


Abstract
An important approach in the design of new environmentally friendly materials is represented by the study of analogous systems already existing in nature. In the search for new water splitting catalysts, the corresponding natural analogue is represented by the oxygen-evolving complex of photosystem II, which is a large membrane protein complex present in photosynthetic organisms. The understanding of the catalytic strategy of its active Mn4CaO5 core is important to unravel the mechanisms of water oxidation in photosynthesis and can serve as an inspiring model for the design of biomimetic catalysts based on largely non-toxic, earth abundant elements. The magnetic interactions between Mn ions are studied in the present work by means of DFT + U broken symmetry ab initio molecular dynamics within a quantum mechanics/molecular mechanics framework. The room temperature dynamics of two different structural models (i.e. with total high-spin and total low-spin ground states) was stable during the simulated time. We observed large fluctuations of the magnetic coupling constants calculated on both the structural models of the complex, causing occasionally instantaneous swapping of the ferromagnetic/antiferromagnetic coupling between the metal centers.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Photocatalytic energy production by direct conversion of solar energy into a chemical fuel, such as hydrogen or hydrocarbons, is a major challenge in the research of new materials for energy. In particular, the discovery of new catalytic materials based on earth-abundant elements that are capable to perform the oxidation of water with high efficiency is a key ingredient for the future development of devices that are able to capture and store solar energy on a large scale [1]. Natural photosynthetic processes efficiently perform this task, capturing solar photons and converting them into chemical energy that can be stored in stable organic molecules. One of the most important steps in this process is represented by the oxidation of water, which is carried out by the oxygen evolving complex (OEC) of photosystem II (PSII), which is a protein/pigments complex that has remained almost evolutionary unchanged since its first appearance about 2.5 billion years ago [2]. The active site of the enzyme is composed of a core of four Mn ions and one Ca ion that are connected together through μ-oxo-bridges in an open cubane fashion [3]. In the last years, an inorganic cobalt-based catalyst film, having favorable working and stability conditions, was shown to be able to catalyze electrochemical water oxidation with small overpotential [4, 5]. The film is an amorphous solid that is self assembled from low cost cobalt salts at the anode. It has been recently combined with a silicon junction and with a hydrogen evolution catalyst to form a prototype 'artificial leaf' [6]. Interestingly, x-ray scattering measurements have revealed that the structure of this inorganic catalyst shares some common features with its natural counterpart [7–9].
Recently, Dau and co-workers [10, 11] have reported an Mn oxide that has also been characterized as water-oxidizing. The design of new materials based on Mn would be desirable since this element, which is that chosen by nature, is earth-abundant, inexpensive and largely non-toxic. Electronic structure calculations and ab initio molecular dynamics (MD) have been successfully used to unravel the structural and functional properties of molecular models of materials for water oxidation based on transition metals [12–14]. These studies are important to understand the similarities and differences of the electronic properties of the catalysts and they might also help in the design of new biomimetic materials, when compared to parallel studies on natural enzymes.
A high-resolution crystallographic structure of the PSII complex has recently revealed the atomistic details of the catalytic Mn4CaO5 core [3]. This structure provides the grounds for parallel computational investigations of the catalytic mechanism of the OEC [15–18]. It also helps the study of natural and synthetic analogous for water splitting reactions [9]. During the catalytic pathways toward oxygen evolution, the natural system cycles through different states, which are the S0–S4 states of the so-called Kok's Cycle [19]. Despite a large amount of data being available for the functional and spectroscopic properties of such states in different organisms and experimental conditions [20–23], the exact reaction mechanism and the strategy adopted by the enzyme to carry on its task are still elusive. An important aspect of the OEC, as well as of their artificial analogues, is represented by the magnetic interactions between the transition metals connected by oxo-bridges. In this respect, electronic paramagnetic resonance is successfully and routinely used to probe the oxidation state and the chemical environment of the PSII core along the Kok's cycle and its intermediates [21]. From the computational point of view, the description of such magnetic states can be conveniently described through broken-symmetry (BS) approach based on density functional theory (DFT) [24–29]. In a recent work, Pantazis et al [18] have proposed two distinct gas-phase (GP) molecular models of the Mn4CaO5 cluster in PSII, differing by the magnetic interactions between Mn ions. These two structures have two different overall spin ground states and their interconversion can explain the presence of two distinct signals observed by EPR spectroscopy in the S2 state [20, 21, 30, 31]. Two important factors should be considered to fully describe the interplay of such magnetic interactions in biologically relevant conditions, which are: the effect of temperature and the role of the protein field. To achieve this goal, ab initio MD simulations coupled to quantum mechanics/molecular mechanics (QM/MM) scheme can be used to unravel both the effect of dynamics and the effect of the environment on the average values and fluctuations of the magnetic properties [32]. To properly describe the electron correlation of the transition metal centers, it is necessary to go beyond the approximations used by the standard DFT, either with the use of hybrid functionals or with the DFT + U scheme [33–37]. Since the latter approach is particularly computationally efficient for ab initio MD calculations, it has been adopted for the present study.
In this work, we present a QM/MM study of the Mn4CaO5 cluster of PSII carried on at the DFT + U level for both the low-spin and the high-spin proposed structures. Our extensive analysis will reveal the role of the temperature and of the protein field in the modulation of the magnetic interactions in the PSII catalytic core.
2. Materials and methods
The portion of the system close to the active site was treated by DFT as an isolated cluster (GP calculations) or immersed into its protein environment (QM/MM calculations). As suggested by Pantazis et al [18], for both calculations we have considered two different configurations of the cluster, which are hereafter referred to as model-A and model-B, differing by the oxidation and spin pattern of the Mn ions as detailed below.
2.1. Gas-phase model
The GP model was defined consistently with the DFT calculations that were previously reported for the S2 state [18, 31, 38]. The system is shown in left panel of figure 1, and it is composed by 238 atoms: the metal core (Mn4CaO5), the side chains of its ligands (Asp170, Glu189, His332, Glu333, Asp342, Ala344, and CP43-Glu354), their neighbors residues (Asp61, Tyr161, His190, CP43-Arg357 and His337), the two water molecules coordinated to the Ca2+, a H2O plus an OH− bound to the terminal Mn4 (see also figure 2), and the closest eight crystallographic water molecules (not shown for clarity in figure 1).
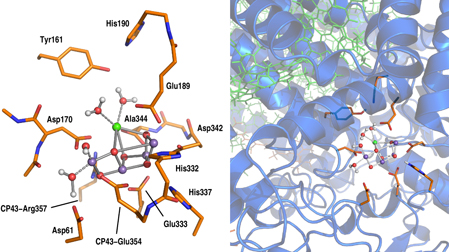
Figure 1. Selected atoms from the molecular models of the catalytic center of the OEC used for the DFT calculations in GP (left panel) and QM/MM calculations (right panel). A detailed description of the atoms considered in both models is reported in section 2. The Mn4CaO5 cluster is shown by a ball-and-stick representation (Mn purple, Ca green, O red and H white), while the other residues included in the QM part are reported with sticks. In the right panel, the portion of the system treated at classical level is shown in cartoon representation (protein residues) and in green sticks (chlorophylls).
Download figure:
Standard image High-resolution image
Figure 2. Structural and magnetic features of model-A and model-B. On the left, the Mn4CaO5 cluster for the two models is shown in a ball-and-stick representation. The right panel describes the configuration of the ground spin state for the two models. The up and down arrows indicate the spin projection, whereas the Roman numerals refer to the formal oxidation state of the four Mn atoms.
Download figure:
Standard image High-resolution imageIn agreement with a previous study [18] we defined and investigated two different conformers of the catalytic core in S2 state (see figure 2), the GP model-A (GP-A) and the GP model-B (GP-B). The geometries of these two models differ from each other by the position of an oxo-bridge (O5) which is bound in one case to Mn4 (model-A) and in the other case to Mn1 (model-B). Consequently, the oxidation states of the Mn1 and Mn4 in the two models differ according to the respective coordination pattern (i.e. Mn1(III) Mn4(IV) in model-A and Mn1(IV) Mn4(III) in model-B).
From the electronic structure point of view, the electron open-shell Mn4CaO5 cluster is composed by one MnIII and three MnIV, with four and three unpaired electrons, respectively. These electrons occupy the quasi-degenerate d-like orbitals, localized close to the metallic center, and they give rise to a local high-spin configuration on each metal center. However, due to the exchange and super-exchange interactions, the alignment (parallel or antiparallel) between the resulting spins of each Mn ion is strongly sensitive to the structural parameters of the Mn4CaO5, thus changing from model-A to model-B. The exchange interactions between the electrons of the Mn ions and the energy spin ladder can be described at a phenomenological level by the Heisenberg Hamiltonian

where  and
and  are the spin operators of the resulting magnetic momentum on the metal center i and j, whereas Jij represents their exchange coupling constant. In order to describe the electronic states using a single Slater-determinant in DFT, we use the BS approach that leads to a good description of the electron density and allows an estimation of the exchange coupling constants Jij [39–41]. In the four-metal core Mn4CaO5, one total high-spin state and seven BS states with different multiplicities have to be calculated in order to evaluate the exchange coupling constants between the four manganese atoms (see the supplementary data, available from stacks.iop.org/NJP/16/015020/mmedia).
are the spin operators of the resulting magnetic momentum on the metal center i and j, whereas Jij represents their exchange coupling constant. In order to describe the electronic states using a single Slater-determinant in DFT, we use the BS approach that leads to a good description of the electron density and allows an estimation of the exchange coupling constants Jij [39–41]. In the four-metal core Mn4CaO5, one total high-spin state and seven BS states with different multiplicities have to be calculated in order to evaluate the exchange coupling constants between the four manganese atoms (see the supplementary data, available from stacks.iop.org/NJP/16/015020/mmedia).
The DFT calculations were performed using the ORCA [42] and CP2K [43] packages. In the ORCA calculations, the all-electron Ahlrichs TZVP basis set [44] and def2-TZVP/J auxiliary basis sets [45] are employed for all atoms, except for the C and H atoms that are described with SVP and SVP/J. We used the B3LYP hybrid functional [46–48], which has already been used to reproduce accurate energetics and magnetic properties for manganese complexes [29]. To investigate the effect of a polarizable environment, we also used an implicit solvent model (with dielectric constant  = 8), as implemented in [49]. In the calculations performed with CP2K, all of the atoms are described by the GTH pseudo-potentials [50, 51]. The DZVP-MOLOPT-SR-GTH basis set, optimized for molecular systems, is employed for all atoms. For the B3LYP calculations, the SZV-MOLOPT-SR-GTH auxiliary basis set is used for the Mn and Ca atoms and the cFIT3 is used for the other atoms [43].
= 8), as implemented in [49]. In the calculations performed with CP2K, all of the atoms are described by the GTH pseudo-potentials [50, 51]. The DZVP-MOLOPT-SR-GTH basis set, optimized for molecular systems, is employed for all atoms. For the B3LYP calculations, the SZV-MOLOPT-SR-GTH auxiliary basis set is used for the Mn and Ca atoms and the cFIT3 is used for the other atoms [43].
In order to reduce the computational cost in ab initio MD simulations, we have adopted the DFT + U scheme [33, 52, 53]. This approach uses a basic density functional (i.e. the generalized gradient corrected PBE functional [54] in our case), including in the KS orbitals calculation an Hubbard correction term (inspired to the Hubbard model) that acts on the d orbitals of the Mn atoms. This Hubbard correction is system dependent and it is defined by the effective parameter U as detailed in the supplementary data. The value of the parameter U was chosen in order to reproduce the Jij values obtained by the B3LYP functional on two reference geometries. The final value U = 1.16 eV was derived from the average of the linear interpolations on the exchange coupling constants Jij, as explained in detail in the supplementary data.
2.2. Quantum mechanics/molecular mechanics model
The starting structure of the oxygen-evolving PSII at 1.9 Å resolution was taken from the protein data bank (PDB ID: 3ARC, [3]). The AMBER99SB force field [55] was used to describe the protein residues whereas the topologies describing the other molecules present in the crystal structure are based on the general AMBER force field [56] and generated using the Antechamber software [57] (for further detail see the supplementary data). The x-ray structure was embedded into a membrane bilayer that was composed of DOPC lipids [58] and solvated in TIP3p water molecules [59]. The total classical system consists of almost 650 000 atoms. Three nanoseconds of MD simulation with harmonic position restraints on the heavy atoms of the protein, cofactors and crystal waters at constant volume and temperature (T = 298 K) were performed, followed by 3 ns of MD simulation with the same harmonic position restraints in NPT ensemble (T = 298 K and P = 1 bar). MD simulations were carried out using the GROMACS software package [60, 61]. Further details are reported in the supplementary data (see the supplementary data, available from stacks.iop.org/NJP/16/015020/mmedia).
Starting from the last step of the position restrained trajectory in NPT ensemble, we cropped a portion of the system consisting of the Mn4CaO5 cluster plus the closest protein domains (i.e. D1, D2 and CP43), and the nearby cofactors and water molecules. The coordinates of the Mn4CaO5 cluster and the four directly coordinated water molecules were modified using the geometries suggested by Patanzis et al [18] for the S2 state of the Kok's cycle (model-A and model-B). Both models were optimized using the CP2K package in a mixed quantum/classical approach [62]. The metal core, its ligands (Asp170, Glu189, His332, Glu333, Asp342, Ala344 and CP43-Glu354), their closest residues (Asp61, Tyr161, His190, His337, Ser169 and CP43-Arg357), the ten closest water molecules plus the four water molecules coordinated to the Mn4CaO5 cluster and the chloride anion next to Glu333 were treated at DFT level, as described in the previous sections, whereas the remaining part of the system was described by a classical force field. The electrostatic coupling between the quantum and the classical portion of the system were treated using fast Gaussian expansion of the electrostatic potential as implemented in the CP2K package [43].
The QM/MM MD were performed in NVT ensemble at a temperature of 298 K using the Nosé–Hoover thermostat [63–65] to couple the system with a double thermal bath (one for the MM system and the other for the QM system), both with a time constant of 0.1 ps. The cutoff for the plane-wave expansion was set to 280 Ryd in the QM cell, with dimensions 26.5, 26.0 and 28.0 Å. QM/MM MD simulations were performed in a PBE + U scheme [33, 52, 53] for the QM part, as described before. The same basis set that was adopted in the GP calculations was employed. Positions of the classical Cα atoms were kept fixed during the dynamic. Both QM/MM MD simulations were set up in their suggested ground spin states (total spin S = 1/2 for the model-A and S = 5/2 for the model-B) with relative spin orientations of the Mn atoms in agreement with previous works [18, 31]. The two simulations ran for 10.0 ps, 7.5 of which have been used for data production.
3. Results and discussion
On the basis of the models proposed by Pantazis et al [18], we built GP models of the two conformers that were representative of the S2 state of the Kok's cycle proposed to be interconvertible in the PSII structure. The two geometries differ mainly by the position of the oxygen O5, which is bound to Mn4 in model-A, whereas in model-B it is bound to Mn1, as shown in figure 2. The two models have different magnetic properties, which are: an overall low-spin configuration for model-A, which is represented by a BS state of multiplicity 2, and an overall high-spin configuration for model-B, which is described by a BS state with multiplicity 6 (see the right panel of figure 2).
Upon geometry optimization, we calculated all the Jij coupling constants using different functionals (table S2 of the supplementary data (see the supplementary data, available from stacks.iop.org/NJP/16/015020/mmedia)). The B3LYP Hybrid functional, which has been shown to fairly reproduce coupling constants in Mn dimers [29], provides a trend that is similar to that obtained following the procedure of [18]. This has been taken as reference for the choice of the parameter U for the Hubbard correction in PBE + U calculations. The optimal choice of the U value has been taken by fitting the Jij parameters on the B3LYP reference for both model-A and model-B, following the procedure detailed in the supplementary data (see also table S3). The resulting value U = 1.16 eV is, therefore, used in all of the following DFT + U calculations.
For both the high-spin(A) and the low-spin(B) states, we report in table 1 the geometrical parameters of the optimized GP models, the optimized QM/MM models, and the averaged values along the QM/MM dynamics. Although it has been pointed out that the cluster geometry is affected by x-ray radiation damage [3, 38], in the last column distances and angles measured in the x-ray diffraction (XRD) structure [3] are shown for comparison. As expected, the main difference between the distances found in the x-ray structure and those of the A and B models arises from the Mn1–O5 and the Mn4–O5 distances. The O5 atom, which is equidistant from the two manganese ions in the crystallographic structure, in our models is clearly bound to only one manganese ion (Mn4 for model-A and to Mn1 for model-B).
Table 1. Comparison of the main distances (Å) and angles (degree) in Mn4CaO5 cluster between the XRD structure [3] and model-A and model-B in GP and in the QM/MM description, optimized in their respective spin ground-state and averaged over 10 ps of simulation MD. The standard deviations are reported in brackets.
| Model-A | Model-B | ||||||
|---|---|---|---|---|---|---|---|
| GP-OPT | QM/MM OPT | QM/MM MD | GP-OPT | QM/MM OPT | QM/MM MD | XRDa | |
| Mn1–Mn2 | 2.78 | 2.82 | 2.79 (0.07) | 2.74 | 2.75 | 2.78 (0.06) | 2.8 |
| Mn1–Mn3 | 3.35 | 3.34 | 3.37 (0.11) | 2.93 | 2.97 | 2.94 (0.07) | 3.3 |
| Mn2–Mn3 | 2.80 | 2.77 | 2.81 (0.07) | 2.80 | 2.76 | 2.82 (0.06) | 2.9 |
| Mn3–Mn4 | 2.82 | 2.78 | 2.77 (0.06) | 3.21 | 3.06 | 3.15 (0.12) | 2.9 |
| Mn1–Ca | 3.62 | 3.58 | 3.61 (0.12) | 3.44 | 3.40 | 3.40 (0.11) | 3.5 |
| Mn2–Ca | 3.39 | 3.37 | 3.39 (0.12) | 3.47 | 3.42 | 3.43 (0.12) | 3.3 |
| Mn3–Ca | 3.50 | 3.36 | 3.58 (0.15) | 3.47 | 3.30 | 3.45 (0.14) | 3.4 |
| Mn4–Ca | 3.98 | 3.59 | 3.83 (0.13) | 4.22 | 3.78 | 3.99 (0.16) | 3.8 |
| Mn1–O5 | 3.16 | 3.05 | 3.07 (0.21) | 1.84 | 1.89 | 1.88 (0.07) | 2.6 |
| Mn4–O5 | 1.93 | 1.89 | 1.87 (0.06) | 3.27 | 3.12 | 3.14 (0.18) | 2.5 |
| Mn1–MMn4 | 5.05 | 4.92 | 4.89 (0.19) | 5.09 | 5.00 | 5.00 (0.18) | 5.0 |
| Mn3–oxo–Mn4 | 99/97 | 96.3/97.5 | 96.3/96.5 (3.1/3.1) | 125 | 114.9 | 117.6 (6.6) | 96/95 |
| Mn1–oxo–Mn4 | 97/92 | 98.2/92.9 | 98.0/92.2 (3.2/3.1) | 95/91 | 96.7/91.5 | 97.7/92.7 (3.0/2.9) | 97/93 |
| Mn1–oxo–Mn3 | 116 | 116.2 | 117.2 (5.6) | 102/97 | 104.0/99.1 | 102.2/97.6 (3.6/3.6) | 118 |
| Mn2–oxo–Mn3 | 99/90 | 97.4/90.4 | 99.2/90.6 (3.1/3.1) | 100/90 | 97.2/90.3 | 99.3/91.0 (3.2/3.0) | 99/91 |
With respect to the GP optimized geometries, the QM/MM relaxed structures display two main differences: a reduced distance between Mn4 and the Ca ion (about 0.4 Å in both models, and a slightly shorter distance between Mn1 and Mn4, which is reflected in a shorter distance between O5 and the Mn that is not bound to it. Whereas the first effect is attenuated by the dynamics, the second remains basically unchanged at finite temperature. As a general remark, we observe that the overall structures remain fairly stable along the simulated time. In particular, the bond distances between the Mn atoms of the cluster remain close to the optimized structure values with fluctuations about 0.1 Å, with the exception of the Mn1–Mn4 distance that experienced rather larger fluctuations due to a reduced rigidity of the Mn4 connection with respect to the cubane structure. Both the enhanced mobility of O5 and its delocalization in QM/MM simulations might suggest a possible role of the protein field to promote the transition from model-A to model-B. This issue can have important implications since the O5 oxo-bridge atom is thought to be a substrate for the molecular oxygen formation catalyzed by the PSII [66, 67]. An improved protein-assisted mobility might, therefore, be relevant to understand the molecular mechanism behind water oxidation.
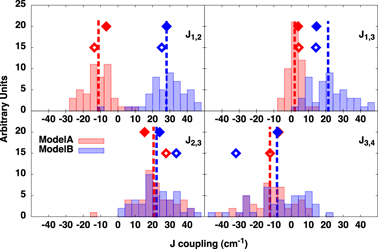
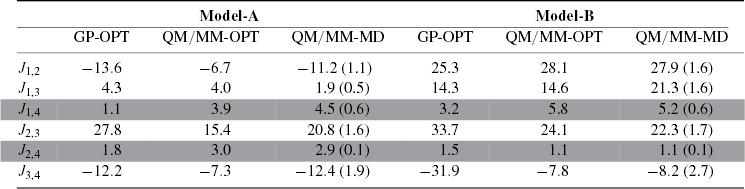
The magnetic coupling constants have been calculated on the QM/MM optimized structures as well as along the MD trajectories. As shown previously [18], the calculated J coupling constants for the two investigated models are consistent with a large amount of EPR experiments. The comparison between the GP and QM/MM single point calculations are reported in table 2. Interestingly, in model-A the physically meaningful Jij values (i.e. J1,2 J1,3 J2,3 J3,4, since the respective atom pairs are connected by oxo bridges) calculated on the QM/MM optimized structure show weaker ferromagnetic and antiferromagnetic interactions with respect to the GP model. Thermal fluctuations act on the magnetic coupling constants, increasing both the ferromagnetic and the antiferromagnetic interactions when compared to the values calculated on the optimized QM/MM structure. In the case of model-B, the differences between the Jij values evaluated on the single QM/MM optimized structure and averaged over the QM/MM trajectory are smaller when compared to the trend found in model-A. In particular, only the J1,3 value differs substantially when the effect of the dynamics at finite temperature is explicitly taken in account. Moreover, in the case of model-B, the QM/MM calculation, independently on the inclusion of the dynamics, decreases the magnetic interactions between Mn2 and Mn3 and between Mn3 and Mn4 with respect to the GP calculations. In particular, as shown in figure 3 and table 2, we found that J3,4 in model-B largely depends on whether the GP models or QM/MM simulations are considered. The reasons for this effect can be attributed to the high mobility of Mn4, which is external to the cubane structure. The small variations of its distance and angle parameters can easily largely affect the value of the J3,4 coupling constant.
{kind=link}
{kind=link}
Figure 3. Histograms of the exchange coupling constants Jij as obtained along the QM/MM MD trajectories at room temperature of model-A (red) and model-B (blue). The average value of each distribution is represented by the dotted lines. The Jij values calculated on the optimized (zero temperature) structures are shown as single color-filled diamonds for the QM/MM system and as empty diamonds for the GP model.
Download figure:
Standard image High-resolution image{kind=link}
Table 2. A comparison of the exchange coupling constants calculated on the optimized GP structure (GP-OPT), on the optimized QM/MM structure (QM/MM-OPT) and averaged over 50 snapshots extracted from the QM/MM MD trajectory (QM/MM-MD). Optimizations of model-A and model-B were carried out in the respective spin ground states. J1,4 and J2,4 (marked in gray) should be considered physically unmeaningful because they refer to Mn ions not linked by a μ-oxo-bridge.
 |
Apart from the average Jij values, another important information available from a dynamical description of the system concerns the fluctuations of the coupling constants due to the thermal motions. An histogram of the coupling parameters calculated along the QM/MM trajectories is shown in figure 3. It is interesting to point out that the fluctuations of the coupling constants are remarkably large, often being about half of the average signal. The points on the tail of the Jij are often rather off their average values, even instantaneously inverting the sign of the magnetic coupling: from antiferromagnetic to ferromagnetic (as in the case of J3,4 in both model-B and model-A) and from ferromagnetic to antiferromagnetic (e.g. J2,3 in model-A, J1,2 in model-B).
These fluctuations, changing instantaneously the kind of the magnetic interaction between manganese atoms, might have important consequences on the interconversion between the two structures and on the catalytic mechanism of the Mn4CaO5 cluster. In particular, it is thought that a specific spin configuration for the manganese atoms, as well as for the oxygen atoms involved in the formation of the O2 molecule, is required in order to allow the water splitting reaction [66]. A proper (dynamical) description of the spin electron density of the Mn4CaO5 cluster in the QM/MM scheme, which takes into account both the biological environment and the thermal fluctuations, is crucial in order to fully characterize the two structures model-A and model-B and to understand their role in the S2–S3 transition. On the basis of similar calculations, in a recent work [31] we have provided a framework in which it is possible to interpret several puzzling EPR experiments that identified metastable states between S2 and S3.
4. Conclusions
In the present work we have investigated the structural and magnetic properties of the Mn4CaO5 catalytic core of PSII. Thanks to the use of DFT + U framework within BS approach, we were able to describe the electronic structure and the magnetic properties of both model-A (low spin) and model-B (high spin) of the PSII in S2 state. The two systems, simulated at room temperature, were structurally stable during the simulated dynamics. The magnetic coupling constants calculated on snapshots along dynamics show large fluctuations in both models, especially for J3,4, which is the most sensitive to structural changes. The thermal fluctuations are so large that they induce instantaneous swapping between antiferromagnetic and ferromagnetic interactions, even in the case of largest  values. These effects might suggest that the relative stabilities of high-spin and low-spin surfaces in both model-A and model-B can be instantaneously inverted; therefore, facilitating surface hopping between the two spin states. Despite this broad distribution, their average values were in general close to that calculated on QM/MM optimized structures. In addition, the present work demonstrates the possibility of performing QM/MM MD simulations with an accurate treatment of the Mn–Mn interactions. The use of such methodologies opens the way to the study of the water splitting mechanisms in PSII within a framework that takes into account the effects of temperature fluctuations and of the protein environment.
values. These effects might suggest that the relative stabilities of high-spin and low-spin surfaces in both model-A and model-B can be instantaneously inverted; therefore, facilitating surface hopping between the two spin states. Despite this broad distribution, their average values were in general close to that calculated on QM/MM optimized structures. In addition, the present work demonstrates the possibility of performing QM/MM MD simulations with an accurate treatment of the Mn–Mn interactions. The use of such methodologies opens the way to the study of the water splitting mechanisms in PSII within a framework that takes into account the effects of temperature fluctuations and of the protein environment.
Acknowledgments
The authors acknowledge funding provided by the European Research Council project no. 240624 within the VII Framework Program of the European Union. Computational resources were supplied by CINECA, PRACE infrastructure and the Caliban-HPC centre at the University of L'Aquila.