



Abstract
Accurate and careful benchmarking of different density-functional approximations (DFAs) represents an important source of information for understanding DFAs and how to improve them. In this work we have studied the lattice constants, cohesive energies, and bulk moduli of 64 solids using six functionals, representing the local, semi-local, and hybrid DFAs on the first four rungs of Jacob's ladder. The set of solids considered consists of ionic crystals, semiconductors, metals, and transition metal carbides and nitrides. To minimize numerical errors and to avoid making further approximations, the full-potential, all-electron FHI-aims code has been employed, and all the reported cohesive properties include contributions from zero-point vibrations. Our assessment demonstrates that current DFAs can predict cohesive properties with mean absolute relative errors of 0.6% for the lattice constant and 6% for both the cohesive energy and the bulk modulus over the whole database of 64 solids. For semiconducting and insulating solids, the recently proposed SCAN meta-GGA functional represents a substantial improvement over the other functionals. However, when considering the different types of solids in the set, all of the employed functionals exhibit some variance in their performance. There are clear trends and relationships in the deviations of the cohesive properties, pointing to the need to consider, for example, long-range van der Waals (vdW) interactions. This point is also demonstrated by consistent improvements in predictions for cohesive properties of semiconductors when augmenting GGA and hybrid functionals with a screened Tkatchenko–Scheffler vdW energy term.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Density-functional theory (DFT) [1] in the Kohn–Sham (KS) [2] framework has proven to be a very useful tool in condensed-matter physics and quantum chemistry [3–7]. Its favorable ratio of accuracy to computational cost makes it a powerful method for calculating ground-state properties of large systems. Hohenberg–Kohn DFT is, in principle, an exact theory but as the form of the exact electronic exchange-correlation (XC) functional is unknown, a wide range of density-functional approximations (DFAs) have been proposed.
Cohesive properties (lattice constant, cohesive energy, and bulk modulus) can be used as probes to identify and understand the performance of each functional applied to solid-state materials, and noticeably different performance has been reported when using different DFAs, with significant deviations possible for a range of properties (see e.g., [8–19]). Therefore, developing reliable and efficient XC functionals that are applicable to a broad range of systems remains an ongoing challenge. It is especially relevant now as DFT calculations are becoming ubiquitous and numerically reproducible [20]. Investigating trends in predictions and deviations from experiment can help us to improve our understanding of DFAs, as well as bulk properties themselves.
In this work, we explore the question of accuracy of DFAs on the first four rungs of Jacob's ladder proposed by Perdew and Schmidt [3, 21], with each rung corresponding to more ingredients and contributions (besides the electron density) forming the DFA, ideally, leading to DFAs with improved accuracy. The local-density approximation (LDA) [2] represents the first rung of Jacob's ladder, considering only the density itself, and therefore it is a successful approximation for systems in which the electron density varies slowly, such as nearly free-electron metals, but is also surprisingly successful for less-uniform systems, such as molecules and semiconductors. However, there are many features that LDA fails to describe, with lattice constants typically being too small, up to 5% shorter than experiment, and cohesive energies (or atomization energies in molecules) that are very inaccurate, typically off by 20%–30% [19].
Functionals based on the generalized gradient approximation (GGA) are on the next rung of the ladder, and represent an extension of the LDA functional by introducing the gradient of the electron density. They are also referred to as semi-local DFAs, and are successful in improving over LDA for certain properties. For example, they cure the overbinding problem of LDA and predict more accurate bond lengths. However, they show a tendency towards underbinding [8–12, 14, 15].
The Perdew–Burke–Ernzerhof (PBE) [22] GGA is one of the most widely used DFAs and many approaches (e.g., revPBE [23] and RPBE [24]) have been explored to improve upon it. A simple modification of PBE is the PBEsol functional [25], which differs from PBE only in two parameters, and is designed to improve upon PBE for equilibrium properties of bulk solids and their surfaces. By restoring the gradient expansion for exchange, PBEsol lattice constants are systematically lower and better than PBE by 1%–2%, however with the drawback of cohesive energies being less accurate [10–14].
The meta-GGA (or MGGA) family [26–28] is on the third rung of Jacob's ladder and makes use of the kinetic-energy density in addition to the electron density and its gradient. While some MGGAs predict cohesive properties with reasonable accuracy for specific systems [8–13, 29, 30], improvements in the lattice constant are often accompanied by a worsening of the bulk modulus or the cohesive energy, or both. The non-empirical Tao–Perdew–Staroverov–Scuseria (TPSS) [28, 31] MGGA functional predicts lattice constants that are slightly smaller than those of PBE, but with improvement in bulk moduli being rather limited. The revised TPSS (revTPSS) [32, 33] alleviates the lattice constant error, but both TPSS and revTPSS worsen cohesive energies compared to PBE [13, 17]. Comparing existing literature benchmarks for the performance of LDA, PBE, PBEsol, and TPSS/revTPSS (see table 1), we see that none of these functionals is able to describe all cohesive properties with high accuracy.
Table 1. The mean absolute error (MAE) in lattice constants a0 (Å), cohesive energies E0 (eV/atom), and bulk moduli B0 (GPa) on testing sets of bulk crystals using different density-functional approximations reported from literature benchmarksa. The mean absolute relative error (MARE) and the maximum absolute relative error (MAX) are given in percentage.
| LDA | PBE | PBEsol | TPSS | revTPSS | HSE06 | ||
|---|---|---|---|---|---|---|---|
| a0 | MAE | 0.071 | 0.061 | 0.030 | 0.054 | 0.039 | 0.033 |
| MARE | 1.5 | 1.2 | 0.6 | 1.1 | 0.8 | 0.7 | |
| MAX | 4.9 | 2.8 | 2.3 | 4.1 | 3.3 | 2.0 | |
| B0 | MAE | 11.5 | 12.2 | 7.8 | 9.6 | 9.6 | 7.3 |
| MARE | 9.4 | 11.0 | 7.0 | 10.3 | 9.4 | 4.0 | |
| MAX | 32.8 | 25.5 | 19.5 | 29.6 | 25.8 | 23.4 | |
| E0 | MAE | 0.77 | 0.19 | 0.31 | 0.20 | 0.22 | 0.25 |
| MARE | 17.2 | 5.0 | 6.9 | 4.9 | 5.1 | 6.5 | |
| MAX | 38.7 | 21.0 | 22.8 | 15.3 | 17.7 | 25.0 |
aThe LDA, PBE, PBEsol, TPSS, and revTPSS data are taken from [19] (44 solids), and the HSE06 data are from [14] (30 solids).
Recently, Sun et al [34] proposed the 'strongly constrained and appropriately normed' (SCAN) meta-GGA. This functional represents a major step in the non-empirical or constraint-based construction of semi-local approximations, as it is the first meta-GGA functional to satisfy all 17 known exact constraints that a meta-GGA can fulfill. Appropriate norms were also added to ensure the functional is accurate for the energies of rare-gas atoms and nonbonded interactions. SCAN has been evaluated for properties of diversely bonded systems, including standard molecular test sets, layered materials, and surface-adsorbate systems [34–38]. It has been shown that SCAN can predict accurate structures and energies for the G3, BH76, and S22 sets [34–36]. In many cases, SCAN yields improvements over other (semi-)local functionals and could be as accurate as a fully nonlocal hybrid functional with decreased computational cost [37, 38].
The weaknesses or deficiencies in current local and semi-local DFAs are often explained in terms of two effects: the presence of self-interaction errors and the lack of long-range van der Waals (vdW) interactions [3–6]. As a result, the fourth and fifth rungs of Jacob's ladder specifically target these effects with hybrid functionals and the random-phase approximation (RPA), respectively. Hybrid functionals add a fraction of exact exchange to conventional GGAs or MGGAs, with the aim of reducing self-interaction errors [39, 40]. With the use of range separation of the exchange interaction, hybrid functionals have become particularly popular in solid-state physics. HSE06 [41, 42] is among the most-widely used screened hybrid functionals, and often shows superior performance over LDA, GGAs, and MGGAs in describing some bulk properties, including lattice constants and band gaps [43–51]. However, both bulk moduli and cohesive energies can be worse than those of PBE [14]. On the other hand, there is a considerable growth of computational cost with the system size N (the number of electrons or basis functions) using a hybrid functional compared to GGAs or MGGAs (sometimes up to 100 times higher [52]). Thus it is often advantageous to use a GGA or a MGGA functional for studies in condensed-matter physics and materials science.
On the fifth rung of Jacob's ladder, the RPA is an approach that treats long-range correlation seamlessly. Recent calculations performed by Harl et al [53] show that the RPA provides a well-balanced prediction of lattice constants and bulk moduli for a diverse range of solids (including semiconductors, ionic crystals, and metals), with relative accuracy of 0.4% and 4%, respectively. However, there are also challenges when using the RPA. Performing self-consistent RPA is computationally very demanding, and therefore most practical RPA calculations are performed in a post-processing manner based on an LDA or GGA reference calculation, with the starting point being crucial for some systems [54, 55]. Furthermore, when the RPA does not start from Hartree–Fock orbitals, there is also a first-order contribution that arises from single excitations [56], which often is omitted.
Given the current performance of DFAs for predicting cohesive properties, it is clear that there is much to be done in developing new functionals. However, the deviations themselves can be useful in this, as they are not random and clear systematic trends can be observed for many systems. For example, Grabowski and co-authors [57] have shown a correlation between the deviations from experiment of the lattice constants and the bulk moduli for nine fcc metals (noble metals, together with Al and Pb), with an increase of the error with the number of d electrons among the 4d and 5d transition metals particularly apparent for PBE.
Analysis of wider trends may point to systematic failures for specific DFAs for specific classes of solids, suggesting approaches to improve functionals and providing new insights in the cohesion of solids. This makes accurate benchmarking of DFAs and exploration of trends in deviations for a large set of solids paramount. In this work, calculations of cohesive properties are reported on the first four rungs of Jacob's ladder for 64 crystalline solids. We focus on assessing the performance of six functionals, particularly from the perspective of examining trends in performance of the different functionals for different classes of solids and the relationships between the deviations in different properties. Particular attention has been paid to convergence of the results, as well as to understanding the different contributions not only to calculated results but also to experimental quantities. All experimental cohesive properties will include some contribution due to vibrational motion, even at 0 K, while the inclusion of zero-point energy (ZPE) has long been considered in computational studies [58, 59]. Thus, any careful comparison of theory and experiment must consider ZPE to ensure a fair and unbiased comparison.
In the present study, the first two rungs are represented by LDA, and PBE and PBEsol, respectively. For the third rung we choose two MGGA functionals: M06-L and SCAN. The M06-L functional [60] was designed to incorporate the spin kinetic-energy density in the XC functional in an empirical way, and has been reported to outperform other semi-local functionals for a combination of thermochemistry, thermochemical kinetics, non-covalent interactions, bond lengths, and vibrational frequencies [60–62]. The present study will provide a benchmark of M06-L for a wide range of solids, including solids of heavier elements, with which the functional has not been widely studied. As noted above, SCAN represents a significant improvement over PBE (and even more so over LDA) when considering different types of bonding in molecules [34, 38], but to-date there has been relatively little benchmarking of it for such a wide range of hard solids. Tran et al [19] have calculated the lattice constants, cohesive energies, and bulk moduli of 44 solids within the SCAN functional. We are extending the benchmark with our larger dataset and the comparison is shown in the supplementary material, available from stacks.iop.org/NJP/20/063020/mmedia. On the fourth rung of Jacob's ladder, we employ the HSE06 functional. Finally, we use the screened Tkatchenko–Scheffler (TS) vdW approach to discuss the role of long-range vdW interactions in the cohesive properties of solids.
The following section (section 2) provides details of the calculations, including the database of solids, computational parameters and the approaches to calculating vdW interactions and ZPE. Section 3 presents the results and discussion, including the general performance of each functional for the solids, a discussion about trends in performance and deviations of cohesive properties, and the role of vdW interactions.
2. Technical Details
2.1. Solid dataset
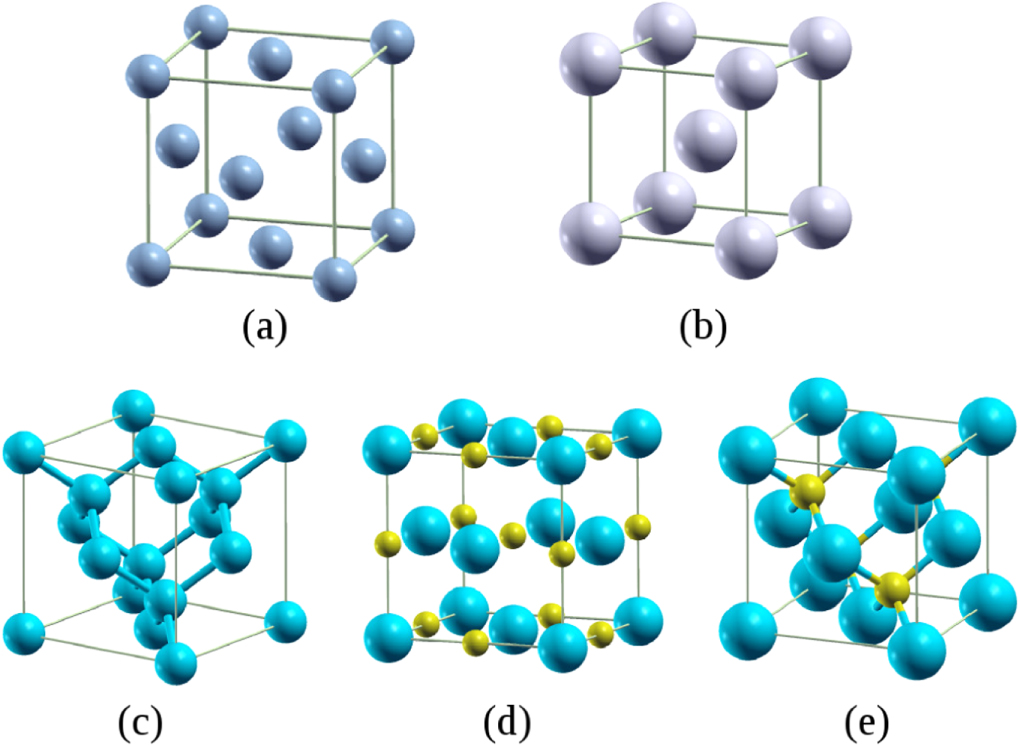
Our dataset comprises 64 crystalline solids with cubic structures: 24 metals and 40 semiconducting and insulating solids. The structures cover: A1 (face-centered cubic (fcc), 13 solids), A2 (body-centered cubic (bcc), 11 solids), A4 (diamond, four solids), B1 (rock-salt, 16 solids), and B3 (zincblende, 20 solids), as shown in figure 1. To explore and understand trends, we divide these 64 solids into five classes, as follows (see table 2): main-group metals (MM), transition metals (TM), semiconductors (SC), ionic crystals (IC), and transition metal carbides and nitrides (TMCN). MM includes groups IA and IIA, together with aluminum and lead; TM covers periods 4–7 of the periodic table; SC represents the solids of groups IVA, IIIA–VA, and IIB–VIA; the six ionic crystals are LiCl, LiF, NaCl, NaF, MgO, and MgS; and there are 10 compounds in the TMCN class (TMC and TMN, TM = Ti, Zr, Hf, V, and Nb). The full list of the solids studied here is shown in the supplementary material, with the calculated and experimental cohesive data tabulated in tables SII–SVII, alongside the relevant literature.

Figure 1. The unit-cell structures of (a) A1-fcc, (b) A2-bcc, (c) A4-diamond, (d) B1-rock-salt, and (e) B3-zincblende.
Download figure:
Standard image High-resolution imageTable 2. The present dataset consists of five classes of solids: main-group metals (MM), transition metals (TM), semiconductors (SC), transition metal carbides and nitrides (TMCN), and ionic crystals (IC).
| Type of solid | Type of structure | Number of solids |
|---|---|---|
| MM | A1, A2 | 9 |
| TM | A1, A2 | 15 |
| SC | A4, B3 | 24 |
| IC | B1 | 6 |
| TMCN | B1 | 10 |
2.2. DFT total energy calculations
All calculations were performed employing the FHI-aims package [63], which implements full-potential, all-electron electronic-structure theory with numeric atom-centered orbital basis functions. These basis functions are grouped together as 'tiers' (levels) in FHI-aims in addition to a minimal basis of occupied orbitals of spherically symmetric free atoms. Besides, there are different sets of numerical defaults for each atomic species, namely light, tight, and really tight settings [63]. For numerical accuracy and computational efficiency purposes, tight setting with tier 2 basis sets are recommended for obtaining cohesive properties of bulk solids [20], and are employed throughout this work. The typical tight setting convergence criteria are 10−5 electrons for the electron density, 10−3 eV for the eigenvalues, and 10−6 eV for the total energy of the system. An meV level of convergence is achieved for the cohesive energy.
For each solid, the equilibrium volume V0 and bulk modulus B0 were determined by fitting cohesive energies per unit-cell at 7–11 different points in a range of V0 ± 20% to a third-order Birch–Murnaghan equation of state (EOS) [64]

where V0, E0, B0, and B0' correspond to the equilibrium unit-cell volume, the cohesive energy, the bulk modulus at V0, and the pressure derivative of the bulk modulus, respectively. The unit-cell volumes V are related to the lattice constants a by the expressions V = a3/4 for the fcc, diamond, and zincblende structures, and V = a3/2 for the bcc structure. The cohesive energy, defined as the energy per atom required to atomize the crystal, was evaluated using the LDA, PBE, PBEsol, SCAN, M06-L, and HSE06 functionals from the energies of the crystal and the constituent free atoms. Note that both SCAN and M06-L calculations were performed using the PBE orbitals, as the functionality to perform calculations with these functionals in self-consistent manner is not yet available within FHI-aims. For SCAN, the effect of non-self-consistency has been assessed by Tran et al [19], and they suggest the difference between self-consistent and non-self-consistent calculations of hard solids should be in most cases below 0.005 Å for the lattice constant and 50 meV/atom for the cohesive energy (see supplementary material).
The spin-restricted formalism was employed for bulk crystals, with two exceptions (the ferromagnetic metals: Fe and Ni), and the spin-unrestricted formalism for open-shell atoms (no fractional occupancies were allowed). The basis sets and k-point meshes in reciprocal space are fully converged. Here we used Γ-centered Monkhorst–Pack [65] k-grids, and the settings for most calculations are outlined in the supplementary material. Relativistic effects are more important for heavy elements; here for consistency a scalar-relativistic treatment using the scaled zero-order regular approximation [66] was employed for all solids. It should be noted that spin–orbit coupling, which can be important in heavy solids, is not included. However, Philipsen and Baerends [67] have compared the scalar-relativistic and fully relativistic calculations of the cohesive properties of 11 solids in four columns of the periodic table and determined that spin–orbit coupling effects barely altered lattice constants, cohesive energies, and bulk moduli, with mean absolute contributions of about 0.005 Å, 0.03 eV, and 1 GPa, respectively. They showed that the spin–orbit coupling contributions were only non-negligible for the cohesive energies and the lattice constants of Au and Bi. Therefore, the scalar-relativistic treatment is sufficient to capture the relativistic effects in the solids considered here.
2.3. Inclusion of the long-range screened vdW interactions in DFT
vdW interactions arise from the correlated motion of electrons and constitute a large part of the long-range electron correlation energy. While most commonly used DFAs lack the ability to describe the long-range correlation energy, they provide a route to the inclusion of the vdW energy in an effective way. Numerous promising ideas have been proposed for including the vdW energy in DFT motivated by the need to treat large and complex molecular and condensed-matter systems [68–71].
One of the most widespread approaches is the pairwise-additive fragment-based one. According to second-order perturbation theory, the vdW energy of a collection of fragments (atoms or molecules) can be expressed as sum of attractive contributions, with the leading nonvanishing term being due to dipole–dipole interactions

where Rij is the interatomic distance between atoms i and j of the system,  is the corresponding dipole–dipole dispersion coefficient, and fdamp is a damping function that is used to avoid singularities at short distances and ameliorate double-counting of correlation at intermediate distances [68–71]. The resulting vdW energy is then augmented to the standard KS-DFT total energy calculated by employing a local, semi-local or hybrid DFA. In principle, the accuracy of any fragment-based DFT-vdW method, referred to as 'DFT+vdW' here, depends solely on the coefficients used and the choice of the damping function.
is the corresponding dipole–dipole dispersion coefficient, and fdamp is a damping function that is used to avoid singularities at short distances and ameliorate double-counting of correlation at intermediate distances [68–71]. The resulting vdW energy is then augmented to the standard KS-DFT total energy calculated by employing a local, semi-local or hybrid DFA. In principle, the accuracy of any fragment-based DFT-vdW method, referred to as 'DFT+vdW' here, depends solely on the coefficients used and the choice of the damping function.
The leading-order C6 term can be expressed as

where the integrand contains the isotropic dipolar polarizability as a function of imaginary frequency. Thus, the polarizability is a central quantity to the description of vdW interactions. The method employed here is based upon the TS scheme [72], which computes the polarizability, the C6 dispersion coefficient and the vdW energy by utilizing Hirshfeld partitioning [73] of the electron density to rescale free-atom values [74] obtained from self-interaction corrected time-dependent DFT calculations. The TS approach yields accurate C6 coefficients for a set of 1225 small molecules (with an accuracy of 5.5%) due to its ability to capture local environmental effects. However, TS lacks the ability to describe the long-range electrostatic screening that extends beyond the range of the exponentially decaying atomic densities. These effects can be included microscopically by modeling the environment as a dipole field of quantum harmonic oscillators (QHOs) and solving the resulting classical electrodynamics self-consistent screening (SCS) equation [75].
In this model, a molecule (or material) is considered as an arrangement of N atoms, each of which is described by a spherical QHO [71, 75, 76]. For a given atom i in the system, one can write

where  is the fully screened atomic polarizability tensor (for a given frequency of the electric field), containing both short-range (via the input atomic polarizability tensor from the TS method) and long-range (via the SCS equation) screening effects. The dipole–dipole interaction tensor, Tij, has the following expression
is the fully screened atomic polarizability tensor (for a given frequency of the electric field), containing both short-range (via the input atomic polarizability tensor from the TS method) and long-range (via the SCS equation) screening effects. The dipole–dipole interaction tensor, Tij, has the following expression

where V(rij) is the Coulomb potential between two spherical Gaussian distributions at the distance rij, with  and
and  representing the QHO positions. The solution of the SCS equation yields the atomic polarizability tensors, from which the isotropic polarizability can be obtained by diagonalizing and tracing the tensor. To calculate the C6 dispersion coefficient (via equation (3)), the Gauss–Legendre approach is utilized [75].
representing the QHO positions. The solution of the SCS equation yields the atomic polarizability tensors, from which the isotropic polarizability can be obtained by diagonalizing and tracing the tensor. To calculate the C6 dispersion coefficient (via equation (3)), the Gauss–Legendre approach is utilized [75].
In this work, we calculate dipolar polarizabilities and C6 dispersion coefficients for a set of 23 semiconductor solids using the method introduced above, and use them via the DFT+vdW scheme to evaluate the role of vdW interactions in the cohesive properties of solids.
2.4. Calculation of zero-point vibrational energy
Zero-point vibrational energy effects have been evaluated using the quasi-harmonic approximation. Phonon calculations were performed using phonopy [77] interfaced with the FHI-aims package. The supercell approach [78] was combined with the quasi-harmonic approximation, i.e., for a set of different volumes, harmonic vibrational frequencies are determined using ab initio calculations, with the zero-point energies being added to the DFT ground-state energies at each volume V. In the FHI-aims package, the finite-displacement method is employed to calculate phonon spectra and the vibrational density of states (DOS), g(V, ω), and ZPE is estimated as the frequency integration over the vibrational DOS

A systematic test of the supercell size was carried out and it was found that a choice of a 2×2×2 supercell with a 8×8×8 k-point grid is sufficient to ensure the convergence of zero-point vibrational energies for all the solids studied here. Within the finite-displacement approach, one may adjust the value of the small finite-displacement δ used to calculate the force response in the three Cartesian directions. This has also been systematically tested, demonstrating that a reasonable variation in δ has only a small impact on the computed ZPE. For example, the ZPE for diamond is about 0.18 eV/atom, and the change in ZPE is less than 1 meV/atom (0.2%) when varying δ from 1×10−3 to 1×10−1 Å. In addition, it was found that the effect of the specific DFT functional on the ZPE is negligible for these systems, and as a result, ZPE was calculated at the PBE level and added on top of the ground-state DFT total energies for all six functionals.
For the analysis of our calculations, we consider statistical quantities: the mean error (ME), the mean absolute error (MAE), the mean relative error (%), and the mean absolute relative error (%), all of which are determined by comparing with reliable experimental values measured at low temperatures and/or extrapolated to 0 K.
3. Results and discussion
3.1. ZPE contributions to the cohesive properties
We start by discussing the contribution of ZPE to the cohesive properties of the solids in our dataset. Figures 2 and 3 show the relative contributions of ZPE to the lattice constants a0, the cohesive energies E0, and the bulk moduli B0 of selected metals and non-metallic solids, respectively, together with available theoretical values reported from previous work [12, 14, 79]. A summary is given in table 3 for all of the solids considered in the present work. The results show that including zero-point vibrations has an even stronger effect on the bulk modulus than on both the lattice constant and the cohesive energy. For 24 metals, the average contributions are 0.2%, 0.7%, and 1.4% for a0, E0, and B0, respectively. In addition, the ZPE effect is, as expected, inversely proportional to nuclear mass (e.g., along the series of Li–Na–Al–K), and is found to be negligible for 'heavy' solids. When only considering the 'light' metals, located on the second to fourth rows of the periodic table, the averaged contribution is almost twice as large as the value calculated for the whole group of metals. In particular, for bulk Li, the lattice constant expands by 0.7% and the bulk modulus reduces considerably by ∼4%.

Figure 2. Zero-point vibrational contributions to the lattice constants a0, cohesive energies E0, and bulk moduli B0 of selected metals. The values are shown as a percentage with respect to the experimental cohesive properties, with the solid curves representing the present ab initio phonon calculations, pink pluses are taken from [79], and green crosses from [12].
Download figure:
Standard image High-resolution imageTable 3. Zero-point vibrational contributions to the lattice constants a0 (Å), cohesive energies E0 (eV/atom), and bulk moduli B0 (GPa) of 64 solids (24 metals, 40 non-metals, including six ionic crystals). The values in parentheses are the corresponding percentage with respect to the reported experimental cohesive properties (%). The 24 metals are divided into two groups: nine elements from periods 2–4, and 15 elements from periods 5–7 of the periodic table.
| a0 | E0 | B0 | |
|---|---|---|---|
| All metals | 0.007 | 0.023 | 1.5 |
| (0.2) | (0.7) | (1.4) | |
| Periods 2–4 | 0.011 | 0.029 | 1.7 |
| (0.3) | (1.1) | (2.1) | |
| Periods 5–7 | 0.004 | 0.020 | 1.4 |
| (0.1) | (0.5) | (0.9) | |
| Non-metals | 0.012 | 0.054 | 3.4 |
| (0.2) | (1.1) | (2.2) | |
| Ionic crystals | 0.026 | 0.044 | 2.9 |
| (0.6) | (1.1) | (4.4) |
For non-metallic solids, ZPE contributions become more pronounced. A set of 40 solids shows average contributions of 0.3% to the lattice constants, 1.1% to the cohesive energies, and 2.2% to the bulk moduli. For ionic crystals, the effect is particularly strong. The largest contribution occurs for LiF, with changes of about 1% (0.04 Å) for a0 and 8% (5.6 GPa) for B0, which are much larger than numerical errors in DFT calculations, and even might be comparable to the contribution due to the long-range electron correlation [80]. This suggests that for non-metallic solids, especially ionic crystals, the inclusion of ZPE becomes vital for an accurate assessment of systems where long-range vdW interactions can be of relevance.
In comparison with existing literature data, our results are in excellent agreement with the calculations reported by Schimka and co-authors [14] using the projector augmented wave method implemented in the Vienna ab initio simulation package [81, 82]. Their dataset comprises 30 solids (including seven metals, six ionic crystals, and 17 semiconductors). For some metals (like Na, Rh, and Cu) and most non-metallic solids, their corrections to both the lattice constants and cohesive energies are essentially identical to the present results, as shown in figures 2 and 3. To the best of our knowledge the evaluation of the ZPE contribution to the bulk moduli has not been performed previously for such a diverse set of bulk solids.

Figure 3. Zero-point vibrational contributions to the lattice constants a0, cohesive energies E0, and bulk moduli B0 of selected non-metallic solids. The solid lines are obtained from the present ab initio phonon calculations, green crosses are taken from [12], and purple stars are from [14].
Download figure:
Standard image High-resolution imageThe present study confirms that ZPE tends to increase lattice constants and reduce bulk moduli, with its inclusion improving the predictions of the LDA functional and worsening those of PBE (see table 1). As a result, neglecting zero-point vibrations will lead to a bias in the appraisal of these and other DFAs. Therefore, in the following sections, ZPE vibrational contributions will always be included in the discussion of functional performance.
3.2. Performance of density functionals along Jacob's ladder
Having assessed the impact of ZPE, we can now study the performance of local, semi-local, and hybrid DFAs for the dataset of 64 solids. The calculated a0, E0, and B0 using the LDA, PBE, PBEsol, SCAN, M06-L, and HSE06 functionals are tabulated in tables SII–SVII of the supplementary material, together with their experimental values, while the MEs and MAEs are shown in table 4. To illustrate the performance of the six functionals for different types of solids, we also plot the relative errors in the bulk moduli and the cohesive energies as a function of those in the lattice constants for the specific groups of solids in figures 4 and 5, respectively. The corresponding MAREs are presented in table 5 in terms of classes of solids, properties, as well as functionals.

Figure 4. Correlation between the deviations of the bulk moduli B0 (%) and those of the lattice constants a0 (%) using the LDA, PBE, PBEsol, SCAN, M06-L, and HSE06 functionals. The definitions of the five classes of solids are given as main-group metals (MM), transition metals (TM), semiconductors (SC), transition metal carbides and nitrides (TMCN), and ionic crystals (IC). All quantities include zero-point vibrational effects.
Download figure:
Standard image High-resolution imageTable 4. The ME and mean absolute error (MAE) in the calculated lattice constants a0 (Å), cohesive energies E0 (eV/atom), and bulk moduli B0 (GPa) of the 64 solids using the LDA, PBE, PBEsol, SCAN, M06-L, and HSE06 density functionals, with respect to experimental values. The mean relative error (MRE, %) and mean absolute relative error (MARE, %) are given in parentheses. The maximum absolute relative error (MAX, %) is shown in the last columns, within the solid marked in boldface using each functional. All quantities include zero-point vibrational effects.
| Functional | ME | MAE | MAX | ||||||
|---|---|---|---|---|---|---|---|---|---|
| a0 | E0 | B0 | a0 | E0 | B0 | a0 | E0 | B0 | |
| LDA | −0.063 | 0.85 | 13.0 | 0.063 | 0.85 | 15.4 | 4.9 | 48.4 | 46.0 |
| (−1.4) | (19.3) | (8.2) | (1.4) | (19.3) | (10.6) | Ba | Fe | Fe | |
| PBE | 0.056 | −0.08 | −9.8 | 0.061 | 0.21 | 13.6 | 2.8 | 19.4 | 33.2 |
| (1.1) | (−2.1) | (−9.6) | (1.2) | (5.5) | (11.6) | Pb | Au | Sn | |
| PBEsol | −0.010 | 0.37 | 3.5 | 0.030 | 0.39 | 10.7 | 2.9 | 31.5 | 32.7 |
| (−0.3) | (8.2) | (−0.5) | (0.6) | (8.9) | (7.4) | Th | Fe | Fe | |
| SCAN | 0.011 | −0.08 | 4.1 | 0.028 | 0.23 | 9.0 | 2.7 | 17.4 | 26.1 |
| (0.1) | (−0.7) | (0.7) | (0.6) | (5.4) | (5.9) | Th | Mo | V | |
| M06-L | 0.031 | 0.18 | −4.3 | 0.075 | 0.27 | 12.2 | 6.1 | 52.8 | 61.3 |
| (0.5) | (6.8) | (−2.4) | (1.4) | (9.0) | (13.1) | Rb | Rb | Rb | |
| HSE06 | 0.036 | −0.37 | 1.6 | 0.042 | 0.40 | 12.2 | 3.2 | 32.6 | 35.0 |
| (0.7) | (−7.5) | (−2.5) | (0.8) | (9.1) | (8.6) | Rb | V | V |
Table 5. The mean absolute relative error (MARE, %) for the lattice constants a0, cohesive energies E0, and bulk moduli B0 of the 64 solids divided into five classes (see main text for definitions).
| LDA | PBE | PBEsol | SCAN | M06-L | HSE06 | ||
|---|---|---|---|---|---|---|---|
| a0 | MM | 3.1 | 0.9 | 1.0 | 0.9 | 3.1 | 1.2 |
| TM | 1.6 | 1.1 | 0.9 | 0.8 | 1.6 | 0.9 | |
| SC | 0.6 | 1.6 | 0.4 | 0.5 | 1.7 | 0.8 | |
| IC | 1.5 | 1.8 | 0.5 | 0.2 | 0.3 | 0.8 | |
| TMCN | 1.2 | 0.5 | 0.5 | 0.3 | 0.6 | 0.4 | |
| E0 | MM | 20.4 | 8.5 | 13.7 | 12.4 | 37.1 | 11.2 |
| TM | 25.1 | 6.5 | 12.5 | 7.4 | 8.3 | 16.8 | |
| SC | 19.3 | 5.0 | 7.6 | 2.4 | 2.6 | 4.6 | |
| IC | 8.1 | 5.1 | 1.4 | 1.7 | 3.3 | 6.0 | |
| TMCN | 15.9 | 2.6 | 6.9 | 5.6 | 3.7 | 7.6 | |
| B0 | MM | 12.1 | 7.0 | 4.5 | 4.4 | 30.5 | 8.3 |
| TM | 17.5 | 8.9 | 9.3 | 8.0 | 10.1 | 11.2 | |
| SC | 4.6 | 16.0 | 6.3 | 4.4 | 13.2 | 5.9 | |
| IC | 9.0 | 14.5 | 6.4 | 3.8 | 4.9 | 7.0 | |
| TMCN | 13.9 | 7.5 | 10.1 | 8.8 | 7.5 | 12.6 |
Let us first discuss the performance of the LDA functional. The present work confirms the well-known trend that LDA underestimates lattice constants, overestimates bulk moduli, and considerably overestimates cohesive energies in most situations. From the MAREs of the three properties summarized in table 5, one can clearly observe that LDA shows a different performance for the different classes of solids. For the SC class, it predicts the lattice constants and the bulk moduli in good agreement with experiment, however at the expense of the cohesive energies, while for ionic crystals it yields moderately accurate results. However, for metals (groups MM and TM) the performance of LDA is unsatisfactory. Note that the performance of LDA for 4d and 5d transition metals is better than 3d and main-group metals.
Turning to the PBE functional, one can see the opposite trend to LDA: lattice constants are too large, and the bulk moduli are too small. Cohesive energies are significantly improved though, with a MARE of ∼6% versus ∼20% for LDA. Overall, PBE improves the agreement with experiment compared to the LDA functional. For the MM and 3d metals, the superiority of PBE over LDA for calculating cohesive properties becomes particularly prominent. For semiconductors and ionic crystals, PBE yields large deviations from experimental results for both the lattice constants and the bulk moduli, while the cohesive energies are reasonably well predicted. A characteristic feature of the PBE functional observed in the current study is the increase of the error with increasing lattice constant, as can be seen, e.g., along the series of C–SiC–Si–Ge–Sn, BN–BP–BAs, and AlP–AlAs–AlSb. For certain systems, the bulk moduli and the cohesive energies follow the same trend. We will return to this observation in section 3.3, when exploring the underlying causes for deviations and trends in the DFAs considered here.
The PBEsol functional was first proposed to improve the description of solids and surfaces compared to PBE [25]. For lattice constants, it does indeed yield improvements, curing the problem of underbinding. In fact, it slightly overbinds bulk crystals. The current study shows significant improvement in the lattice constants using PBEsol for semiconductors, insulators, and fcc metals (except the alkaline-earth elements, Ni, Cu, and Th). Consistently, the bulk moduli are much better predicted with PBEsol compared to PBE for the aforementioned systems. For instance, the MAREs are 0.4% and 0.5% respectively for the lattice constants of the SC and IC classes by PBEsol (versus 1.6% and 1.8% by PBE). The corresponding MAREs for the bulk moduli are very similar for the same solids, both about 6% (versus 14.5% and 16%, respectively, by PBE). For the rest of the solids, i.e., the bcc metals (except K and Rb) and TMCN, both lattice constants and bulk moduli calculated with PBEsol are only marginally better or even worse than with PBE. This agrees with the finding by Janthon and co-authors [83] in a study of 30 transition metals. The substantial change from PBE to PBEsol is a consistent shift of the bonding nature towards tightening: decreasing of the bond lengths (lattice constants) and increasing of the bulk moduli. Overall, PBEsol shows overbinding of solids. One 'expected' problem of using the PBEsol functional, which is observed in the present data, is that the calculated cohesive energies are inaccurate, only halving the LDA error and twice as large as the PBE ones. This can be traced back to inaccurate total energies of the individual atoms [25].
Turning to the meta-GGA functionals, the overall performance of M06-L for cohesive properties lies between that of LDA and PBE. However, no consistent trend can be identified. The current work suggests that M06-L significantly improves the performance on the cohesive properties of ionic crystals in comparison with LDA, GGAs, and hybrid functionals. For the six solids, it slightly overestimates the lattice constants, and reasonably predicts the cohesive energies and the bulk moduli, with resulting MAREs of 0.3%, 3.3%, and 4.9%, respectively. On the other hand, for most semiconductors, M06-L overestimates lattice constants, and underestimates cohesive energies, as well as the bulk moduli. In particular, for main-group metals, M06-L severely overbinds: the lattice constants are too small, and both the cohesive energies and the bulk moduli are too large. For transition metals, M06-L improves the lattice constants of 3d elements in better agreement with experiment, but worsens the other two properties; a significant improvement in cohesive energies is found only for the coinage metals (Pd, Pt, Ag, and Au), however at the expense of both the lattice constants and the bulk moduli. Overall, the M06-L functional performs less accurately than PBE for the TM group, as can be seen from table 5.
The recent SCAN functional is a non-empirical meta-GGA, unlike M06-L, and is the first functional to fulfill all known constraints that a meta-GGA can fulfill. The current study reveals that SCAN predicts remarkably accurate cohesive properties for a diverse set of bulk crystals and tends to strike a good balance between PBE and PBEsol. For non-metallic systems, significant and consistent improvement is found for the predictions of all three cohesive properties using the SCAN functional: the lattice constants are very accurate, with a MARE of less than 0.5% for each class of solids, while both the bulk moduli and the cohesive energies are reasonably well predicted. Our work also suggests that particularly for ionic and semiconductor solids, SCAN remains the best method of choice among the six density functionals tested here, with MAREs within 0.5%, 3%, and 5% for a0, E0, and B0, respectively. For the MM group, SCAN performs equally as well as PBE does for the prediction of the lattice constants, but yields a better prediction of bulk moduli, while for the TM group, the performance of SCAN is as good as PBEsol. Overall, SCAN overestimates the bulk moduli of the bcc metals and underestimates those of the fcc metals. Especially for the 4d and 5d coinage metals (Pd, Ag, Pt, and Au), SCAN cures the underbinding problem of PBE to a large extent. To our knowledge, a theoretical assessment on the reliability of the SCAN functional was first performed by the SCAN authors themselves [34], for the lattice constants of 20 solids. To compare the present results to their data, we consider the same set of 20 solids. The mean absolute deviation and mean deviation of the lattice constants are calculated to be 0.01 Å and 0.002 Å, respectively, between the two studies. This suggests our post-SCF procedure is a good approximation for the current work. A further evaluation of SCAN for other cohesive properties should be performed in the future.
The final DFA tested in the present work is the HSE06 hybrid functional. HSE06 predicts reliable lattice constants and bulk moduli for non-metallic crystals and its performance is only slightly worse than SCAN and PBEsol. For both the ionic and semiconductor solids, the general trend that HSE06 overestimates the lattice constants, underestimates the bulk moduli, and predicts reasonably good cohesive energies is confirmed. While HSE06 improves agreement with experiment for the lattice constants of TMCN, it underestimates the cohesive energies, and considerably overestimates the bulk moduli. Our study indicates that HSE06 exhibits an overall tendency to underbind solids, and this can be pronounced for metals. The overestimation of the lattice constants is found for nearly all of the metals (with the exception of V). Especially for the main-group metals (except Al and Pb), the HSE06 lattice constants are even larger than the PBE values, with a corresponding decrease in bulk moduli. For the TM group, a large difference is seen between the fcc and bcc metals: HSE06 overestimates the bulk moduli for nearly all bcc-TM (except for Fe), whereas it typically underestimates those for fcc-TM (except for Rh). In addition, the cohesive energies show significantly increased errors compared to the PBE results. This can be explained by the fact that hybrid functionals generally increase the stability of the spin-polarized atom, thus reducing stability of bulk systems [48, 49].
Figures 4 and 5 show comparisons of the density functionals tested here for describing the cohesive properties of all five classes of solids. Among the six functionals, LDA predicts accurate lattice constants and bulk moduli for semiconductors, however, the cohesive energy remains a considerable issue at this level, with a MARE of ∼20% for the 24 solids in this group. While PBE outperforms LDA on the prediction of the cohesive energies for semiconductors, it worsens both the lattice constants and the bulk moduli. In fact, the improved performance of PBE upon LDA can be mainly seen in its better description of the cohesion in the systems having a (semi-)metallic nature. As depicted in figures 4 and 5, all the cohesive properties obtained using PBE are in much better agreement with experiment for the MM, TM and TMCN groups, compared to the LDA results. Among the six functionals, PBE remains the best for the 3d transition metals. The PBEsol functional cures the underbinding of PBE, however it somewhat overbinds, but yields reliable lattice constants and bulk moduli for ionic crystals, semiconductors, and fcc metals. Employing PBEsol, the MARE of semiconductors is reduced by a factor of four for lattice constants and two and a half for bulk moduli respectively, compared to the PBE errors. Overall, the PBEsol cohesive energies show systematic overestimation, halving the LDA deviation, with the exception of ionic crystals for which PBEsol yields accurate predictions. The HSE06 functional corrects the lattice constants, and improves agreement with experiment for ionic and semiconductor solids, however it still shows a tendency to underbind. The increased errors in cohesive energies, together with the demanding computational cost, remain significant issues for using the HSE06 functional.

Figure 5. Correlation between the deviations of the cohesive energies E0 (%) and those of the lattice constants a0 (%) using the LDA, PBE, PBEsol, SCAN, M06-L, and HSE06 functionals. Some outliers are explicitly marked.
Download figure:
Standard image High-resolution imageIn comparison, the M06-L MGGA functional improves on LDA for TM and TMCN (though its performance is slightly worse than PBE), however it yields significant deviations from experiment for the main-group metals, as seen in table 5. This is consistent with the study by Zhao and Truhlar [61]. Compared to PBE, M06-L performs better for semiconductors with lighter elements. For C, Si, BN, BP, and AlP, the cohesive properties obtained from M06-L are in good agreement with experiment. The bulk moduli calculated with M06-L are as good as the LDA values, whereas PBE yields the worst performance among the six functionals, while the M06-L cohesive energies are greatly improved compared to the LDA results. However, when considering heavy-element systems, there is barely any improvement to be gained by using M06-L over PBE. This can be seen for the cases of GaSb, InSb, ZnTe, CdTe, etc. Similar behavior is also observed for ionic crystals. The M06-L functional significantly improves the description for the six ionic compounds that have medium mass, with a MARE of only 0.3% for the lattice constants versus those of 1.5% and 1.9% by LDA and PBE, respectively. Turning to the non-empirical MGGA functional, our study suggests that SCAN can be regarded as the method of choice for non-metallic solids and it agrees remarkably well with experiment for all the three cohesive properties. The SCAN lattice constants are considerably smaller than those of PBE, with an increase of the bulk moduli and larger cohesive energies. For metals, no significant improvement is found on the lattice constants and bulk moduli, while SCAN worsens the cohesive energies compared to PBE. For the 4d and 5d coinage metals, SCAN substantially improves on PBE.
In summary, our study shows that all of the functionals considered have varying performances in predicting the cohesive properties of solids, which strongly depend on the bonding nature of the solid. To understand these trends and behavior in more detail, we now move to discussing the relationships between pairs of cohesive properties, that is, a0/B0 and a0/E0.
3.3. Correlations between cohesive properties
Much attention has been paid in the literature to understanding the relationship between geometrical parameters and properties of various classes solids and the cohesive energy and bulk modulus. It has been found that both E0 and B0 are inversely related to a0 (or the nearest-neighbor distance d) [84–89]. This inverse relation can be explained by the volume dependence of the total energy (the EOS) causing a monotonic decrease of the bulk modulus with increasing volume (see equation (1)). There are several analytical expressions reported in the literature for different families of cubic solids [84–89], e.g., semiconductors (groups IVA, IIIA–VA, and IIB–VIA), ionic crystals (groups IIA–VIA and IA–VIIA), and metals (groups IA and IIA, and noble metals), based upon empirical approaches. While such empirical methods are often not able to yield highly accurate results, they can still be very useful, particularly for illustrating trends in properties of a wide variety of materials.
Anderson and Nafe [84] first proposed an empirical relationship between bulk modulus and crystal volume of the form  , where x depends on the bonding type of solid. Cohen and co-authors [85, 86] studied a number of covalent and ionic crystals, and deduced an analytical expression between the bulk modulus and the nearest-neighbor separation d for diamond and zincblende structures
, where x depends on the bonding type of solid. Cohen and co-authors [85, 86] studied a number of covalent and ionic crystals, and deduced an analytical expression between the bulk modulus and the nearest-neighbor separation d for diamond and zincblende structures

where λ is an empirical ionicity parameter accounting for the ionic character of the bonds. This relationship was found to be appropriate for group IVA (λ = 0), IIIA–VA (λ = 1), and IIB–VIA (λ = 2). For the IA–VIIA rock-salt compounds, Cohen proposed

A similar scaling of B0d3.5 for the rock-salt structure was suggested by Schlosser [87, 88], and the cohesive energy was discussed in terms of d as well. Aresti et al [89] studied the cohesive energy of zincblende solids using the form

According to the empirical expressions given by equations (7)–(9), an overestimation (underestimation) of the lattice constant corresponds to an underestimation (overestimation) of the bulk modulus or the cohesive energy. In fact, for the present dataset all of the DFAs considered broadly follow this trend.
Figures 4 and 5 show the relative errors in the bulk moduli and the cohesive energies as a function of the lattice-constant errors, using the LDA, PBE, PBEsol, SCAN, M06-L, and HSE06 functionals for the 64 solids divided into five classes. It can be seen that most data points are in the quadrants II and IV of the Cartesian plane, reflecting that the shorter the lattice constants, the larger the bulk moduli and the cohesive energies, and vice versa. This indicates that those DFAs can predict the observed experimental trends (the inverse relationships of bulk moduli and cohesive energies with lattice constants). If one further looks into the errors concerning a given type of solids, large differences can be found for specific functionals. For SC and IC, a nearly monotonic dependence is observed for the deviations of the bulk moduli from experiment upon those of the lattice constants using all the functionals tested, that is, the overestimated lattice constants is accompanied by the underestimated bulk moduli. It can also be observed that there is a better ('linear') correlation between the errors from the PBE calculations, in comparison with, e.g., LDA, applied to the SC compounds, where half of the values fall in the quadrant III, in disagreement with the empirical study based on experimental observations. Overall, the PBE functional yields the most consistent results with experiment, and it is the method that reproduces systematic trends in the cohesive properties of solids better than the other functionals.
Turning to metals (the MM and TM groups), a noticeable scatter of the errors is found, e.g., from the LDA, PBEsol, and M06-L calculations, with the inversely correlated behavior being somewhat captured by the PBE, HSE06, and SCAN functionals. This also reflects the poor performance of LDA and M06-L, particularly in the description of main-group metals. Finally, for the TMCN group, there is no clear trend shown by any method used here. We note that no clear experimental correlations have been found between any pair of cohesive properties for this class of solids (see figures 6 and 7), mainly due to the mixed nature of the metallic, covalent, and ionic bonding in these solids [90, 91].

Figure 6. The experimental lattice constant as a function of the cohesive energy for selected semiconductors, ionic crystals and metals. The definitions of the five classes of solids are given as main-group metals (MM), transition metals (TM), semiconductors (SC), transition metal carbides and nitrides (TMCN), and ionic crystals (IC).
Download figure:
Standard image High-resolution imageThe above findings suggest that the errors in different cohesive properties obtained from DFT calculations are correlated and system dependent. In general, PBE shows superior performance to LDA, due to the inclusion of the density gradient term for describing nonlocality in realistic systems. The improvement of PBE over LDA is significant for main-group and 3d metals. For some semiconductors and 'heavier' 4d, 5d transition metals, the cohesive properties are better predicted by LDA than by PBE (except for the cohesive energies). When it comes to the M06-L functional, the present study shows that it can perform well for small and medium-sized solids. This can be ascribed to the fact that the parameters in the M06-L functional are obtained from fits to molecular systems, allowing it to describe mid-range interactions to some extent, which can be seen from its good performance for the ionic crystals considered here.
Furthermore, the current work suggests that investigating the relationships between errors in cohesive properties can help us to understand performance and capability of DFAs. In particular, it is found that the system-dependent behavior of the cohesive properties is best reproduced at the PBE level. Indeed, by using the PBE functional an increase of errors in bulk moduli is accompanied by that in lattice constants for many solids in the current dataset. This is consistent with the finding by Grabowski and co-authors, who discussed the dependence of deviations given by the LDA and PBE functionals for fcc metals [57]. The behavior of the increasing errors with mass, the 'sizable error' [92], can also be observed in the lattice constants. In figure 8, the deviations of the calculated lattice constants from experiment are plotted as a function of their experimental values for 24 metals and 40 non-metals using the six functionals. One can see that the monotonic relationship, indicating that the systematic error increases as the crystal unit-cell volume increases, is better reproduced by PBE and HSE06, rather than LDA, PBEsol, M06-L or SCAN. The trend is more pronounced in non-metals than metals, in good agreement with experiment (see figures 6 and 7). For 40 non-metallic solids, a nearly monotonic behavior is captured using PBE, while two separate regions are found using M06-L. Our analysis suggests the size-related error issue can be related to the lack of the long-range electron correlation in density approximations, which is discussed further below.

Figure 7. The experimental lattice constant as a function of the bulk modulus for selected semiconductors, ionic crystals and metals. The dashed lines serve as a guide to the eye.
Download figure:
Standard image High-resolution image
Figure 8. Deviations of DFT lattice constants from experiment (Å) for 24 metals (crosses) and 40 non-metals (solid squares) versus their experimental lattice constants (Å) using the LDA, PBE, PBEsol, SCAN, M06-L, and HSE06 functionals. Some outliers are explicitly marked.
Download figure:
Standard image High-resolution image3.4. Challenges for meta-GGA and hybrid functionals
The accurate prediction of both structural and energetic properties of bulk solids remains a difficult task for current DFAs. The SCAN meta-GGA and HSE06 hybrid functionals, which belong to the third and the fourth rungs of Jacob's ladder, were specifically developed to improve upon conventional LDA and GGAs. As we have seen, semiconductors (24 solids) and insulators (six solids) benefit greatly from SCAN and HSE06 for predicting structural properties (a0 and B0): SCAN yields remarkably accurate lattice constants (MAREs: 0.5% and 0.2% versus 1.6% and 1.8% by PBE), bulk moduli (MAREs: 4.4% and 3.8% versus 16% and 14.5%), while HSE06 performs reasonably well for both properties, with the errors reduced by more than a factor of two compared to the PBE results. The SCAN functional improves on PBE for predicting all the three properties of semiconductor and insulator solids simultaneously and this is clearly a substantial improvement for a semi-local DFA.
However, for transition metals meta and hybrid functionals generally yield no improvement in accuracy over GGA functionals [28, 31, 92]. For the lattice constants, PBE yields fairly accurate results for 3d metals, but systematically overestimates them for 4d and 5d metals, consistently underestimating both bulk moduli and cohesive energies. In particular, PBE fails to describe the cohesion in coinage metals including Pd, Ag, Pt, and Au. The averaged errors are about 2% larger in the lattice constants, 17% in the bulk moduli, and 11% in the cohesive energies for those four metals. Note that the size-related issue discussed in section 3.3, the increase of the error with increasing nuclear mass, is more pronounced here. The origin of this size-related error is not fully understood, but it is likely that this can be related to the presence of self-interaction errors and/or the lack of the long-range electron correlation in semi-local DFAs. As vdW interactions arise from the correlated motion of electrons, they are larger in magnitude for larger systems (i.e., with more electrons), and constitute a significant part of the long-range electron correlation energy. The inclusion of the attractive vdW energy helps shorten bond lengths and stabilize the crystal.
The SCAN functional (as well as M06-L) captures mid-range interactions to a certain extent, which can be seen from its improved description for the interaction energies of the S22 dataset [34]. When applied to solid transition metals, our results suggest that SCAN achieves a major improvement over PBE for 4d and 5d coinage metals where the electron correlation becomes more important between the almost filled d shells. M06-L also performs better for medium-sized unit cells, but for those solids with a large unit-cell volume (heavy nuclear mass), it performs worse than PBE for cohesive properties. This is also an indication that vdW interactions are responsible for part of the deviations of modern DFAs, with the meta-GGA form of SCAN and M06-L allowing them to capture mid-range vdW interactions. This likely accounts for some of the better performance of these functionals for some systems. However, the long-range vdW interactions are still missing from these functionals [36, 35], with the present study showing noticeable deviations from experiment for solid systems including 3d metals, alkali and alkaline-earth metals.
Part of these deviations will also stem from self-interaction errors [93]. The HSE06 hybrid functional, partially counters self-interaction errors by admixing a fraction of exact exchange. As already seen in section 3.2 and other studies [48, 49], HSE06 yields improved performance on the cohesive properties of molecules, semiconductors, and insulators. While for metallic systems HSE06 performs worse than PBE, particularly for the cohesive energies of transition metals, the HSE06 results deviate significantly from experimental values, mainly due to the incorrect treatment of localized d electrons. This illustrates both the impact of self-interaction errors and the challenge of accounting for them. Therefore, given the different contributions that DFAs need to account for (i.e., adding vdW interactions and correcting self-interaction errors), it is not surprising that different semi-local and hybrid functionals underbind bulk solids and yield increased errors with increasing system size (nearest-neighbor distance, lattice constant, or nuclear mass).
3.5. vdW interactions in bulk solids
As noted previously, one potential source of the size-related issues is the lack of a full description of vdW interactions in the six functionals studied, particularly, the lack of long-range vdW interactions, which form part of the correlation energy. The importance of such interactions for bulk solids has become clearer in recent years [80, 94]. In our previous study of six ionic and semiconductor solids, long-range vdW interactions were included using C6 coefficients derived from accurate dielectric functions. The inclusion of long-range vdW interactions on top of the PBE and HSE06 functionals lead to a significant improvement for the cohesive properties of the six solids compared to the results without vdW interactions, and as a result the characteristic underbinding of PBE and HSE06 was largely remedied [80].
Here we extend this study to 23 semiconductors, comprising all of the SC group in the present study, apart from tin. The vdW interactions have been modeled using the method proposed by Tkatchenko et al [75], which accounts for both short-range hybridization and long-range screening effects, seamlessly. As briefly discussed in section 2.3, recent studies demonstrated that screening effects become important in large molecules and materials [71, 75, 76]. Figure 9 shows the MAREs in the cohesive properties (a0, E0, and B0) obtained for the 23 semiconductors using the six functionals considered in the present study, alongside the results where three of the functionals are augmented with long-range vdW interactions, specifically, PBE+vdW, M06-L+vdW, and HSE06+vdW (corresponding data are shown in table SVIII). Employing PBE+vdW leads to a consistent improvement in all three cohesive properties compared to PBE alone, reducing deviations from experiment significantly, especially for the lattice constants and bulk moduli. This is unsurprising, as the trends for PBE showed a size-related dependence for the deviations from experiment, with PBE performing worse for systems with heavier elements and, hence, larger lattice constants. As such larger systems will have a larger contribution from vdW interactions, augmenting PBE with a vdW term clearly and understandably leads to improvement.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. The mean absolute relative error (MARE, %) obtained within LDA, PBEsol, PBE, PBE+vdW, M06-L, M06-L+vdW, SCAN, HSE06, and HSE06+vdW compared to experimental data for the lattice constants a0, cohesive energies E0, and bulk moduli B0, as an average over 23 semiconductor solids (including groups IVA, IIIA–VA, and IIB–VIA).
Download figure:
Standard image High-resolution image{kind=link}
Turning to HSE06, again, all three cohesive properties are in better agreement when the functional is augmented with a vdW term. Overall, HSE06+vdW yields the most accurate results among the DFT+vdW approaches employed, solving the underbinding issue of HSE06, and in fact leads to slight overbinding, with overestimation of both cohesive energies and bulk moduli. In contrast, augmenting M06-L with a vdW term improves lattice constants and bulk moduli but worsens the MARE in the cohesive energies, and as a result gives mixed performance. This likely stems from the fact that the functional form of M06-L contains many empirical parameters that are determined through fits to molecular data. This will partly capture some vdW interactions, but effects and contributions outside or beyond the dataset will not be captured, leading to limited transferability. Similar to M06-L, the ability to capture some mid-range vdW interactions of SCAN, which has been seen from the improved performance on e.g., the S22 set [34] and the bulk solids in this work, makes it difficult to further develop DFT+vdW methods. One of our co-authors [95] has recently illustrated this particular issue of coupling vdW interactions with semi-local functionals, pointing out that the effective range (at which distance fluctuations are correlated) of SCAN depends on system size. Capturing new or higher-level contributions will always be a challenge for empirical functionals (MGGAs or GGAs). Developing new functionals and augmenting existing ones with physically motivated contributions, such as long-range vdW interactions, may often be easier starting from a non-empirical functional that is not fitted to specific types of systems or interactions.
4. Conclusions
In this work the cohesive properties of 64 solids have been studied using DFAs from rungs 1–4 of Jacob's ladder (LDA, GGA, MGGA, and hybrid functionals). As a precursor to comparing the calculated values to experiment, the importance of zero-point vibrational contributions has been explored and found to be significant for certain solids and properties, with the bulk moduli, in particular, have a larger contribution from ZPE than lattice constants or cohesive energies.
For the cohesive properties, the present study broadly reproduces the well-known trends of the LDA and PBE functionals. LDA delivers fairly good predictions on the lattice constants and bulk moduli of covalently-bonded systems, however, the cohesive energies are considerably overestimated with a MARE of ∼20%. PBE predicts overall accurate cohesive energies for a diverse type of crystals and for metals and TMCN the improvement over other existing functionals is pronounced. While PBE underbinds significantly, PBEsol and HSE06 both improve on this, with PBEsol overbinding and HSE06 underbinding but to a lesser extent than PBE. In comparison, the MGGA M06-L gives a better description than PBE for certain semiconductors and ionic crystals, but yields poor prediction of bulk moduli, while the SCAN functional is superior for the description of cohesion in non-metallic solids.
By comparing the DFT results to experimental studies and empirical observations, trends in the relationships between pairs of cohesive properties have been explored with all six functionals being able to broadly reproduce the experimental trends. However, systematic differences are observed in the deviations of the DFT results from the experimental data. As the experimental trends point to a clear relationship between the lattice constant and both the bulk modulus and the cohesive energy, it would be expected therefore, for deviations of the bulk moduli and the cohesive energies to be inversely related to those of the lattice constants. The PBE functional reproduces this behavior better than other functionals, suggesting that PBE has clear and systematic reasons for its deviations from experiment.
Overall, none of the functionals tested here can be considered as a universal method that is better than the others when applied to a broad range of solids. Considering the trends in deviations for different classes of solids and different quantities (and between different quantities) can be fruitful for developing new DFAs. It is noticeable that deviations from experiment increase for a number of functionals with larger unit-cell sizes (and hence heavier atoms). Undoubtedly, some of the deviation stems from the lack of long-range vdW interactions in these functionals, which will be more significant for heavier atoms. For 23 semiconductors, augmenting the PBE and HSE06 functionals with a vdW correction leads to consistent improvements. However, augmenting the empirical MGGA M06-L leads to improvements in lattice constants and bulk moduli, but worsens cohesive energies. The current work reinforces the need to go beyond (semi-)local approaches for obtaining accurate cohesive properties of bulk solids. It can also be concluded that some of the deviation stems from the lack of long-range vdW interactions. PBE represents a natural starting point to add such long-range vdW interactions, as its deviations from experiment are systematic and more consistent than other functionals, and also because of its computational efficiency.
Acknowledgments
AT acknowledges support from the European Research Council (ERC Consolidator Grant BeStMo). GXZ would like to acknowledge support from the National Natural Science Foundation of China (Grant No. 21503057) and the Fundamental Research Funds for the Central Universities (Grant No. HIT.NSRIF.2017032).