [84]: in clusters consisting of atoms of different species, especially in a case where the involved atoms differ strongly in their energetics, a situation can occur, where the double ionization potential of one atom lies below the inner-valence threshold of the other. In such a case, an inner-valence ionized atom can deexcite as an electron is transferred from its neighbor filling the inner-valence vacancy. The excess energy is used to further ionize the electron donor atom. This scenario was termed ETMD. A sketch of the process is shown in figure 10. While ETMD involves an electron transfer mechanism, just as the exchange contribution to ICD, it differs from ICD, as the initially excited atom is neutral after ETMD and the electron donor atom is doubly charged. Thus, ETMD can only occur in cases where the inner-valence ionization potential of one of the atoms of a heteronuclear cluster exceeds the double ionization potential of another atom of the cluster.
In later work it was predicted, that this process can cover not only two atoms, but extend to a case where three atoms are involved in the decay [85, 86]. In such a case the energy gained from the electron transfer is used to ionize the third atom. The initial terminology was extended by naming the latter process 'ETMD(3)' and the decay involving only two atoms 'ETMD(2)'. A sketch of ETMD(3) is shown in figure 11. Possible systems to investigate ETMD were pointed out in [87, 88] to be for example ArKr or ArXe raregas clusters.

Figure 11. Sketch of ETMD(3). Top: an atom of a heteronuclear trimer is ionized in an inner-valence shell. Middle: the vacancy is filled by an electron from a neighboring atom. The excess energy is transferred to a third atom yielding an outer-valence ionization of that atom. Bottom: after ETMD(3) the left and the right atoms are ionized with a vacancy in each outer-valence shell. This configuration will typically lead to a Coulomb explosion of the trimer.
Download figure:
Standard image High-resolution imageOn the side of experiments, the detailed study of Kreidi et al on the decay of neon dimers after Ne( -ionization found a possible signature of ETMD in ion/ion/electron-coincidence data. After the 1s-ionization a local Auger decay occurs, creating a doubly charged dimer with both holes being located at one atom (
-ionization found a possible signature of ETMD in ion/ion/electron-coincidence data. After the 1s-ionization a local Auger decay occurs, creating a doubly charged dimer with both holes being located at one atom (![${\rm N}{{{\rm e}}^{2+}}(2{{{\rm s}}^{-2}}){{[}^{1}}S]/{\rm Ne})$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn34.gif) . These states are able to decay via ETMD to the
. These states are able to decay via ETMD to the ![${\rm N}{{{\rm e}}^{2+}}(2{{{\rm p}}^{-2}}){{[}^{3}}P]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn35.gif) /
/![${\rm N}{{{\rm e}}^{1+}}(2{{{\rm s}}^{-1}}){{[}^{2}}S]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn36.gif) triply charged dimer states. However, at similar internuclear distances and energetics an exchange-ICD is possible, as well, drowning the experimental signature of a pure ETMD. A further evidence of ETMD was reported in [89] after examining the ionization of ArXe clusters. In 2011 the occurrence ETMD(3) and ETMD(2) was finally unambiguously measured using two different experimental approaches [90, 91]. After ionizing the Ar
triply charged dimer states. However, at similar internuclear distances and energetics an exchange-ICD is possible, as well, drowning the experimental signature of a pure ETMD. A further evidence of ETMD was reported in [89] after examining the ionization of ArXe clusters. In 2011 the occurrence ETMD(3) and ETMD(2) was finally unambiguously measured using two different experimental approaches [90, 91]. After ionizing the Ar  -shell of ArKr clusters using synchrotron radiation, ETMD(3) was observed in an electron/electron-coincidence experiment by the group of U Hergenhahn [90]. The ETMD-electrons observed in this measurement occurred at very low kinetic energies below 2 eV in coincidence with emitted photoelectrons. In the work by Sakai et al [91] a COLTRIMS setup [92–94] was employed to perform measurements on Ar2 after creating triply charged dimers with a
-shell of ArKr clusters using synchrotron radiation, ETMD(3) was observed in an electron/electron-coincidence experiment by the group of U Hergenhahn [90]. The ETMD-electrons observed in this measurement occurred at very low kinetic energies below 2 eV in coincidence with emitted photoelectrons. In the work by Sakai et al [91] a COLTRIMS setup [92–94] was employed to perform measurements on Ar2 after creating triply charged dimers with a  -vacancy in one of the argon atoms using synchrotron radiation of an energy of
-vacancy in one of the argon atoms using synchrotron radiation of an energy of  eV. A manifold of breakup channels was observed, i.e. the breakup into Ar
eV. A manifold of breakup channels was observed, i.e. the breakup into Ar Ar+, Ar
Ar+, Ar Ar2+, Ar
Ar2+, Ar Ar3+ and Ar
Ar3+ and Ar Ar2+. The latter was assigned to be created in an ETMD after a decay of the triply charged dimer states Ar
Ar2+. The latter was assigned to be created in an ETMD after a decay of the triply charged dimer states Ar ![$^{3+}(3{\rm s}3{{{\rm p}}^{4}}){{[}^{2}}S]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn44.gif) /Ar and Ar
/Ar and Ar ![$^{3+}(3{\rm s}3{{{\rm p}}^{4}}){{[}^{2}}D]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn45.gif) /Ar. Furthermore, a weak signal of ETM-decaying satellite-states as e.g. Ar
/Ar. Furthermore, a weak signal of ETM-decaying satellite-states as e.g. Ar ![$^{3+}(3{{{\rm p}}^{2}}4{\rm s}){{[}^{2}}D{{,}^{2}}P]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn46.gif) /Ar was observed.
/Ar was observed.
3.2. ICD after electron capture
Along with the prediction of ICEC [82, 83] Müller et al extended their previous work [95] and investigated electron capture in loosely bound systems in more detail [96]. As a free electron approaches an atom it may be captured into a bound state by emitting its excess energy in form of a photon. Apart from this radiative recombination (which is the inverse of photoionization), a time-reversed Auger decay is possible, as well: the excess energy of the captured electron is employed to excite another electron of the atom. This process is termed dielectronic recombination in the literature.
With ICEC, Gokhberg et al demonstrated, that the energy gained in an electron capture can be transferred to a loosely bound neighbor as in ICD, ionizing that neighbor. Müller et al investigated, in line with these findings, theoretically the occurrence of a two-center dielectronic capture (2CDR), where the atom receiving the excess energy is not ionized, but resonantly excited. They concluded, that this process indeed might be eminent: the recombination probability can be enhanced by several orders of magnitude due to a neighboring atomic center in the vicinity of the capturing atom. Its resonance character should cause 2CDR to be much more efficient than single center radiative recombination, even though a distant atom is involved in this process.
3.3. ICD-like processes after core-ionization of aqueous solutions
Already in 2008 pioneering work by Aziz et al illuminated the decay processes that occur after core excitation of OH− solvated in water [46] employing photoelectron spectroscopy and a liquid jet [97]. Follow-up investigations of liquid water and hydrogen peroxide(aq.) decaying after core excitation [98, 99] demonstrated very complex de-excitation schemes: apart from local Auger decays and ICD, other decay channels occurred that are possible only if a proton transfer between the molecules takes place. These nuclear rearrangements of the intermediate core-hole states happen on the timescale of the O core-hole lifetime of approximately 4 fs. By comparing results from decay of liquid H2O and D2O the features attributed to proton migration are visible or suppressed due to the larger mass (and thus slower nuclear motion) of the migrating deuteron. The proton migration-mediated decay mechanisms identified consist of the decay of a 'Zundel'-like cation
core-hole lifetime of approximately 4 fs. By comparing results from decay of liquid H2O and D2O the features attributed to proton migration are visible or suppressed due to the larger mass (and thus slower nuclear motion) of the migrating deuteron. The proton migration-mediated decay mechanisms identified consist of the decay of a 'Zundel'-like cation ![$[{\rm H}{{{\rm O}}^{*}}\cdots {\rm H}\cdots {{{\rm H}}_{2}}{\rm O}]_{({\rm aq})}^{+}$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn48.gif) involving different decay routes. Firstly, a proton-transfer mediated Auger process (PMT-Auger) may occur:
involving different decay routes. Firstly, a proton-transfer mediated Auger process (PMT-Auger) may occur:
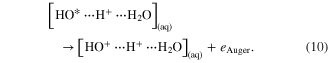
The state on the right-hand side will finally form a charge separated species of the type ![${{[{\rm O}{{{\rm H}}^{+}}\;\cdots {{{\rm H}}_{3}}{{{\rm O}}^{+}}]}_{({\rm aq})}}$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn49.gif) . Secondly, a proton-transfer mediated ICD (PMT-ICD) was observed, that generates final state species of the kind
. Secondly, a proton-transfer mediated ICD (PMT-ICD) was observed, that generates final state species of the kind ![${{[{{{\rm H}}_{2}}{{{\rm O}}^{+}}\;\cdots {{{\rm H}}_{2}}{{{\rm O}}^{+}}]}_{({\rm aq})}}$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn50.gif) :
:
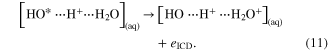
Very recently, these findings were supported by detailed simulations of the 1s Auger electron spectra of liquid H2O and D2O [100].
Kryzhevoi and Cederbaum investigated in theoretical work the decay of small ammonia clusters after core-ionization [101]. In this case a similar increase of the diversity of electronic decay processes was found. The energy gained as the core vacancy is filled, can be transferred to a neighboring molecule, as illustrated by the high kinetic energy part of the computed Auger spectra. Additionally, due to the decrease of the triple ionization threshold by charge separation in an extended system, ICD occurring subsequent to an initial local Auger decay becomes a major decay route in clusters. For the case of dimers approximately 23% of the cations created after the Auger decay undergo subsequent ICD. In the case of trimers even a fraction of approximately 43% of these states decays by ICD. Further work on inner-valence ionization of ammonia clusters [102] demonstrated, that the efficiency of ICD changes strongly, as the clusters are protonated or deprotonated, i.e. that the efficiency of ICD depends on the pH value of the decaying matter.
A series of publications on core excitation and core ionization of aqueous solutions was published by Pokapanich et al [47, 103, 104]. In the latter article it was shown that the salvation of metal-halide salts considerably influence the occurrence of ICD and ICD-like decay channels in core-ionized H2O in aqueous solutions [104]. Primarily, the alkali cations and the halide anions act as a suppressant for the O(1s) core-hole decay as the number of (otherwise open) ICD channels is reduced. The halide anions, however, participate in ICD-like decays, as the halide charge density is 'pulled towards' the core-ionized water molecule. This effect was mainly found for larger halides, i.e. Cl−, Br−, and I−, as the efficiency of these decay channels increases with increasing polarizability of the anion.
3.4. Two-center resonant photoionization
A series of publications by Müller, Najjari and Voitkiv [95, 105–108] retriggered the interest in resonant ICD [109] in 2010. It was investigated theoretically in great detail how the presence of a neighboring atom affects photoionization. In small systems consisting of only two atoms, the photoionization probability of one atom can increase by orders of magnitude, if a resonant excitation of the other atom and a subsequent ICD is energetically possible [105]. By adding a third atom to the system, the ionization probability is even further enhanced, additionally affecting the temporal evolution of the ionization probability [108].
Experimentally, studies of argon/water clusters showed an increased ionization probability of the water molecules upon excitation of the argon atoms [110]. Further experimental studies of HeNe dimers revealed an enhanced creation (by a factor of approximately 35) of HeNe+ ions after exciting different levels of the He atom of the dimer [111]. The latter publication described the process in a picture of one atom being an antenna for absorbing the light and ICD being the process of transferring that energy to the other atom of the compound.
3.5. Three-electron ICD
In very detailed theoretical work by Stoychev et al [112] on ICD following  –Auger decay of neon dimers a weak contribution due to a further possible decay scheme was identified. A doubly ionized dimer state of the type
–Auger decay of neon dimers a weak contribution due to a further possible decay scheme was identified. A doubly ionized dimer state of the type ![${\rm N}{{{\rm e}}^{2+}}(2{{{\rm s}}^{-2}}){{[}^{1}}S]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn52.gif) /Ne may deexcite to
/Ne may deexcite to ![${\rm N}{{{\rm e}}^{2+}}(2{{{\rm p}}^{-2}}){{[}^{1}}D]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn53.gif) /
/![${\rm N}{{{\rm e}}^{+}}(2{{{\rm p}}^{-1}}){{[}^{2}}P]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn54.gif) or
or ![${\rm N}{{{\rm e}}^{2+}}(2{{{\rm p}}^{-2}}){{[}^{1}}S]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn55.gif) /
/![${\rm N}{{{\rm e}}^{+}}(2{{{\rm p}}^{-1}}){{[}^{2}}P]$](https://content.cld.iop.org/journals/0953-4075/48/8/082001/revision1/jpb509541ieqn56.gif) in a decay involving three electrons. The two inner-valence holes are filled simultaneously by two 2p-electrons of the same atom. The energy gained in this de-excitation ionizes a 2p-electron of the neighboring Ne atom. Extensive theoretical studies of NeAr dimers proposed the occurrence of this decay scheme in this species [113], as well, and a corresponding process of simultaneous creation and transfer of two virtual photons in krypton heterotrimers was suggested [114]. The '
in a decay involving three electrons. The two inner-valence holes are filled simultaneously by two 2p-electrons of the same atom. The energy gained in this de-excitation ionizes a 2p-electron of the neighboring Ne atom. Extensive theoretical studies of NeAr dimers proposed the occurrence of this decay scheme in this species [113], as well, and a corresponding process of simultaneous creation and transfer of two virtual photons in krypton heterotrimers was suggested [114]. The ' ICD' (as it was termed in literature) was finally experimentally observed in NeAr dimers employing the COLTRIMS-method, as a decay from Ne/Ar
ICD' (as it was termed in literature) was finally experimentally observed in NeAr dimers employing the COLTRIMS-method, as a decay from Ne/Ar to
to  /Ar
/Ar  was identified [115, 116].
was identified [115, 116].
3.6. ICD of multiply excited clusters
In the regime of multi-photon ionization another ICD-like phenomenon was proposed in 2010 by Kuleff et al. If a homoatomic cluster is irradiated with photons of an energy belonging to a resonance below the ionization threshold of the cluster constituents, obviously two things may happen: firstly, the atoms of the cluster are excited, secondly (and much less probable) depending on the intensity of the irradiating photon beam, a two photon ionization may occur. As shown in [117] (and, as well in work by Müller et al examining other aspects of the process [107]) the cluster environment gives rise to another reaction route. The excitation energy of one atom might be transferred to an excited neighbor causing an ionization of that neighbor (see figure 12 for a sketch).
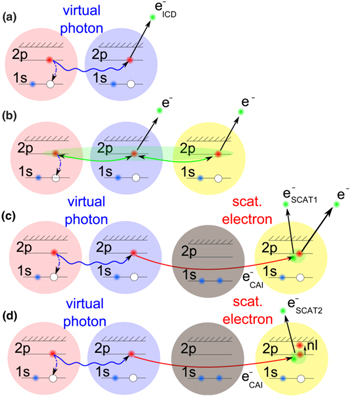
Figure 12. Depending on the amount of (excited) surrounding atoms many different interatomic de-excitation mechanisms are possible and were observed in [121]: (a) ICD of the multiply excited constituents, (b) 'collective autoionization' (CAI) of three or more excited atoms and (c), (d) a CAI involving scattering of the emitted electron. Reprinted with permission from [121]. Copyright 2014 by the American Physical Society.
Download figure:
Standard image High-resolution imageAfter first evidence in neon cluster [118, 119], the process was finally found in experiments performed on helium droplets being excited by an intense photon beam from an free electron laser [120, 121]. These publications coined the term 'collective autoionization' (CAI) for their findings and examined the different types of this ICD-like-process (as depicted in figure 12) in great detail. ICD of the multiply excited cluster (figure 12(a)) was observed mainly for small clusters. As the cluster size increases, processes involving more atoms occurred. These processes were identified to consist of either a single step (as depicted in figure 12(b)):
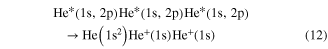
or of multiple steps involving scattering of the electron emitted due to CAI (figures 12(c) and (d)):
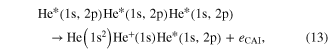
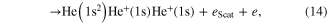
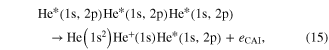
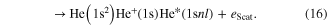
4. The role of nuclear dynamics
4.1. Theoretical groundwork
A particularly interesting aspect of ICD is the timescale on which the decay occurs. As the number of atomic or molecular neighbors increases, it becomes even more efficient (i.e. faster) than a local Auger decay. This was demonstrated theoretically in earlier work by Santra et al [16] for neon clusters and later by Averbukh et al in work on endohedral fullerenes [122]. For smaller systems ICD is typically slower than a local Auger decay, but outpaces photon emission by orders of magnitude. In such system, however, another interesting facette of ICD emerges: ICD takes place within the same time frame as typical nuclear motion of the decaying system. As the efficiency of ICD strongly depends on the internuclear distance R of the two atoms or molecules involved, the nuclear dynamics that are triggered in many cases after the excitation or ionization of (e.g.) a dimer strongly affect the decay. This was already pointed out in pioneering work by Santra et al [13] and forthcoming articles [14, 15]. The latter articles predicted that the role of nuclear dynamics may be visible in a measurement: as the system is excited, the intermediate  /Ne-state that will decay by ICD is created. The intermediate vibrational wave packet (in [15] actually only the decay of the
/Ne-state that will decay by ICD is created. The intermediate vibrational wave packet (in [15] actually only the decay of the  electronic state was investigated) starts to propagate on the intermediate potential energy surfaces. During this propagation ICD occurs, with the system decaying to the repulsive doubly charged two-site states
electronic state was investigated) starts to propagate on the intermediate potential energy surfaces. During this propagation ICD occurs, with the system decaying to the repulsive doubly charged two-site states  /
/ ) of the dimer (see figure 13 for a sketch of the process). As time-dependent calculations in [15] demonstrate, a fingerprint of the nodal structure of the intermediate vibrational wave packet could be found in the ICD electron kinetic energy spectrum (or correspondingly the KER spectrum). As a nodal structure of such a kind needs some time to evolve and to built up, the pattern becomes more clearly visible for longer decay times, as depicted in figure 14.
) of the dimer (see figure 13 for a sketch of the process). As time-dependent calculations in [15] demonstrate, a fingerprint of the nodal structure of the intermediate vibrational wave packet could be found in the ICD electron kinetic energy spectrum (or correspondingly the KER spectrum). As a nodal structure of such a kind needs some time to evolve and to built up, the pattern becomes more clearly visible for longer decay times, as depicted in figure 14.

Figure 13. Visualizing the role of nuclear dynamics of ICD: a neon dimer is ionized from the ground state ( ) to the intermediate state (
) to the intermediate state ( ) that will undergo ICD (lower panel). After population the intermediate wave packet starts to propagate and decay to the final state (
) that will undergo ICD (lower panel). After population the intermediate wave packet starts to propagate and decay to the final state ( ) (upper panel). Reprinted with permission from [15]. Copyright 2003, AIP Publishing LLC.
) (upper panel). Reprinted with permission from [15]. Copyright 2003, AIP Publishing LLC.
Download figure:
Standard image High-resolution image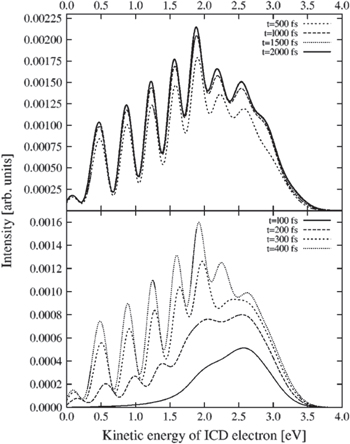
Figure 14. Temporal evolution of the kinetic energy spectrum of the ICD electron. After a certain time the nodal structure of the decaying intermediate vibrational wave packet becomes clearly visible with the spectrum converging at times of approximately 2 ps. Reprinted with permission from [15]. Copyright 2003, AIP Publishing LLC.
Download figure:
Standard image High-resolution imagePioneering experiments on ICD of neon dimers [3], however, did not observe these nodal features. Even though the underlying physics were assumed correctly and the calculations were performed accurately, the final state potential energy surface in [15] was only considered as an average of the different states possible. A followup publication that resolved this issue by including all possible states to the decay produced the correct ICD electron spectrum [123].
With these theoretical groundworks it became obvious, that the temporal evolution of ICD should be of great interest. ICD was, for example, expected to be a process, where a non-exponential decay behavior could be cleanly observable in time-resolved measurements, which became more and more feasible (see e.g. pioneering work by Drescher, Krausz et al and the group of P Corkum [124, 125]). In further theoretical work by Kuleff and Cederbaum a very intriguing yet intuitive demonstration of the process of ICD was performed in 2007. In [126] the electron dynamics of ICD were examined in real time and space taking electron correlation among all electrons fully into account. As a model system the NeAr dimer was chosen in order to trace the evolution of the electron cloud during ICD. The results are shown in figure 15. The distribution of the electron hole starts as a 2s-vacancy in the neon atom of the dimer. The s-character of the hole yields a single-ridge feature at the neon atom, just as expected for a s-shell. After a few femtoseconds the distribution located at the neon atom changes: a minimum occurs at the center of the neon atom, as the 2s-vacancy is filled by a 2pz-electron. Accordingly, at the argon atom, the hole density increases. It has, as well, mainly the p-character of the emitted 3pz-ICD-electron. The findings nicely demonstrate that the decay is dominated by the ICD channel consisting of  Ar decaying into
Ar decaying into  Ar
Ar confirming similar experimental findings on Ne2 reported in [127]: by measuring the ICD-electron angular emission distribution in the dimer-fixed frame, a preferred emission along the the molecular axis of the dimer was found. This was attributed to mainly the 2pz-electron being involved in ICD. The emission pattern is shown in figure 16. A similar behavior was later observed by Kreidi et al and Semenov et al [39, 128] and Yamazaki et al [40].
confirming similar experimental findings on Ne2 reported in [127]: by measuring the ICD-electron angular emission distribution in the dimer-fixed frame, a preferred emission along the the molecular axis of the dimer was found. This was attributed to mainly the 2pz-electron being involved in ICD. The emission pattern is shown in figure 16. A similar behavior was later observed by Kreidi et al and Semenov et al [39, 128] and Yamazaki et al [40].
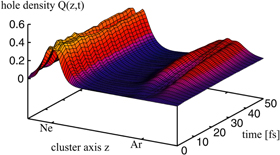
Figure 15. Evolution of the electron hole density in real space and time after Ne(2s)-ionization of a NeAr dimer. The electron hole is initially located at the neon atom occurring in a s-contribution (i.e. as a single ridge). As time evolves the s-character of the hole at the neon atom changes into a double-ridge structure, as expected from the a 2p-electron filling the 2s-hole. Accordingly a 3p-hole is created at the argon atom, as the ICD electron is emitted. Reprinted with permission from [126]. Copyright 2007 by the American Physical Society.
Download figure:
Standard image High-resolution image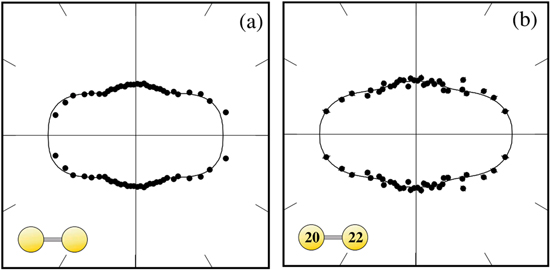
Figure 16. Angular emission pattern of the ICD electron in the dimer frame. The dimer is aligned horizontally, as depicted by the icon. (a) shows the emission distribution for 20Ne20Ne and (b) for 22Ne20Ne. Reprinted with permission from [127].
Download figure:
Standard image High-resolution image4.2. Nuclear dynamics driven ICD
In 2010 a theoretical and experimental co-study of the HeNe dimer unveiled another possible influence of the nuclear dynamics on ICD [129]. Sisourat and Sann et al demonstrated the existence of an ICD-channel that is energetically closed at the ground state mean internuclear distance of the HeNe dimer. The decay of these states is therefore driven by the nuclear motion of the ground state and the intermediate decaying state. Figure 17 shows the corresponding results obtained from ab inito calculations [130–132]. The different states depicted in figure 17 are the intermediate 2s-ionized  He
He (state 1) and the shakeup states
(state 1) and the shakeup states  He
He (state 2a:
(state 2a:  and state 2b:
and state 2b:  ) and
) and  He
He ,
, (state 3). While the tail of the vibrational wave function of the ground state extends to internuclear distances R up to
(state 3). While the tail of the vibrational wave function of the ground state extends to internuclear distances R up to  a maximum occurs at
a maximum occurs at  . The different intermediate states are energetically open at distances
. The different intermediate states are energetically open at distances  (state 1),
(state 1),  (states 2a, 2b) and
(states 2a, 2b) and  (state 3). Regarding ICD of state 1, which is only open at largest internuclear distances, the following conclusion was made: initially a small portion of the vibrational ground state can be found at these very large internuclear distances. After population of state 1, the dimer starts to contract, reaching a regime of internuclear distances in which ICD is no longer possible. After a period of ∼1.5 ps the dimer's internuclear distance is large enough again to allow for ICD. Therefore, as the dimer stretches and contracts due to its nuclear dynamics, the probability for ICD changes. This is visible in figure 17, where the time derivative of the decaying wave packet divided by the radiative decay rate is shown. In that representation a normalized instantaneous rate of 1 corresponds to a case where ICD and photoemission are equally efficient. The figure shows on one hand, that even for the weak states ICD is able to outpace radiative decay and on the other hand the oscillations of the decay rate corresponding to the vibrational period of the dimer can be seen. The behavior of state 2 is similar, however, the oscillations occur at a different period of ∼1 ps. As this is in line with the vibrational periods of the two vibrational states involved in the decay, the effect of nuclear motion can be traced explicitly in these cases, as well. State 3 is different: here ICD is open for all vibrational levels over all internuclear distances. The oscillatory structure observed for the other states vanishes for state 3, as several vibrationally excited states having different vibrational periods are involved. Furthermore, the dramatic drop in the normalized instantaneous rate occurs due to the fact, that after a short time already most of state 3 is depopulated by ICD.
(state 3). Regarding ICD of state 1, which is only open at largest internuclear distances, the following conclusion was made: initially a small portion of the vibrational ground state can be found at these very large internuclear distances. After population of state 1, the dimer starts to contract, reaching a regime of internuclear distances in which ICD is no longer possible. After a period of ∼1.5 ps the dimer's internuclear distance is large enough again to allow for ICD. Therefore, as the dimer stretches and contracts due to its nuclear dynamics, the probability for ICD changes. This is visible in figure 17, where the time derivative of the decaying wave packet divided by the radiative decay rate is shown. In that representation a normalized instantaneous rate of 1 corresponds to a case where ICD and photoemission are equally efficient. The figure shows on one hand, that even for the weak states ICD is able to outpace radiative decay and on the other hand the oscillations of the decay rate corresponding to the vibrational period of the dimer can be seen. The behavior of state 2 is similar, however, the oscillations occur at a different period of ∼1 ps. As this is in line with the vibrational periods of the two vibrational states involved in the decay, the effect of nuclear motion can be traced explicitly in these cases, as well. State 3 is different: here ICD is open for all vibrational levels over all internuclear distances. The oscillatory structure observed for the other states vanishes for state 3, as several vibrationally excited states having different vibrational periods are involved. Furthermore, the dramatic drop in the normalized instantaneous rate occurs due to the fact, that after a short time already most of state 3 is depopulated by ICD.
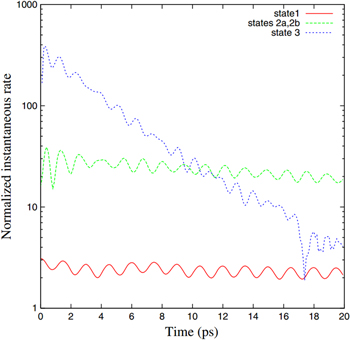
Figure 17. The normalized instantaneous rate of the decay states 1, 2a, 2b and 3 (see text) as a function of time. Reprinted with permission from [129]. Copyright 2010 by the American Physical Society.
Download figure:
Standard image High-resolution image4.3. The helium dimer
Havermeier et al were able to finally observe the nodal structure of the IC-decaying vibrational state, that was initially predicted to occur in the neon dimer [15], in a very exotic rare gas system: the helium dimer [31, 133].
For a long time, even the existence of that rare gas dimer was under dispute by itself. After it was predicted already in 1928 by Slater [134] that a very weakly bound compound of two helium atoms may be stable, an ongoing dispute among theoreticians was triggered [135]. All other elements in the periodic table have some electrons which are more loosely bound than the two electrons of the helium atom, making helium the most unpolarizable element. This lack of polarizability yields a very weak van der Waals interaction between helium atoms, allowing—in the case of a helium dimer—for only a single vibrational state. Even though the potential minimum, which is located at an internuclear distance  , has a depth of almost 1 meV [136], the dimer is only bound by a binding energy in the order of 10−7 eV [136–138]. This reflects the fact, that the zero point motion of the atoms of the dimer is close to 1 meV, explaining the long fought debate on the dimer's existence. Already substituting one of the helium atom of He2 with its isotope 3He causes the system to became unbound due to the increased zero point motion [139]. Finally, the dispute on the existence was put to an end by the detection of the dimer employing a matter-wave diffraction method [137, 140].
, has a depth of almost 1 meV [136], the dimer is only bound by a binding energy in the order of 10−7 eV [136–138]. This reflects the fact, that the zero point motion of the atoms of the dimer is close to 1 meV, explaining the long fought debate on the dimer's existence. Already substituting one of the helium atom of He2 with its isotope 3He causes the system to became unbound due to the increased zero point motion [139]. Finally, the dispute on the existence was put to an end by the detection of the dimer employing a matter-wave diffraction method [137, 140].
Apart from being the most weakly bound atomic system in the universe other properties of the helium dimer, are intriguing, as well. Along with its very low binding energy comes its huge size. Experimentally the mean internuclear distance was determined by Grisenti et al to be  [137]. The tail of the vibrational wave function, however, extends almost into the macroscopic regime of several
[137]. The tail of the vibrational wave function, however, extends almost into the macroscopic regime of several  [138]. Therefore, the helium dimer can be considered (just as many other van der Waals bound systems) as basically empty, as the internuclear distance is orders of magnitude larger than the diameter of the helium atom. Furthermore, the helium dimer exists in a so-called 'quantum halo state' [141]: as the vibrational wave function extends far beyond the potential well into to the classically forbidden region, the helium atoms of the dimer reside mostly 'in the tunnel'. The probability density of internuclear distances is therefore very well approximated by an exponential decay function for internuclear distances larger than
[138]. Therefore, the helium dimer can be considered (just as many other van der Waals bound systems) as basically empty, as the internuclear distance is orders of magnitude larger than the diameter of the helium atom. Furthermore, the helium dimer exists in a so-called 'quantum halo state' [141]: as the vibrational wave function extends far beyond the potential well into to the classically forbidden region, the helium atoms of the dimer reside mostly 'in the tunnel'. The probability density of internuclear distances is therefore very well approximated by an exponential decay function for internuclear distances larger than  [136]. A recent review on helium clusters and their interesting properties can be found in [142].
[136]. A recent review on helium clusters and their interesting properties can be found in [142].
In order to trigger ICD in a helium dimer, shakeup ionization was used:
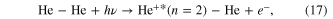
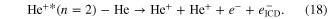
By measuring the emitted ions and electrons in coincidence, the typical fingerprint of ICD, i.e. the 45°-diagonal feature when displaying the dependence of the electron kinetic energy and the KER [3], was obtained. The corresponding distribution published in [31] is shown in figure 18. Two features are visible: a horizontal line corresponding to the  -shakeup photoelectron (a photon energy of
-shakeup photoelectron (a photon energy of  eV was chosen for ionization) and the aforementioned diagonal belonging to events where the ICD electron was measured in coincidence with the two He+-ions.
eV was chosen for ionization) and the aforementioned diagonal belonging to events where the ICD electron was measured in coincidence with the two He+-ions.
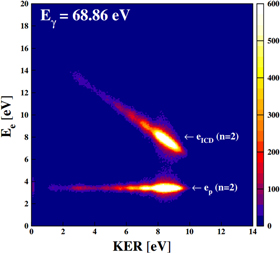
Figure 18. Kinetic energy release versus electron kinetic energy of a Coulomb exploding He dimer. Reprinted with permission from [31]. Copyright 2010 by the American Physical Society.
Download figure:
Standard image High-resolution imageFor large systems (as most van der Waals bound clusters), the KER-distribution gives information on the internuclear distance of the particles involved in the Coulomb explosion. The two ions can be treated in good approximation as two point charges that are repelling each other by the Coulomb force. Within the 'reflection approximation' [143] the relation of KER and internuclear distance R is given (in atomic units) simply as:  . Figure 18 therefore reveals, that ICD occurs in dimers with internuclear distances up to
. Figure 18 therefore reveals, that ICD occurs in dimers with internuclear distances up to  For larger internuclear distances the competing de-excitation by photon emission is similarly or more effective limiting the observability of ICD, whose probability drops with
For larger internuclear distances the competing de-excitation by photon emission is similarly or more effective limiting the observability of ICD, whose probability drops with  .
.
A further, closer look at figure 18 reveals a modulation of the KER, which was examined in more detail in [144–146]. Figure 19 shows the KER-distribution. In order to obtain the theoretical KER-spectrum, the different partial contributions from decaying electronic states with different symmetries were added up. Figure 20 explains in more detail the origin of the nodal structure observed. In the top panel the probability density of the vibrational wave function of the electronic state that is mainly contributing to ICD is shown as a red slash dotted line. In order to account for the dependence of ICD on the internuclear distance of the atoms involved in ICD, a weight factor of  needs to be considered. Correspondingly, the black slash-dotted line depicts the vibrational wave function multiplied by
needs to be considered. Correspondingly, the black slash-dotted line depicts the vibrational wave function multiplied by  . As the decay occurs, the structure of the weighted vibrational wave function is reflected at the final state potential energy curve, which is shown in the middle panel. On the right the resulting KER-distribution of that decaying state is plotted, visualizing that the nodal features indeed belong to nodes and maxima present in the vibrational wave function. In the experiment, however, the superposition of all possible decaying states is measured. Surprisingly, this does not completely wash out the structures, even though the positions of the maxima and minima differ strongly for each state.
. As the decay occurs, the structure of the weighted vibrational wave function is reflected at the final state potential energy curve, which is shown in the middle panel. On the right the resulting KER-distribution of that decaying state is plotted, visualizing that the nodal features indeed belong to nodes and maxima present in the vibrational wave function. In the experiment, however, the superposition of all possible decaying states is measured. Surprisingly, this does not completely wash out the structures, even though the positions of the maxima and minima differ strongly for each state.

Figure 19. Kinetic energy release of the Coulomb exploding He dimer after ICD. The theoretical spectrum (black line) was obtained by adding up all partial spectra (colored lines). Reprinted with permisson from [144]. Copyright 2010, rights managed by Nature Publishing Group.
Download figure:
Standard image High-resolution image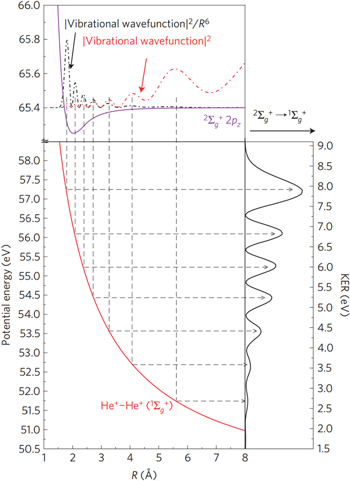
Figure 20. Top: probability density of the vibrational wave function of the electronic state that is mainly populated in the initial shakeup process ( ) (red dotted line). The black dotted line shows the latter probability density multiplied with
) (red dotted line). The black dotted line shows the latter probability density multiplied with  taking into account the decay behavior. Middle: potential energy curve of the final state of the decay. Right: kinetic energy release after the decay. Reprinted with permisson from [144]. Copyright 2010, rights managed by Nature Publishing Group.
taking into account the decay behavior. Middle: potential energy curve of the final state of the decay. Right: kinetic energy release after the decay. Reprinted with permisson from [144]. Copyright 2010, rights managed by Nature Publishing Group.
Download figure:
Standard image High-resolution image4.4. Examining the temporal evolution of ICD in an experiment
Already one of the three articles, that are typically referred to as the ones that experimentally confirmed the existence of ICD [2–4], elucidated the efficiency of the process by measuring the decay width of ICD in neon clusters. In pioneering work Öhrwall et al showed that the lifetime of a 2s-vacancy in neon atoms being condensed to clusters is much shorter than that of the latter vacancy in isolated neon atoms [4]. Further publications measuring the lifetime of IC-decaying states by assuming an exponential decay function followed: Trinter et al performed vibrationally resolved measurements of ICD of HeNe heterodimers that yielded lifetimes that differed depending on the decaying state from 100 fs to 1 ps [111]. Another method that was applied to infer the timescale of ICD is the so called 'core-hole clock'. By comparing the ratio of ICD and a competing Auger decay, the IC-decay times can be extracted if the Auger lifetimes are known. This approach was, for example, employed by Pokapanich et al to examine ICD of aqueous ions [103]. Furthermore, by comparing the structure of the KER spectrum of a certain IC-decaying state in NeAr obtained in an experiment to different theoretical predictions, Ouchi et al were able to determine the lifetime of that state [116].
While it was experimentally possible to measure the decay times of ICD, a truly time-resolving measurement depicting the non-exponential decay behavior was on the agenda of many ultrafast laboratories for a long time (see for example [147] for a short review). Most recently two experiments approached this goal employing different techniques to gather information on the timing of individual ICD events in a coincidence measurement.
Schnorr et al used a COLTRIMS setup attached to the Hamburg free electron laser facility (FLASH) to measure ICD after 2s-ionization of neon dimers [148]. By using a pump/probe scheme employing a split mirror, the decay time of single ICD events was probed. A sketch of the involved potential energy curves is shown in figure 21: in a first step the FLASH-light ( eV) is used to create the 2s-vacancy to trigger ICD populating the
eV) is used to create the 2s-vacancy to trigger ICD populating the  –Ne-state located in the middle of figure 21. After a delay, which is set by slightly shifting one half of the split mirror used for focussing the FLASH-beam, the probe-pulse arrives. In case the intermediate
–Ne-state located in the middle of figure 21. After a delay, which is set by slightly shifting one half of the split mirror used for focussing the FLASH-beam, the probe-pulse arrives. In case the intermediate  –Ne-state is not yet depopulated by ICD, a further ionization to Ne
–Ne-state is not yet depopulated by ICD, a further ionization to Ne Ne or Ne
Ne or Ne Ne+ can occur. If, however, ICD took place before the probe-pulse occurs, the dimer is already in a (Coulomb exploding) Ne
Ne+ can occur. If, however, ICD took place before the probe-pulse occurs, the dimer is already in a (Coulomb exploding) Ne Ne+-state, thus enabling an ionization into a triply charged Ne
Ne+-state, thus enabling an ionization into a triply charged Ne Ne+-state. By measuring the two ions that are created during the process in coincidence the temporal decay function of ICD can be deduced from the dependence of the intensity of the Ne
Ne+-state. By measuring the two ions that are created during the process in coincidence the temporal decay function of ICD can be deduced from the dependence of the intensity of the Ne Ne+-state on the split mirror delay. It is obvious, that the intensity of the photon beam needs to be chosen carefully for this approach to work: on one hand the experiment needs to be operated at an intensity where direct multi-photon processes are rare in order to avoid the direct creation of the Ne
Ne+-state on the split mirror delay. It is obvious, that the intensity of the photon beam needs to be chosen carefully for this approach to work: on one hand the experiment needs to be operated at an intensity where direct multi-photon processes are rare in order to avoid the direct creation of the Ne Ne+-state. One the other hand the pump/probe-approach requires a certain saturation of each ionizing step in order to gather sufficient statistics. Within the error bars of this challenging experiment an exponential decay behavior was found with
Ne+-state. One the other hand the pump/probe-approach requires a certain saturation of each ionizing step in order to gather sufficient statistics. Within the error bars of this challenging experiment an exponential decay behavior was found with  fs.
fs.
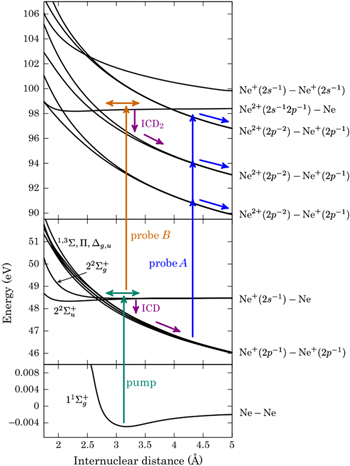
Figure 21. Potential energy surfaces of the neon dimer that are involved in the pump/probe scheme by Schnorr et al to extract the lifetime of single ICD events. Reprinted with permission from [148]. Copyright 2013 by the American Physical Society.
Download figure:
Standard image High-resolution imageAt the same time Trinter et al came up with a different approach to measure the decay time of individual ICD events [149]: in streaking experiments the timing information is typically mapped to a measurable kinetic energy, e.g. of a photoelectron (see e.g. work by Drescher et al [124]). It turns out, that nature itself does something very similar in case of a decay that involves emission of an electron, that is much faster than the photoelectron emitted afore. This effect is known already for a long time [150–152]. In early work by Niehaus [150], autoionization after inner-shell photoionization was examined:
The influence of the Coulomb field of a receding (slow) photoelectron on the autoionzation process was termed 'post collision interaction' (PCI). In a simplified (and classical) view the following happens, as PCI occurs: if an Auger electron emission happens while a photoelectron emerges from its parent ion, the charge of the parent ion changes. Thus, the photoelectron is suddenly exposed to the Coulomb field of a doubly charged ion. As a result the photoelectron kinetic energy decreases (and accordingly the energy of the Auger electron increases). It is understandable, that the amount of energy loss of the photoelectron depends on the decay time of the parent ion. If the decay is fast, the sudden change of the potential happens at small distances between the photoelectron and the ion, i.e. at distances where the Coulomb interaction is still very strong. For longer decay times (and thus larger distances between the photoelectron and the ion) the effect weakens [153–155]. As the energy loss is a measurable quantity, an experimental access to the decay time of single decay-events is possible. This is especially interesting in a coincidence measurement where, following that approach of 'PCI-streaking', the temporal evolution of a quantity measured in coincidence with the energy loss can be obtained.
In figure 22(a) the conversion function mapping a measured electron kinetic energy to decay time is plotted for four different initial photoelectron energies. It was obtained using a simple one dimensional classical model. In a simulation an electron of a kinetic energy of Ei = 140 meV is emitted. After a delay time tICD a second electron (the ICD electron) with a kinetic energy of 10 eV (which is the most typical kinetic energy for an ICD electron in He2) is launched. At some distance Rp from the origin, the second electron reaches the photoelectron. The energy difference Ed between a Coulomb potential of charge two and a Coulomb potential of charge one at Rp is the amount of energy the photoelectron is decelerated. Plotting tICD versus  yields figure 22(a). The conversion function is (as expected) strongly nonlinear and the range of accessible decay times is determined by the initial energy of the photoelectron prior to PCI. Considering the most upper curve, which belongs to a photoelectron being emitted at an energy of
yields figure 22(a). The conversion function is (as expected) strongly nonlinear and the range of accessible decay times is determined by the initial energy of the photoelectron prior to PCI. Considering the most upper curve, which belongs to a photoelectron being emitted at an energy of  meV: if ICD happens, for example, at a time of 100 fs, the photoelectron is decelerated and occurs at an energy of only 60 meV. The actual experiment was performed similarly to the one by Havermeier et al. Helium dimers were ionized and excited using synchrotron radiation in order to trigger ICD. As the ICD electron has a mean energy of 10 eV, the photoelectron needed to be sufficiently slow in order for PCI to occur. The photon energy was chosen accordingly to create a shakeup photoelectron of E = 140 meV. In figure 22(b) the results of the coincidence measurement are shown in a graph showing the dependence of the KER on the photoelectron energy. The latter is converted to decay times, employing the procedure just mentioned, yielding the time axis on the right side of panel (b). As the figure reveals, the KER strongly depends on the time at which ICD takes place. For short decay times mainly lower KERs occur. This (making use of the reflection approximation described in the previous section) can be understood intuitively, as the helium dimer is a huge system and large internuclear distances lead to small KER values. As nuclear motion evolves after the shakeuop-ionization of the dimer, it starts to contract, causing the main peak at around KER = 8 eV to occur. This KER-values corresponds to the inner turning point of the vibrational motion of the dimer. For longest decay times, i.e. smallest energy losses of the photoelectron, the vibrational features observed in [31] build up, which is consistent with the fact, that these sharp characteristics need sufficient time for the dimer to vibrate in order to form.
meV: if ICD happens, for example, at a time of 100 fs, the photoelectron is decelerated and occurs at an energy of only 60 meV. The actual experiment was performed similarly to the one by Havermeier et al. Helium dimers were ionized and excited using synchrotron radiation in order to trigger ICD. As the ICD electron has a mean energy of 10 eV, the photoelectron needed to be sufficiently slow in order for PCI to occur. The photon energy was chosen accordingly to create a shakeup photoelectron of E = 140 meV. In figure 22(b) the results of the coincidence measurement are shown in a graph showing the dependence of the KER on the photoelectron energy. The latter is converted to decay times, employing the procedure just mentioned, yielding the time axis on the right side of panel (b). As the figure reveals, the KER strongly depends on the time at which ICD takes place. For short decay times mainly lower KERs occur. This (making use of the reflection approximation described in the previous section) can be understood intuitively, as the helium dimer is a huge system and large internuclear distances lead to small KER values. As nuclear motion evolves after the shakeuop-ionization of the dimer, it starts to contract, causing the main peak at around KER = 8 eV to occur. This KER-values corresponds to the inner turning point of the vibrational motion of the dimer. For longest decay times, i.e. smallest energy losses of the photoelectron, the vibrational features observed in [31] build up, which is consistent with the fact, that these sharp characteristics need sufficient time for the dimer to vibrate in order to form.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 22. (a) Conversion of measured photoelectron energy to decay time: the four lines show four examples of initial (undisturbed) photoelectron energies of (top to bottom) 140, 100, 70 and 30 meV. if, for example, an electron that is expected to occur at an energy of 140 meV is decelerated due to PCI (see text) and measured at an energy of 60 meV the corresponding decay time was 100 fs. (b) KER versus measured electron energy. The time-axis on the right was obtained according to the most upper curve (initial energy of 140 meV). Reprinted with permission from [149]. Copyright 2013 by the American Physical Society.
Download figure:
Standard image High-resolution image{kind=link}
By integrating over the KER release the decay function of ICD in the helium dimer was retrieved, finally unveiling the non-exponential decay behavior that occurs due to the nuclear dynamics during ICD in an experiment.
5. Summary
During the 10 years since the experimental verification of Interatomic Coulombic Decay a broad field of experimental and theoretical studies has emerged. In this review a small subset of the topics investigated was presented with the goal to give an impression on the diversity of the field and its achievements.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft within the ICD-research unit FOR-1789. I am indebted to L Cederbaum, K Gokhberg, N Sisourat, K Ueda, U Hergenhahn, H Schmidt-Böcking and R Dörner for all the discussion, ideas and support during the last years. I would like to express my thanks for all the effort and hard work that the members of the experimental atomic physics group in Frankfurt spend every day.