

Abstract
Objective. Given the rapid expansion of the field of neural stimulation and the rigorous regulatory approval requirements required before these devices can be applied clinically, it is important that there is clarity around conducting preclinical safety and efficacy studies required for the development of this technology. Approach. The present review examines basic design principles associated with the development of a safe neural stimulator and describes the suite of preclinical safety studies that need to be considered when taking a device to clinical trial. Main results. Neural stimulators are active implantable devices that provide therapeutic intervention, sensory feedback or improved motor control via electrical stimulation of neural or neuro-muscular tissue in response to trauma or disease. Because of their complexity, regulatory bodies classify these devices in the highest risk category (Class III), and they are therefore required to go through a rigorous regulatory approval process before progressing to market. The successful development of these devices is achieved through close collaboration across disciplines including engineers, scientists and a surgical/clinical team, and the adherence to clear design principles. Preclinical studies form one of several key components in the development pathway from concept to product release of neural stimulators. Importantly, these studies provide iterative feedback in order to optimise the final design of the device. Key components of any preclinical evaluation include: in vitro studies that are focussed on device reliability and include accelerated testing under highly controlled environments; in vivo studies using animal models of the disease or injury in order to assess efficacy and, given an appropriate animal model, the safety of the technology under both passive and electrically active conditions; and human cadaver and ex vivo studies designed to ensure the device's form factor conforms to human anatomy, to optimise the surgical approach and to develop any specialist surgical tooling required. Significance. The pipeline from concept to commercialisation of these devices is long and expensive; careful attention to both device design and its preclinical evaluation will have significant impact on the duration and cost associated with taking a device through to commercialisation. Carefully controlled in vitro and in vivo studies together with ex vivo and human cadaver trials are key components of a thorough preclinical evaluation of any new neural stimulator.
Export citation and abstract BibTeX RIS
1. Introduction
Neural stimulators are active implantable devices that provide therapeutic intervention, sensory feedback or improved motor control via electrical stimulation of neural or neuro-muscular tissue in response to trauma or disease. Because of their complexity, regulatory bodies typically classify these devices in the highest risk category (Class III), and they are therefore required to go through a rigorous regulatory approval process before progressing to market [1, 2]. The pipeline from concept to commercialisation of these devices is long and expensive [3, 4]; careful attention to both device design and its preclinical evaluation will have significant impact on the duration and cost associated with commercialising a new device.
Since the introduction of the first commercial implantable stimulators in the late 1950s, there have been many devices approved for clinical use, resulting in a dramatic impact on the quality of life of millions of people around the world [5, 6]. Five devices currently dominate neural stimulation from a clinical perspective: cochlear implants that stimulate the auditory nerve to treat profound hearing loss; spinal cord stimulation to treat severe chronic back pain; vagal nerve stimulation to treat epilepsy and depression; deep brain stimulation to alleviate the motor disorders associated with Parkinson's disease and essential tremor; and sacral nerve stimulation to provide bladder control in patients with spinal cord injury [2, 7, 8]. These five devices have a market size of $7.6 billion (2016) with a projected growth of 7–17% percent compound annual growth rate [2, 9].
In addition to these well-established devices, there are a large number of devices in development or at an early stage of commercialisation [7]. Recently the field has experienced an exciting new phase of innovation generated by the National Institutes of Health's Stimulating Peripheral Activity to Relieve Conditions (SPARC) program, the Defense Advanced Research Projects Agency's ElectRx, SUBNETS, RAM and HAPTIX programs, and Galvani Bioelectronics bioelectronic therapies program [10–12]. These initiatives call for significant interdisciplinary collaboration and include the development of detailed anatomical and physiological maps of neural circuits associated with disease and the implementation of excitatory or inhibitory neuromodulation techniques for therapeutic stimulation.
Given the rapid expansion of the field and the rigorous regulatory approval requirements, it is important that there is clarity around the safety requirements and preclinical studies associated with the development of neural stimulators. Moreover, the next generation of neural stimulators are likely to be far smaller than current commercial devices [6, 13]; miniaturization brings with it additional constraints that influence device safety, reliability and efficacy. The present review examines basic design principles associated with the development of a safe neural stimulator and reviews the typical preclinical safety studies that need to be considered when taking a device to first-in-human (FIH) or Phase I clinical trials. While aspects of this review will also be relevant to recording-only implantable devices, there are a number of excellent reviews covering the design, preclinical studies and clinical application of recording electrodes (e.g. [14–17]) and are therefore not specifically considered here.
2. Design principles
A neural stimulator must be designed for a specific clinical application. Its features will be dependent on multiple factors including the underlying anatomy and physiology of the target site and surgical access to that site. There are a number of fundamental design requirements that are underpinned by the need to be safe, efficacious and robust, common to all neural stimulators that include: (a) Surgical insertion: Surgical insertion of the neural stimulator must be achieved with minimal damage to the electrode array or the target neural population and surrounding tissue; (b) Biocompatibility: The implant must be biocompatible—the assembled device implanted in the target site must demonstrate long-term biocompatibility; (c) Mechanical stability: The electrode array and cable must be designed to minimise movement relative to their target neurons. They must not damage neurons, other tissue or organs in the vicinity of the implant or result in adverse systemic effects. The implant must be designed to withstand repeated movement, and if it is designed for use in children the device must accommodate growth related changes; (d) Electrical stability: The electrode array must be electrically stable over long-term implantation and the insulation must not delaminate or allow fluid ingress resulting in inter-channel crosstalk; (e) Electrical stimulation: Stimulation of the electrodes must be efficacious in activating the target neural pathway and do so well within safe stimulation limits. The electrode array should be designed to ensure the electrical stimulus can be localized to discrete groups of neurons. Chronic electrical stimulation must not cause tissue damage or neural loss at the electrode-tissue interface, or dissolution or delamination of the electrode surface; (f) Infection: The implant must be designed to minimize the risks of infection. Minimizing infection may include smooth implant surfaces, the elimination of cavities and careful selection of biomaterials; (g) Replacement: The implant should be designed to allow replacement without damage to the neural substrate or surrounding tissue. Although these devices should be designed for long-term use, the design criteria must incorporate device removal as a result of a depleted primary battery, technological upgrade or infection [7, 18].
Poor design or manufacturing decisions and/or inappropriate preclinical evaluation can result in device failure in patients [19, 20]. Device failure can have severe adverse mental and physical effects on the patient, add significantly to healthcare costs and have a serious negative impact on the neural stimulator industry [21].
The successful development of active implantable devices is achieved through close collaboration between many disciplines including engineers, scientists and the surgical/clinical team. It is also important to engage potential end users of the technology during device development and to obtain feedback from implant subjects following their participation in early clinical trials [22]. To this end, the US Food and Drug Administration (FDA) Center for Devices and Radiological Health have established a Patient Preference Initiative designed to drive more patient-centric device innovation [23].
3. Overview of preclinical studies
Carefully controlled in vitro and in vivo studies together with ex vivo and human cadaver trials form the key components of a thorough preclinical evaluation of any new neural stimulator. In this section we briefly introduce these three components of preclinical evaluation; they are expanded upon in subsequent sections (in vitro, section 4; in vivo, section 5; and Human cadaver studies, section 6). Figure 1 illustrates this pipeline of device development from concept to commercialisation with a deliberate emphasise on preclinical studies. Importantly, preclinical studies not only serve to demonstrate safety and efficacy, they also provide ongoing feedback to the design of the device before it is locked into a final form.
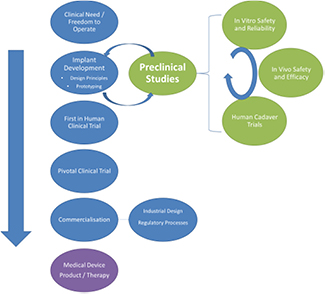
Figure 1. Medical device development pipeline with the preclinical activity expanded. Design of a neural stimulator is an iterative process with feedback opportunities at many stages of development. Preclinical evaluation is typically undertaken on prototype components rather than a fully manufactured device.
Download figure:
Standard image High-resolution imageMany aspects of the preclinical research illustrated here can be undertaken in parallel although initial in vitro screening studies for safety and reliability would typically precede any extensive in vivo study. Initial human cadaver studies are often performed early in a project while more extensive research is undertaken later in the development following final agreement on the form factor of the device.
Key aspects of the preclinical research are typically performed by an academic interdisciplinary research team with the relevant expertise. There are, however, additional preclinical studies that are generic and have been clearly defined in the International Organization for Standardization (ISO) document 10993-1 'Biological evaluation of medical devices' covering general biocompatibility testing of a device [24, 25], and ISO 14708 'Active Implantable Medical Devices' covering the evaluation of a device for mechanical and electrical integrity. This work is typically performed by experienced commercial research organizations after the final design specifications have been locked in and will be briefly described in section 7.
While many new implants will incorporate materials with long, successful histories of clinical use (e.g. polydimethylsiloxane (PDMS); platinum (Pt)) devices using these materials must nevertheless be carefully evaluated for each new application. These studies may include a suite of in vitro and chronic in vivo tests, however it is also appropriate to leverage earlier work by others if it is relevant to the safety and efficacy of the device under investigation.
Novel materials with little or no prior history of clinical application will require more extensive testing. In addition, new materials may require commercially relevant studies designed to ensure that any novel properties are maintained following packaging, sterilization and long-term storage.
Finally, it is important to emphasize that there is no specific roadmap outlining a sequence of preclinical studies that must be performed in order to obtain regulatory approval for a device. The studies required will vary from device to device and will be influenced by factors including the novelty of: (a) the materials used; (b) the neural target; (c) the surgical approach; and (d) the proposed stimulator. For example, a novel device designed for a new application will require significantly greater preclinical testing than an approved device being re-purposed for another application where it can cite a significant history of preclinical and clinical safety. While the responsibility for the study design rests with the clinical, scientific and engineering team developing the technology, regulatory bodies encourage early dialogue with the development team before initiating preclinical studies. For example the FDA provide an early interaction program ('Q-Sub') where preclinical testing strategy, development of a clinical study protocol and guidance regarding regulatory pathway for novel devices can be discussed [26]. The FDA will also often develop specific guidelines to assist with the design process and include both in vitro and in vivo tests. For example the Guidance for Retinal Prostheses includes preclinical tests that should be considered before initiating any clinical evaluation [27].
3.1. In vitro studies
In vitro studies provide relatively rapid feedback on: (a) the performance of electrodes, cables and insulation under both passive conditions and in response to repeated electrical stimulation [28–38]; (b) mechanical failure modes associated with accelerated movement [39]; (c) the evaluation of material adhesion and delamination of the electrical insulation creating inter-channel crosstalk [5, 25, 32, 40]; (d) the efficacy of hermetic sealing techniques [41–43]; (e) the study of insertion forces associated with electrode implantation in order to optimise the design of the electrode array [44–47]; and (f) the in vitro assessment of the electrode array for biological cell–electrode interactions [48], glial scarring [49] and cytotoxicity [50–52].
In vitro studies are often performed under conditions of accelerated aging including the use of elevated temperatures, harsh electrolyte environments and accelerated mechanical testing [25, 32, 49]. For example, there is an approximate doubling of reaction rates for every 10 °C increase in temperature based on the Arrhenius equation [25]. These tests are generally performed at an early stage in the evaluation of a device, providing a mechanism for screening prior to chronic in vivo studies. Some in vitro studies can be readily extended for long periods of time (years) to provide estimates of a device's lifetime [25, 43, 53].
Finally, in order to reduce the ethical and financial burden of animal studies, significant effort is being directed towards expanding the repertoire of in vitro studies to more accurately replicate the in vivo environment thereby improving the efficacy of in vitro studies used to screen novel materials [4, 32, 48–51]. In addition there is considerable interest in developing in silico models of clinical trials that includes evaluation of medical devices [54].
3.2. In vivo studies
In vivo studies include early proof-of-principle research performed in an animal model of the disease or injury in acute anesthetised animals, through to chronic safety and efficacy studies in awake, freely moving animals. Chronic preclinical studies should be designed to address the mechanical and electrical integrity of the implant (abiotic) while in the same animal addressing the effect of the implant on its biological environment (biotic, figure 2; [25, 32, 55]). Abiotic failure includes loss of optimal device function including high impedance electrodes, low impedance cable crosstalk, delamination or corrosion of electrode contacts, or delamination of insulation. In contrast, biotic failure includes any adverse inflammatory cell response, extensive fibrous tissue encapsulation or neural degeneration associated with the implant or the effects of electrical stimulation, or functional reductions in neural activity measured electrophysiologically.

Figure 2. Timeline of major events following chronic implantation in vivo. These events are divided into abiotic and biotic changes that need to be carefully monitored in any carefully controlled preclinical study. Reproduced from [75]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageAcute in vivo studies include: (a) studies designed to examine the efficacy of stimulation for a specific electrode array and stimulation site [56–58]; (b) research designed to understand the underlying physiological mechanisms associated with the specific application [59–61]; (c) the response properties of the target neural pathway to electrical stimulation in order to optimise electrical stimulation parameters [59, 62–68]; and (d) the response of the target neural population to short-term electrical stimulation [69–74]. A number of these acute studies are important in establishing device design (e.g. the location and size of electrode contacts), while other studies provide insight into the physiological mechanisms associated with the electrotherapy.
Few preclinical in vivo device studies examine the recovery period covering the first ~14 days post-implantation (figure 2) but rather evaluate the biotic environment after the majority of the inflammatory response has resolved (the chronic period).
Chronic in vivo studies can be divided into passive (no stimulation) and active (stimulation) studies. Passive studies include: (a) the evaluation of electrode insertion trauma [76, 77]; (b) the effects of movement on both the implant and the tissue environment [78–84]; (c) the long-term performance and biocompatibility of electrodes as well as implant and cable materials (e.g. [18, 30, 83–91]; (d) the ability to safely replace an electrode array [76, 92, 93]; and (e) safety issues specifically related to growth in children fitted with a device [94–98]. Chronic active studies are designed to evaluate the safety, and potentially the efficacy, of chronic electrical stimulation at the intended stimulation site and on the adjacent biological environment [38, 80, 99–105] and the electrodes [29, 38, 83, 105, 106].
Although there are useful guidelines defining the boundary between damaging and non-damaging electrical stimulation [107], these guidelines were derived from experimental data based on acute stimulation using large surface area Pt electrodes in direct contact with cortical neurons and stimulated at a fixed pulse rate of 50 pulses per second [108]. These stimulus parameters represent a small subset of the parameters used in clinical devices and are therefore not suitable to define safety levels for applications outside that subset [107].
Important parameters including stimulus rate, electrode area, near/far field, duty cycle, electrode material [10] as well as the target neural population, will influence the boundary between safe and damaging electrical stimulation. This large and mainly unexplored parameter space requires that chronic active safety studies are necessary for almost all new device applications.
While key components of the evaluation of device safety can be performed in normal animals, if possible it is important to also demonstrate therapeutic efficacy through preclinical stimulation studies. These efficacy studies require well developed animal models of the disease or injury. The careful selection of appropriate animal models for preclinical safety and efficacy studies will be discussed in section 5.1.
In designing preclinical studies, it is important to note that the evaluation is not typically undertaken on a fully manufactured device, rather prototype components that have a similar form and are manufactured from the same materials as the proposed final product are sufficient. The use of carefully designed prototypes in preclinical testing ensures the design process is an iterative one that allows ongoing modifications to be made throughout the device development (figure 1).
At a later stage of development the fully assembled neural stimulator may undergo preclinical assessment for safety and reliability prior to clinical trial [109]. These studies normally involve the implantation of the device in a subcutaneous tissue pocket.
Although preclinical evaluation of neural stimulators typically centre on the safety and efficacy of the electrode-neural interface, it is important that any external components of the device are also designed and assessed for safety. For example the technical standards for general medical device safety, including electromagnetic compatibility, are described in IEC 60601 [110]. In addition it is necessary to ensure that the telemetry link between the external device and the implant conforms to radiation guidelines for exposure developed by the International Commission on Non-Ionizing Radiation Protection (ICNIRP) [111–113], and that MRI compatibility of the device is carefully assessed. This type of evaluation is typically performed by a commercial contractor and will not be considered further in this review.
3.3. Human cadaver and ex vivo studies
Studies using human cadaver material also play a very important role in developing safe implants (figure 1). These studies include research designed to: (a) develop the form of the implant compatible with the anatomy of the target site; (b) optimise the surgical approach; (c) develop specialist surgical instruments; (d) evaluate electrode insertion trauma; and (e) practice the surgical procedure prior to commencing the clinical trial.
4. In vitro evaluation of device safety and reliability
This section reviews the typical in vitro testing undertaken during the development of a neural stimulator. The evaluation of the electrode array and cable assembly is the major focus of this testing.
In vitro evaluation of neural stimulators includes a broad range of tests that can be used to validate the safety and reliability of these devices. These tests focus on specific chemical, electrical or mechanical properties and are evaluated under highly controlled environments. In vitro testing allows fast turnaround and can be the primary source of results to guide the early stages of device development. Evaluation techniques cover both (a) bench-top testing designed to evaluate physical properties; and (b) a simulated biological environment designed to evaluate performance under conditions similar to the intended application (figure 3). The suite of in vitro tests to be performed should be carefully planned to complement a necessarily limited set of in vivo experimental cohorts.
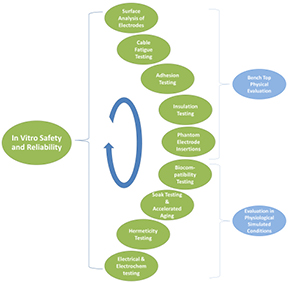
Figure 3. Typical suite of in vitro techniques that can be used to evaluate the safety and reliability of a neural stimulator.
Download figure:
Standard image High-resolution image4.1. Bench-top physical evaluation
4.1.1. Surface analysis of electrodes.
Analysis of the surface features of electrode arrays can be used to evaluate the quality and repeatability of the electrode/implant surface treatments and fabrication methods, and to provide feedback on the stability of the electrode surface following long-term implantation and electrical stimulation. Careful analysis of the surface features of electrodes should follow both in vitro and in vivo testing. Surface features that can be evaluated include mechanical degradation, surface deposits, roughness and evidence of corrosion such as surface pitting (figure 4). These features can be assessed using a variety of tests for both: (a) surface topography using optical profilometry, scanning electron microscopy (SEM) or atomic force microscopy [28, 29, 32, 38, 50, 75, 114, 115]; and (b) elemental composition using energy dispersive x-ray analysis, x-ray photoelectron spectroscopy or Raman spectroscopy [28, 116–118].

Figure 4. Scanning electron microscopy is used extensively to evaluate the surface of electrodes and to assess the status of insulators following chronic in vitro or in vivo studies. This example illustrates the surface of two Pt electrodes following long-term in vivo implantation. (a) Extensive pitting corrosion following electrical stimulation using charge balanced biphasic current pulses at a charge density of 250 µC/cm2/phase delivered via a stimulator using electrode shorting for charge recovery. (b) Unstimulated control electrode exhibited no evidence of Pt corrosion although manufacturing marks are evident. Higher magnification images of (a) and (b) are illustrated in (c) and (d) respectively. Scale bars: (a) and (b) = 100 µm; (c) and (d) = 10 µm.
Download figure:
Standard image High-resolution image4.1.2. Cable fatigue testing.
Cable or electrode lead fracture and/or insulation breakdown are common failure modes associated with neural stimulators and are particularly problematic in devices associated with inherent movement [20, 31, 39, 84, 119–121]. In vitro evaluation of fatigue resistance must be designed to model the anticipated extent of flexion and strain profiles expected in the clinical setting [119, 122]. There are published industry standards that provide guidelines for the testing of electrode cables (e.g. for cardiac pacemakers [123] and cochlear implants: [124, 125]). These studies are typically performed under accelerated conditions to achieve high cycle numbers (i.e. long equivalent implant years) over months of testing [122] or are run to cable failure [121].
A rotary bending fatigue test can be used to evaluate the reliability of cables bent at a specific radius of curvature [126]. This test abstracts the torsion and bending movements experienced by implanted cables into a simple test where the cable is rotated around its axis while held in a bent position. Usually both ends will be rotated together while holding the cable in a fixture with fixed radius. This provides continuous stress to the wires from the torsion moment. Axial fatigue testing is an important method for evaluating wires in a process that involves stresses throughout the volume of the cable and not just the surface [127].
There are a number of methods that can be used in concert to carefully assess the extent of cable and insulation damage induced by fatigue testing. These include electrical tests for continuity and current leakage using Electrochemical Impedance Spectroscopy (EIS; [83, 128]), optical inspection of cables under magnification and mechanical tests of conductor fatigue resistance [121, 122].
4.1.3. Adhesion testing.
Many potential electrode materials are being developed with properties that include increased mechanical compliance with neural tissue and a significant increase in charge injection capacity over conventional electrode materials such as Pt [40, 89, 129–134]. These materials can be directly coated onto the metal substrate. While promising, many of these coatings delaminate from the metal substrate during long-term electrical stimulation [133, 135, 136] resulting in a loss of their electrochemical advantage. It is important to evaluate the adhesive properties of novel coatings using well controlled mechanical tests before committing resources to extensive in vivo testing.
Suitable tests include measuring adhesion by tape test [137, 138], the scratch adhesion test [139–141] and an ultrasonication test [133]. Using the American Society for Testing and Materials standard as a guideline, the adhesion test requires two incisions in an 'x' to be cut into the coated electrode to expose the underlying metal using a surgical blade. Adhesive tape is placed over the incision for a defined time and then removed. The site is then examined under a microscope and image analysis techniques are used to calculate the loss of coating versus a control material such as IrO2 (figure 5). Multiple trials are used to establish statistical significance. The scratch adhesion test involves scratching the coating surface at a fixed rate with a diamond stylus using a range of fixed loads. The scratches are examined under magnification for evidence of failure modes including the nature and extent of spallation, conformal cracking and tensile cracking [139, 141]. The critical load is recorded and the test repeated several times to achieve statistical significance. Adhesion efficacy can also be readily measured against control coatings by comparing the extent of coating loss measured using image analysis techniques, after a defined duration of ultrasonication [133].

Figure 5. The adhesion tape test is an efficient and effective method for evaluating the adhesion properties of electrode coatings. In this example, Pt squares were coated with (a) IrO2 or (b) a conductive polymer. An incision is made in the coating in the form of an 'x' using a new No. 11 stainless steel surgical blade to expose the underlying Pt. Adhesive tape (3M 2080) was placed over the coating for 5 minutes and then removed. There was minimal loss of IrO2 along the incision line. In contrast the polymer lifted along the incision line (arrows) and ~25% of the coating was removed with the removal of the adhesive tape. Scale bar = 1 mm.
Download figure:
Standard image High-resolution imageFinally, it is important to ensure that novel electrode coatings are not adversely affected by the sterilization process; they do not absorb and slowly release sterilization agents such as ethylene oxide that could leach into the biological environment; they survive standard shipping and storage protocols without losing any advantage; and surgical teams are carefully advised regarding any specific handling requirements.
4.1.4. Insulation testing.
Although most emphasis in both in vitro and in vivo safety and efficacy studies focus on the performance of the electrode-neural interface, the long-term performance of the insulation must also be carefully evaluated as its breakdown can have major implications for the efficacy of neural stimulators [5]. These effects are most critical in devices associated with significant movement resulting in abrasion of the insulation [120], and in small implants where insulation is by necessity very thin [19]. Because the lifetime of many insulating films are adversely affected by the electric field strength, evaluating the efficacy of new insulating materials should be performed under electrically active conditions [25, 43, 142].
In vitro studies under controlled elevated temperatures with longitudinal EIS recordings and pre- and post-stimulation imaging is an effective way of evaluating the performance of insulation materials [32, 35, 36, 106]. Insulation failure following completion of chronic in vivo studies has also been detected using microscopy [120] and SEM [75, 83, 106, 143].
Fluid ingress under the insulation can produce conductive pathways or crosstalk between adjacent cable channels, significantly reducing the efficacy of stimulation. This is particularly problematic using high impedance microelectrodes. The extent of the crosstalk can be measured on completion of an in vitro study by measuring the impedance of adjacent electrodes using EIS or pulsatile stimuli with the electrodes removed from the electrolyte. Crosstalk impedances approaching an order of magnitude more than the impedance of the electrodes in the electrolyte are considered unacceptable (Seligman, personal communication).
4.1.5. Phantom electrode insertions.
A change in the physical properties of an electrode array can adversely affect its insertion properties and result in a significant increase in insertion trauma. For example, a reduction in the stiffness of an array as a result of a reduction in size may result in increased trauma as a result of the array buckling during insertion [47, 144], while an increase in stiffness due to changes in cable properties can also lead to increased trauma [145]. A useful in vitro test to assess the performance of new electrode designs is to perform phantom insertions under appropriate conditions while measuring the insertion force and imaging the electrode array during insertion. For electrodes designed to be implanted in brain tissue this evaluation can be performed using agar [44], while in applications such as cochlear implants ex vivo or model cochleae are used and insertion force measured [45–47, 146] or visualised via fluoroscopy [147, 148].
4.2. Evaluation in physiological simulated conditions
4.2.1. Biocompatibility testing.
Implantable materials that are exposed to tissue or body fluids must be biocompatible. The FDA recommends that all constituent materials that will be in permanent contact with tissue or fluid should be tested or should have a documented history of safe use in devices. Importantly, in addition to evidence of safety for the component materials, evaluation of the assembled device is also required. These tests include cytotoxicity, sensitization, irritation or intracutaneous reactivity, acute systemic toxicity, material-mediated pyrogenicity, subacute/subchronic toxicity, genotoxicity, chronic toxicity and carcinogenicity. The FDA has developed guidelines to assist industry in preparing medical devices that come into direct or indirect contact with the human body [52]. If developing a device using components with long histories of biocompatibility, these tests, as noted above, would be undertaken late in the device development pipeline and would be typically performed by a contract research organisation.
Of the in vitro biocompatibility tests listed above, cytotoxicity is the one most commonly described for the initial evaluation of new materials [50, 51]. Specific neural and non-neural cell lines are cultured directly onto the surface of the material undergoing testing. Cell survival is quantified at two time points. Analysis is performed using simple cell count quantification, qualitative scoring of cell adhesion, or quantification of morphological cell aspects such as neurite length or number [149]. Test materials are evaluated against both positive and negative control materials [50].
In addition to the screening for cytotoxicity, cell-culture studies provide an opportunity to compare the extent of glial scarring for candidate biomaterials [49], as well as studying the effects of cell growth on electrode impedance in a highly controlled environment [34]. It should be noted, however, that this type of testing does not replicate the complex milieu of a biological system.
4.2.2. Soak testing and accelerated aging.
Neural implants need to be reliable over the long-term in the biological environment. The use of harsh in vitro environments or reactive accelerated aging [32, 37] can be a valuable tool for screening prototype materials and devices in the early stages of device development [25]. The simplest evaluation of device stability consists of soaking the assembly passively in saline, preferably at body temperature or higher, over many months. This allows validation of the stability of the components or assemblies under a corrosive fluid environment [30, 36, 41].
Accelerated ageing techniques are useful in reducing the time to failure and validating material selection, fabrication methods or a combination of the two. Identifying quickly the limitations and failure methods of a neural interface will speed up the development cycle. However the accelerated aging parameters must be selected with care; too harsh an environment may generate failure modes that would not be experienced in vivo. Ideally a device should experience similar rates of deterioration and failure in the accelerated in vitro environment and in vivo [25].
Electrode corrosion has been an active concern from the early days of the development of neural prostheses from both a toxicology [150] and electrode life [151] perspective. Corrosion resistance to electrical stimulation can be investigated by direct measurement of metal or material concentration in the electrolyte and/or inspection of the electrode surface using SEM. For example, inductively coupled mass spectroscopy has been used to quantify trace amounts of iridium following in vitro electrical stimulation of iridium oxide coated electrodes [35]. The corrosion behaviour can also be inferred from measurements including open-circuit potential, potentiostatic control, EIS, and linear voltammetry techniques [35–38, 55, 152, 153].
The controlled in vitro environment is useful for characterising the dissolution properties of the electrode, but it is a much simpler environment than the body where reactive oxygen species and other ions abound [154]. The corrosion level of electrode materials such as Pt exhibit much greater dissolution in inorganic saline than saline containing small amounts of protein [155, 156] or in vivo [29, 157]. In vitro studies evaluating the effects of electrical stimulation on electrodes can therefore serve as a guide for a worst-case limit for charge delivery to avoid corrosion. While in vitro studies with added protein such as fetal bovine serum can be informative and performed at low cost, it remains essential that the long-term performance of novel electrodes or stimulation regimes are also evaluated in vivo [30, 33].
4.2.3. Hermeticity testing.
A key design criteria associated with the long-term reliability of any implant is the efficacy of its hermetic seal [43]. Modern neural prostheses can have more than 60 electrical feed-through connections, although higher numbers will be required as higher-density electrode arrays are developed for clinical use. The clinical gold standard for hermetic encapsulation of implants remains the use of titanium cans sealed using a laser welder [158]. Other materials that have been used in implantable devices include (in decreasing order of hermeticity) ceramics, glass, epoxy, and silicone [5, 43]. While having good hermetic properties, ceramic and glass are brittle, increasing the risk of breakage associated with an impact injury. An epoxy-based hermetic seal is typically confined to use in prototype devices, while silicone is relatively permeable to water molecules. The use of polymers for hermetic encapsulation is attractive because many have excellent biocompatibility, ease of fabrication, flexibility, electromagnetic transmission, and cost compared with titanium. In addition because they are thin, polymers provide an attractive alternative for the next generation of small implants. The main technical challenge is ensuring long-term effective hermetic bonding. Finally, novel high density hermetic feedthroughs are under development for neural prostheses using doped nanocrystalline diamond channels within an insulating polycrystalline diamond substrate [159].
The most common technique to test the hermetic seal with current encapsulation technologies is the helium leak detection test [158–160]. This test is well characterised and yields a well-accepted model of expected durability of the hermetic seal, e.g. 10 or 50 equivalent years. The test uses a mass spectrometer in a vacuum to measure the leak rate of the tracer gas as it escapes after placing the device to be tested into a high pressure helium environment [161]. The test has been adapted to evaluate components of the capsule by placing the device on the spectrometer vacuum intake using an o-ring seal, and testing the leakage from a helium chamber through the tested components.
The main drawback for helium-leak testing technology is the need for void space within the hermetic capsule to allow measurable volumes of helium to flow. For instance, testing a capsule with 1 cm3 internal volume for an equivalent 100 year hermeticity requires a sensitivity of 2 × 10−13 mol s−1 of helium, which is at the sensitivity limits of spectrometers [43]. Smaller internal volumes would not be able to be tested with current technology. This presents a challenge with shrinking electronic manufacturing technologies and a desire for chip-scale hermetic packaging (applied directly on the silicon wafer). Alternatives such as on-chip water detectors are being developed for future applications in neural stimulators. Long-term soak testing at room or an elevated temperature provides an alternate technique [42, 161, 162], however its obvious drawback is the duration of the testing procedure.
4.2.4. Electrical and electrochemical performance.
Electrodes in contact with neural tissue form the only active interface between the implant and the body. The electrode-tissue interface must be carefully evaluated to ensure that the electrochemical behaviour is always within safe limits. The electrochemical function of the electrodes is usually tested by a combination of cyclic voltammetry (CV), EIS and Voltage Transient analysis. While subsets of these tests can be safely performed in vivo, in vitro testing can be more comprehensive and allow verification of the limitations imposed by the choice of electrode area, surface treatment, material, or even substrate configuration (e.g. recessed versus protruding electrodes).
The setup for these electrochemical tests is usually a 3-electrode cell with phosphate buffered or physiological saline and a reference electrode of Ag|AgCl, saturated calomel or even Pt. These tests are best used for comparing different electrode materials [115]. Due to the simplicity of the electrolyte compared to the biological environment, the measured values will be at best an approximation of the electrode behaviour in vivo.
The charge storage capacity of the electrode interface is one of the important parameters to characterise. This refers to the maximum charge than an electrode-electrolyte interface can hold while remaining within the limits of non-reversible oxidation or reduction, although it is important to note that there are other potentially damaging reactions, due to irreversible Faradaic reactions, that can occur within the water window.
The charge storage capacity can be estimated from a slow CV plot (50 mV s−1), measured as the area within the curve between the hydrolysis limits [163]. For neural interface applications, a saline solution (typically buffered) is used as the electrolyte for this measurement although it is acknowledged that the in vivo electrochemical environment will behave differently [33]. The charge storage capacity is intended as an indication of the inherent properties of the material and is useful for comparing electrodes of different materials (figure 6), or different morphologies of the same material (e.g. comparing sputtered versus activated iridium oxide electrodes [164]).

Figure 6. Example of in vitro electrochemical comparison of conductive hydrogel versus platinum electrodes having the same geometric surface areas. Cyclic voltammetry (CV; left) illustrates an increased charge storage capacity for the hydrogel electrode (16.8 mC cm−2) versus the platinum electrode (2.04 mC cm−2) as indicated by the larger area of the CV curve. The electrochemical impedance spectroscopy (EIS; right) illustrates reduced impedance of the conductive hydrogel electrode compared with platinum across the frequency range tested.
Download figure:
Standard image High-resolution imageA different test is necessary to quantify the charge injection capacity that is accessible during electrode use. This will be the sub-portion of the storage capacity which can be achieved when injecting charge with the intended stimulus waveform (e.g. charge-balanced biphasic current pulses). The charge injection capacity is the limit where the charge delivery waveform polarises the electrode within voltage limits for irreversible reactions (such as hydrolysis). This can be measured from the voltage transient during charge delivery where the electrode polarisation is evident at the end of the first phase (figure 7; [38, 128, 165]). In physiological saline, the quantification can also be evaluated via chemical analysis of the solution for signs of metal oxides [166].
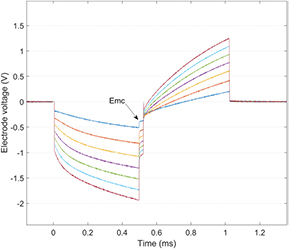
Figure 7. Voltage transients on a platinum disc electrode (600 µm diameter) from 500 µs/phase biphasic current pulses incremented in 100 µA steps from 100 (blue) to 700 µA (red). The electrode polarisation observed at the most cathodic potential (Emc) after the first phase, indicates a charge injection limit of ~53 µC cm−2, since 300 µA takes the electrode voltage to the hydrolysis limit for platinum. These recordings were made using a two Pt electrode configuration with the reference a large surface area Pt electrode.
Download figure:
Standard image High-resolution imageCharge injection capacity can be altered by modifying an electrode's surface either by applying coatings or by increasing the real surface area. Laser roughening of Pt electrodes is an example of modifying the surface of an electrode to change its electrochemical behaviour [167]. However, it is important to demonstrate that any advantage associated with modifying an electrode surface is maintained in vivo [168].
Recent electrochemical studies examining electrical stimulation of Pt electrodes have provided new insights into their charge injection capacity. These studies also contribute to the development of improved in vitro environments for the evaluation of electrodes under conditions of electrical stimulation [169, 170].
The reversibility of the electrochemical charge injection RedOx reactions is inferred from the shape of the CV curve. The curve should also repeat itself over several cycles to indicate no net polarisation of the interface. The hydrolysis limits are represented in a CV curve as large current peaks (dissolved gas) and indicate the range for the working polarisation voltage. Typically the CV is repeated at the limited no-hydrolysis range to study the electrochemical behaviour (figure 6).
Understanding the electrochemical behaviour of the electrode material is important to ensure complete charge recovery during stimulation [171]. Residual levels of direct current (DC) as low as 0.4 µA has been shown to result in tissue damage [172]. It is therefore important to ensure that any stimulator used in chronic preclinical safety studies are closely based on the design intended for clinical application. Specifically the level of symmetry of the charge balanced current pulses and the techniques used to recover residual charge (e.g. electrode shorting; capacitive coupling) should be accurately modelled. Finally, it should be ascertained that the charge injection processes used are limited to double layer charging (capacitive) and reversible surface reactions [166]. When creating polymeric (or bioactive) electrodes, these measurements characterise the electrodes at different stages of fabrication to yield optimal surface conditions [173].
Perhaps the most important performance test is measuring the impedance of the electrodes [173], which can then repeated in vivo during the life of the implant. The impedance is determined by the specific configuration of the electrodes including the material, electrode area, surface characteristics and electrolyte accessibility (electrode perimeter and substrate shape) [174]. Once in vivo, the changes in impedance typically relate to the extent and nature of the tissue environment surrounding the electrodes.
The impedance can be measured at the linear low-polarisation region using EIS (figure 6). Alternatively, a non-linear electrode impedance at stimulation-relevant amplitudes can be measured from the voltage transients (figure 7). Measuring the voltage transient incursion with stimulation pulses is a simple technique which is easily deployed within implantable stimulator electronics, it is relevant for specifying stimulator parameters, and provides a clear indication of whether or not the stimulator is operating within its voltage compliance.
5. In vivo preclinical safety and efficacy studies
Here we review the typical in vivo testing undertaken during the development of a neural stimulator. The safety and efficacy of the implant in both passive and active conditions is the major focus of this testing (figure 8).
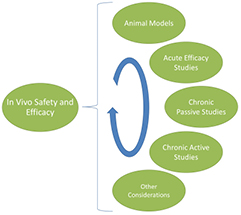
Figure 8. Preclinical in vivo studies cover a broad range of techniques that are used to evaluate the safety, efficacy and reliability of a neural stimulator.
Download figure:
Standard image High-resolution imageIn vivo evaluation of neural stimulators covers a broad range of techniques designed to validate the safety, efficacy and reliability of these devices. While there is no defined template for the conduction of in vivo preclinical studies, experienced researchers will combine an early acute efficacy study with carefully considered chronic passive and active studies to provide a comprehensive preclinical evaluation of the device. Finally, when designing these studies it is important that consideration is given to the choice of the most appropriate animal model(s). The results of in vivo preclinical studies will impact the design of the neural stimulator and play an important role in gaining regulatory approval for use of the technology in clinical trials.
5.1. Animal models
Regulatory bodies require the device to be evaluated in an animal model prior to granting approval to go to clinical trial. Careful consideration and selection of an animal model(s) is an important component of any successful preclinical study; their contributions have been reviewed in the case of a number of devices [175, 176]. It is important to demonstrate early in the development pipeline that electrical stimulation provides a significant therapeutic advantage for a particular disease or injury. This is typically evaluated in small animals that accurately model the disease or injury (table 1). This work can be performed in either acute [177–179] or chronic [180–185] studies.
Table 1. Examples of small animal models used to evaluate the safety and efficacy of neural stimulators.
| Species | Condition | Device | References |
|---|---|---|---|
| Mouse | Hearing loss | Cochlear Implant | [192, 193] |
| Mouse | Inflammatory disease | VNS | [194] |
| Mouse | Postoperative ileus | VNS | [195] |
| Rat | Bladder dysfunction | SRS | [196] |
| Rat | Urinary incontinence | PNS | [178, 197] |
| Rat | Depression | DBS | [198, 199] |
| Rat | Type 2 diabetes | CSNS | [184] |
| Rat | Inflammatory bowel disease | VNS | [180, 200] |
| Rat | Parkinson's disease | DBS | [201] |
| Rat | Hearing loss | Cochlear implant | [202] |
| Rat | Blindness | Retinal implant | [203, 204] |
| Rat | Neuropathic pain | DBS | [205] |
| Rat | Myocardial ischemia | VNS | [61, 177] |
| Rat | Hypertension | VNS | [206] |
| Rat | Spinal cord injury | ISMS | [82] |
| Rat | Spinal cord injury/amputation | ISMS | [106] |
| Rat | Provide sensory feedback | ICMS | [105] |
| Guinea pig | Hearing loss | Cochlear implant | [207, 208] |
| Guinea pig | Myocardial infarction | VNS | [181] |
Notes: DBS, deep brain stimulation; SRS, sacral root stimulation; VNS, vagal nerve stimulation; ISMS; intraspinal microstimulation for gait assistance; PNS, pelvic nerve stimulation; CSNS, carotid sinus nerve stimulation; ICMS intracortical microstimulation.
In addition to playing an important role in evaluating the efficacy of neural stimulation, an accurate disease/injury model should closely reflect the condition of the target neural population likely to be experienced clinically. This is important because neural degeneration, as a result of the disease or injury, is likely to affect the stimulus levels required to evoke a therapeutic effect. Furthermore, it is possible that a compromised neural population will respond to implantation and electrical stimulation in a manner that differs from that of normal tissue. Finally, although not critical to regulatory approval, acute studies using animal models of the disease or injury are important for studying the mechanisms underlying any therapeutic effects delivered by electrical stimulation.
While small animal models play an important role in efficacy studies they are not generally considered suitable for the long-term evaluation of device safety and/or optimising the surgical approach. Large animal models more accurately reflect the anatomy, surgical access and the mechanical environment that a neural stimulator would experience clinically. Moreover, the use of larger animals allows investigators to evaluate electrode arrays and cables that are similar in size to the proposed clinical device (table 2). Numerous preclinical studies performed by device manufacturers also highlight the importance of using large animal models in establishing device safety [12]. Finally, large animal models are often used without modelling the disease state. Under these conditions the emphasis of the study is on device safety and reliability rather than efficacy.
Table 2. Examples of large animal models used to evaluate the safety and efficacy of neural stimulators.
| Species | Condition | Device | References |
|---|---|---|---|
| Rabbit | Blindness | Retinal implant | [183, 210, 211] |
| Cat | Blindness | Retinal implant | [80, 212] |
| Cat | Hearing loss | Cochlear implant | [99, 213, 214] |
| Cat | Hearing loss | Auditory brainstem/midbrain implant | [103, 215] |
| Cat | Parkinson's disease | DBS | [44, 191] |
| Cat | Bladder dysfunction | Sacral neuromodulation | [179] |
| Dog | Spinal cord injury | FES | [98] |
| Dog | Blindness | Retinal implant | [216] |
| Dog | Multiple conditions | VNS | [217] |
| Dog | Blindness | Retinal implant | [182] |
| Minipig | Blindness | Retinal implant | [218] |
| Minipig | Parkinson's disease | DBS | [187] |
| Pig | Hearing loss | Cochlear implant | [219] |
| Pig | Blindness | Retinal implant | [109, 162, 220] |
| Pig | Spinal cord injury | ISMS | [81] |
| Pig | Parkinson's disease | DBS | [221] |
| Sheep | Parkinson's disease | DBS | [222] |
| Sheep | Chronic back pain | SCS | [63] |
| Monkey | Balance disorder | Vestibular prosthesis | [223] |
| Monkey | Hearing loss | Cochlear implant | [96, 224] |
| Monkey | Sensory feedback | ICMS | [105] |
| Monkey | Parkinson's disease | DBS | [225] |
Notes: DBS, deep brain stimulation; SRS, sacral root stimulation; VNS, vagal nerve stimulation; ISMS; intraspinal microstimulation for gait assistance; FES, functional electrical stimulation; ICMS, intracortical microstimulation.
5.2. Acute efficacy studies
The suite of preclinical in vivo studies often include acute studies used to establish the optimal location and form factor for the electrode array relative to the neural tissue; to demonstrate that the device can readily evoke neural activity well within safe stimulus limits for the given electrode material [10, 107, 186]; and to demonstrate that the electrical stimulation can provide therapeutic efficacy. Although these acute studies typically do not require the use of a clinical grade stimulator (e.g. [178, 184]), the use of such a stimulator informs design features including the surface area of stimulating electrodes (and therefore electrode impedance), providing confidence that the proposed stimulator can deliver the required stimulus within its voltage compliance [58] and current density (and therefore stimulation efficacy).
5.3. Chronic passive studies
The implantation of an electrode array will evoke a host response designed to isolate the foreign body [17, 80, 89, 187–189]. The extent of this reaction depends on many factors including surgical technique, biocompatibility, and the mechanical characteristics of the implant relative to the host environment. The nature of the tissue response will also vary over time as acute oedema and the proliferation of inflammatory cells are gradually replaced by a mature tissue capsule around the implant (figure 2; [55]). Chronic in vivo studies evaluate the extent of this tissue response and any associated neural loss that could significantly affect the performance of a neural stimulator. As noted above, chronic passive studies are important for examining the biological response of the electrode-neural interface as well as any electrical and mechanical/material failure modes associated with the electrode array and cable assembly [84, 87]. These results can also be used as control data when assessing the effects of electrical stimulation per se as part of a chronic stimulation study.
The implant duration in chronic passive studies varies considerably and while the acute inflammatory response in an implant site typically resolves over a few weeks [55, 112, 190], the rate of transition from an acute to chronic phase is dependent on a number of factors including the physical and mechanical properties of the electrode-neural interface and the biocompatibility of the implant. Periods of 1–12 months are typically used [55, 66, 76, 87, 89, 188, 189, 191]. In addition to assessing the chronic tissue response it is also important to examine for evidence of neural loss, if any, associated with the implant procedure. Neural loss may take several weeks to become apparent, it is therefore common for these studies to involve implantation periods of several months in order to ensure a mature tissue response and a stable electrode-neural interface. Longer implantation periods also provide a more extensive evaluation of the electrode array and cable assembly.
Finally, chronic passive studies have also been used to evaluate the feasibility and safety of the surgical removal and replacement of an electrode array, an important consideration in the design of neural stimulators for clinical application [76, 92, 209].
5.4. Chronic active studies
Safe electrical stimulation is achieved via capacitive and reversible Faradaic reactions that are dependent on the use of charge-balanced stimulation, the efficacy of the charge recovery technique and the electrode material. For excellent reviews the reader is directed to [10, 186, 226]. Stimulus parameters vary significantly across neural implants reflecting the wide variation in peripheral and central nervous system targets and the desired therapeutic endpoint (table 3). In addition there are wide variations in both the design of electrodes and stimulators that can impact on long-term stimulation safety. This great variation in neural targets, stimulation waveforms, electrode design and stimulator specifications require chronic active in vivo safety studies to be performed for each specific neural application.
Table 3. Examples illustrating the large range of stimulus parameters employed by neural stimulators in clinical settings.
| Device | Source | Stimulus level (peak) | Pulse width (µs) | Stimulation rate (Hz) | Reference |
|---|---|---|---|---|---|
| Cochlear implant | Current source | 0.1–0.9 mA | 10–50 µs | 500–3000 | [236] |
| DBS (STN, GPi & Th) | Voltage source | 1.5–5 V | 60–450 | 130–185 | [237, 238] |
| DBS (STN) | Current source | 2.3 mA | 74 | 150 | [239] |
| SCS | Current source | 0.5–5.0 mA | 30 | 10 000 | [240] |
| VBLOC | Current source | 1.0–6.0 mA | N/A | 5000 | [241] |
| SCS | Current source | 1.8–3.0 mA | 160 | 30–300 | [242] |
| VNS (Seizure) | Current source | 0.25–2.5 mA | 250 | 20 | [243] |
| VNS (IBD) | Current source | 0.25–1.25 mA | 500 | 10 | [244] |
| Retinal prosthesis | Current source | 0.1–3.0 mA | 150–500 | 50–500 | [245] |
aMean setting across 136 subjects; DBS, Deep brain stimulation; STN, subthalamic nuclei; GPi, globus pallidus; Th, thalamas; SCS, spinal cord stimulation; VBLOC, vagal nerve block for weight control; N/A, data not available; IBD, inflammatory bowel disease. Note that all studies used macroelectrodes and charge balance stimuli however the charge recovery technique, the dimensions of the electrodes and the range of charge densities used were often not described.
A key component of any preclinical chronic active study includes the use of the electrode array implanted in the intended anatomical site; and the stimulator, its charge recovery mechanism and the proposed 'worst case' stimulation parameters designed for the specific clinical application (e.g. [185]). Even then, it should be noted that stimulus levels producing a therapeutic effect in an animal model may not be sufficient to produce a similar effect in humans as a result of issues of scale. For example, targeting a particular fascicle via an epi-neural cuff electrode may require significantly higher stimulus levels clinically than the levels used to demonstrate safety in preclinical animal studies.
These studies should also include longitudinal electrode impedance and electrophysiological and/or behavioural monitoring to demonstrate that the electrode array/cable assembly is viable, the stimulator is operating within its voltage compliance and the stimulus levels employed are evoking appropriate neural activity. Longitudinal electrophysiological monitoring also provides insight into the functional changes that may occur to the target neural population over the course of the stimulation program [80, 83, 99]. It is also useful to monitor chronically implanted animals with longitudinal clinically relevant outcome measures. For example, in chronic preclinical studies of retinal prostheses standard clinical monitoring was used, including Intraocular Pressure, Fundus imaging, Electroretinography, Optical Coherence Tomography and clinical observations from a specialist [80]. Finally, it is important to monitor chronically stimulated animals for evidence of stimulus induced side effects. Often these can be measured using standard physiological techniques (e.g. heart rate; blood pressure; respiration rate), however not until early clinical trials is it possible to clearly determine whether any adverse side-effects will limit the neurotechnology under development.
As noted above, the use of an animal model of the disease or injury state is considered the gold standard; however normal animal models are usually suitable to assess stimulation safety. Ideally, bilateral implantation with unilateral electrical stimulation should be employed as it provides a within animal control, increasing statistical power while keeping animal numbers to a minimum. Regulatory bodies typically recommend implant durations of at least 3–6 months for active preclinical studies (e.g. [27]).
In the great majority of studies it is necessary to remove the electrode array before tissue preparation and histological analysis. The removal of the electrode array must be undertaken carefully to ensure that there is no disruption to either the underlying tissue or the electrode array, and there is no histological artefact introduced due to bleeding or trauma as a result of the dissection. Fixed neural tissue is typically less susceptible to damage during electrode removal than unfixed tissue. Dissection at this stage also ensures there is no bleeding artefact. Removed electrodes are available for SEM inspection as described in section 4.1.1.
The ability to analyse the electrode tissue interface without removing the electrode array can provide important insights that are not apparent when the electrode array has been removed. Recent immunohistochemical techniques describe the evaluation of the tissue response with microelectrodes in situ [227, 228]. Techniques are also available for the in situ inspection of macroelectrodes, although the approach is more limited in terms of resolution and tissue staining [229].
Quantitative histological analysis is a key component of any chronic preclinical study. Such techniques are readily available by combining appropriate histological and immunohistochemical techniques with image analysis software such as NIH's ImageJ (figure 9). Specific staining for neural tissue, astrocytes, microglia/macrophages and fibrous tissue will provide a good overview of the histological status of the electrode-neural interface [56, 73–75, 80, 104, 105, 182, 185, 188, 228, 230, 231]. Quantitative data must be compared statistically with appropriate control tissue—ideally implanted, unstimulated tissue from the same animal.
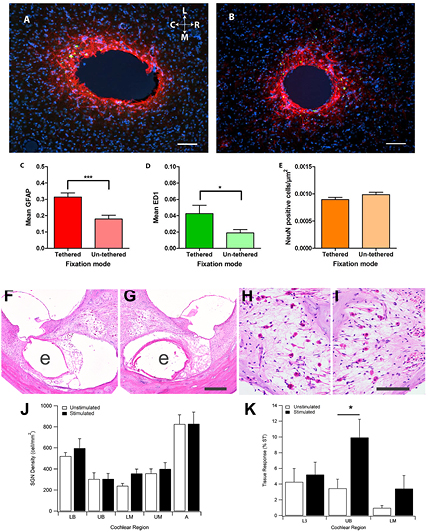
Figure 9. Examples of quantitative histological analysis techniques undertaken following 12 weeks of implantation of electrodes in the rat brain (A-E) and chronic implantation and electrical stimulation of electrodes implanted in the deafened cat cochlea (F-K). (A) Tissue reaction to a 200 µm diameter microelectrode tethered to the skull. Sections were immunohistochemically labelled for GFAP (red), macrophages (ED1; green) and DAPI (blue). (B). Tissue reaction to an un-tethered implant of the same design as (A) showing a reduction in GFAP and ED1 reaction products. (C) Quantified GFAP density surrounding (0–200 µm) the implants with respect to the fixation mode (mean and standard error of the mean). ***p,0.001. (D). Quantified ED1 density surrounding (0–200 µm) the implants with respect to fixation mode (mean and standard error of the mean). *p,0.05. (E). Number of neurons surrounding (0–200 µm) the implants with respect to fixation mode (mean and standard error of the mean). (F) Stimulated and (G) the contralateral control cochlea following 28 weeks of implantation. This image illustrates the upper basal (UB) cochlear region that was subject to the most extensive electrical stimulation. Tissue response is evident in the basal turn scala tympani (ST) of both cochleae ('e' illustrates the location of each electrode array). (H) and (I) the extensive loss of auditory neurons observed in both cochleae is a feature of the deafening technique and the duration of deafness associated with the study. (J) There was no difference in the auditory neuron population between stimulated control cochleae throughout all regions of the cochlea. (K) Small regions of fibrous tissue (expressed as a percentage of the ST) were evident in the lower basal (LB), UB and lower middle (LM) cochlear turns. There was a small but statistically significant increase in tissue response localised to the UB turn of the stimulated cochleae. *p,0.05. Scale bars: A and B 100 µm; G 200 µm and I 100 µm. A-E reproduced from [231] CC BY 3.0; F-K Reproduced from [185]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageIt is also advisable to seek an experienced clinical pathologist to review the histology using appropriate blind techniques [38, 80, 90]. In addition to providing an important independent assessment, such a review can provide a useful clinical perspective on the degree of inflammation. A pathologist can also provide recommendations for additional staining options should the tissue response contain any unidentified inflammatory cells.
Correlating these histological changes with longitudinal electrophysiological and electrode impedance data recorded from the same animal provides a comprehensive data set for the preclinical evaluation of a neural stimulator.
5.5. Other considerations
5.5.1. Heating effects associated with active implants.
Heating effects that may result in tissue damage is a safety consideration for any active implantable device. Heating of tissues can occur as a result of power dissipation at the electrode array associated with the electrical stimulus [25, 232]; the neural stimulating circuitry [113, 233, 234]; and the telemetry link [113, 235]. While research has been undertaken using modelling, acute animal studies and cadaver tissue, this work is most accurately performed in chronic studies using large animal where factors that influence power dissipation including the size of organs, blood perfusion and tissue encapsulation of the implant, most accurately model the clinical situation.
Heating of tissue from electrical stimulation is rarely considered a problem for all but the highest powered devices [25]. In contrast, heat generation via neural stimulating circuitry becomes an issue for devices where the stimulator is placed close to the target neural population such as retinal prostheses [232–235] and fully implanted cortical devices where the stimulator/telemetry circuitry is mounted into the electrode array [113]. While device standards require the surface temperature of the implanted electronics to be <2 °C above body temperature for the general tissue environment [124, 125], higher tolerances are adopted when the implant is located close to neural tissue [113]. Measuring the extent of thermal change in animal studies is a non-trivial task requiring hardware specifically designed for the research [233, 235].
Issues of heat generation and other safety concerns associated with the telemetry link in wireless devices, comes under the highly regulated guidelines for non-ionizing radiation developed by ICNIRP [111–113]. With a long, successful history of devices operating within these guidelines there is little call for undertaking preclinical safety studies to assess this aspect of wireless devices.
5.5.2. Issues associated with paediatric devices and growth.
Cables under tension are significantly more susceptible to fatigue based damage [121] and show an increased risk of migration of the electrode array. This is a particularly significant issue in the development of devices designed for use within the paediatric population with a cable system designed to accommodate growth.
It is important to clearly understand the growth that occurs between an electrode array and its implanted stimulator/pulse generator from birth to adulthood. For example, while the cochlea is fully grown at birth, there is a 25 mm growth in the human temporal bone between the cochlea to the site of the stimulator from birth to adulthood [95]. A cable for a paediatric cochlear implant must accommodate for this growth.
Chronic passive preclinical studies designed to examine the effects of growth on both the cable and the adjacent tissue have been performed for both spinal cord stimulators [98] and cochlear implants [96, 97]. This work also developed techniques designed to protect excess cable from tissue growth [94, 219, 246].
5.5.3. Ethical considerations.
The use of animals in research is governed by specific ethical and regulatory guidelines to ensure that the specific research is necessary and is performed under conditions that both minimise the number of animals used while ensuring statistical significance, and the experiments are designed to minimise distress to the animals. It is important that these experiments are designed and conducted in collaboration with experienced veterinary input. Institutional animal ethics committee approval is required before commencing any studies involving animals, and the committee, the animal facility and the researchers must operate under clear regulatory guidelines managed under local state jurisdictions. Under some circumstances approval to undertake animal studies is also required from the funding agency (e.g. U.S. Department of Defence).
6. Cadaver and ex vivo studies
Once proof of principle has been established in preclinical in vivo models, development of the implant for clinical application requires engineers and clinical partners to work closely to marry the priorities of implant design with those of best surgical practice. Depending on the preclinical model used to show validity for the device, many of the design criteria established in the preclinical phase of the project will need to be assessed and validated for clinical application (figure 10). Ideally aspects of design such as surgical technique, electrode array design and the routing and fixation of the cable can be adopted from the preclinical research described above. Identification of the variances between the preclinical model and the human application will be critical to refining and optimising surgical approaches, developing any specialist surgical tooling and evaluating the extent of mechanical robustness testing the human implant requires. It is particularly important for both surgeons and engineers to see first-hand the biomechanical implications of the position of an implant, the texture and 'feel' of the tissue, the tissue planes where cabling will run, how much surgical exposure of the implant site is required, and what surgical instruments are required.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Typical development activities for human cadaver studies prior to FIH clinical trial.
Download figure:
Standard image High-resolution image{kind=link}
At the most basic level, the overall size and shape of an implant can be defined and refined through measurements and trials with explanted cadaveric target organs. The surgical access can be identified and evaluated for feasibility. At this point, the engineering and clinical team members can identify anatomical fixation sites for the electrode array and cable to ensure mechanical stability. It is important that the implant does not exert excessive static forces on the surrounding tissue, as this will lead to tissue erosion and potentially result in the extrusion of the device. Likewise, the implant needs to be protected from external trauma and designed to absorb dynamic forces associated with movement of the surgical site in which it is located. Cable migration is a common adverse event in clinical settings, particularly associated with highly mobile regions of the body [247]. Techniques to stabilise the implant may include the addition of suture points, hardware such as screws, clips or patches, or the excavation of underlying bone to create a recess and/or a site of fixation for the implant. Knowledge of tissue planes aid in the determination of the implant site and can be used to secure and protect the implant. Cable routing should also be optimised to minimise wire flexion and potential damage to organs along its route. Appropriate preclinical models can then be identified that best approximate the human anatomy and physiology for in vivo testing.
Note that early human cadaver work does not require full body specimens. In the initial stages of feasibility analysis and clinically-oriented design, an ex vivo cadaveric organ may be all that is required. This tissue can be an excellent medium for trialling prototype designs with the surgical team for fast turnaround go-no-go decisions.
Once initial design decisions have been made preclinical safety and efficacy studies using animal models are initiated as described above. These studies are used to build the body of evidence needed to show that the concept is sound, safe and efficacious. The design of the implant is typically refined during preclinical studies where physiological and histological feedback can be gathered to support the suitability of the device (figure 1). The advent of rapid prototyping technologies such as 3D printing has dramatically reduced the iteration cycle time for implant design and development. Additionally, some features will need alteration to fit with specifics of the target subject's anatomy (e.g. [248]). Obviously, the design decisions made during the preclinical experiments need to be undertaken with the proviso that they are subsequently translated back to a clinical device. In some instances, investigators may be presented with the opportunity to acutely assess implant designs in altruistic patients willing to volunteer or donate organs or tissue either during [249], or immediately after, resection surgery. In these cases, the preclinical data can be validated against living or freshly excised human tissue. This has the ultimate benefit of trialling human grade prototypes to assess the feasibility and extent of trauma associated with the insertion of the electrode array or to assess electrophysiological parameters to refine preclinical models, and to provide the surgical team with feedback on the feasibility of the surgical approach.
6.1. Develop common language
Communication between surgeons and engineers is essential at the early stages of a project where members of a team coming from a diverse range of backgrounds may have unique terminology and perspectives on the process ahead. In highlighting differing perspectives, priorities and scope of work for team members can be clearly defined within the team [250]. Efficiency of device implantation, adherence to safe surgical practices and long-term safety for the patient are the main priorities for the surgical team. Safety, efficiency, efficacy and long-term reliability of the device are typically the main priorities for the engineering team. Patient health, device efficacy and quality of trial data being collected are typical priorities of the clinical team. While the priorities of project team members have considerable crossover, ensuring articulation and acknowledgement of these priorities at the appropriate time can be a challenge. Once common language is established with a development team, design criteria for a human system can begin. Setting the design criteria with the endpoints in mind allows for the implant development process to be mapped out.
6.2. Development and optimisation of surgical procedures and tools
The development of novel surgical procedures is often required when interfacing new technologies with the human body. These procedures are developed to minimise surgical trauma to the recipient, while safely locating the implant securely in the surgical site [144, 148, 251–256]. Furthermore, the cable trajectories and connectors may be located distal to the primary implant location. Therefore, customised approaches and new surgical tools may be required to facilitate the surgery. These include: trocars to guide the implant and cabling safely through tissue to its final position; pneumatic insertion tools to overcome surface tension in implanting electrode arrays into neural tissue; stereotactic frames to accurately guide electrodes to specific neural targets; and imaging techniques to assist or guide electrode insertion [47, 148, 254, 257]. The development of new surgical tools is a process that occurs in tandem with the development and optimisation of the surgical approach.
6.3. Preparation for clinical trial
To capstone the surgical development and preclinical programs of novel devices, human cadaver studies are revisited in order to confirm and practice the surgical procedures in preparation for a clinical trial. Investigators may choose to conduct these end-stage studies in whole cadaver specimens in order to more accurately mimic conditions in theatre. Viewed as a mock run through of the implantation procedure, surgeons have the opportunity to trial their new approaches and tools, engineers can perform various system checks of the implants while clinical support staff have the opportunity to ensure the workflow meets clinical standards and takes inventory of tools and consumables required for surgery. The whole procedure can be photographed and documented in order to prepare surgical operating procedures and risk assessment documentation.
6.4. Other considerations
6.4.1. Ethics and OH&S considerations.
The use of human tissue is governed by specific ethical and safety guidelines. These are in place for the wellbeing of the investigators, as well as to protect the dignity and family of the person from where the ex vivo or cadaveric specimen originated. Institutional ethics committee approval is required before commencing any studies involving human tissue. Ethically, researchers have a responsibility to protect the identity of the donor. From a safety perspective, all instruments that are used with human tissue should be segregated from standard laboratory stock equipment and cleaned and sterilised according to standard operating procedures for dealing with human tissue. The overarching rationale for performing cadaver or ex vivo studies is to generate the confidence and data to show that design principles developed in preclinical models are translatable to clinical application. This data forms part of the body of work required for clinical trial approval and contributes to subsequent commercial and regulatory pathways.
6.4.2. Alternatives to cadaver and ex vivo studies.
The use of ex vivo tissue and cadavers particularly applies to the development of a new medical device where there is no substantially equivalent predicate. The pros and cons of undertaking cadaver and ex vivo studies are summarised in table 4. In some instances, modelling and computer-aided design may provide valuable tools to develop the form and fit of a device [258]. Modern rapid prototyping techniques, such as 3D printing have led to the development of lifelike anatomical models which can be used in place of ex vivo or cadaver samples, while in silico based simulations may help accelerate the clinical trial process for medical devices in the future [54].
Table 4. Pros and Cons of undertaking cadaver and ex vivo studies.
| Pros | Cons |
|---|---|
| Provides accurate data on the dimensions of the target structure in humans required to scale the electrode array | Not a perfect model of a patient in vivo |
| – No bleeding | |
| – No other natural fluids | |
| – Tissue can be partially degraded/rigor mortis | |
| – No movement; cannot assess mechanical strain | |
| Provides first-hand experience of surgical anatomy | Safety assessments are limited |
| – No physiology/no functional outputs | |
| Validates and improves preclinical models - which may result in lower animal usage | Expense |
| Useful for defining and practicing new surgical procedures | Availability |
| Ethical constraints (e.g. privacy) | |
| OH&S constraints (e.g. safety) | |
7. Evaluation of fully assembled neural stimulators for clinical application
The preclinical studies described above form an important component of the medical device development pathway (figure 1) and are typically undertaken by an interdisciplinary research team working within an academic research institute. When the device design is locked-in for clinical trial and is manufactured within a suitable facility, the fully assembled device must undergo additional testing to ensure it is safe for human implantation. The majority of the testing followings specific standards and will be only briefly reviewed here.
The device design must be reviewed to ensure it is manufactured from known biocompatible materials suitable for long term implantation in compliance with ISO 10993 Series—Biological evaluation of medical devices [52] and ISO 14708 Active Implantable medical Devices [124], covering the evaluation of a device for mechanical and electrical integrity. The sterility of the device must also be established, typically via a chemical method such as Ethylene Oxide sterilization due to the embedded electronics and batteries in neural stimulators (e.g. ISO 11135 [259]). This routinely includes such testing as bacterial endotoxin, bioburden, ethylene oxide/ethylene chlorohydrin residuals as well process challenge devices in fractional, half and full sterilisation cycles to establish the required sterility assurance level (SAL) for the device. ISO 11135 (Annex E) also specifies the requirements for release of devices from a sterilization process where there is only sufficient product to comprise a single sterilization load, such as devices for a clinical trial, avoiding the need to perform a full process validation for the device.
The suitability of the device's sterile packaging must be assessed to ensure the packaging can be sealed yet allow the contents to be sterilized, as well as maintain the device in a sterile state during shipping, handling and storage (ISO 11607-1 [260], ISTA 2A [261]). Typically this includes the use of a double sterile barrier packaging system. Environmental testing of the device, to simulate its use in vivo as well as conditions experienced during shipping, handling and storage, are required to ensure it will maintain functionality for the intended design life (e.g. IEC 60068-2-1—Low Temperature; IEC 60068-2-2—Dry Heat, IEC 60068-2-14—Thermal Cycling).
More extensive testing is typically required for commercial devices undergoing regulatory approval for commercial sale, outside the closely controlled and monitored conditions of a clinical trial. Much of this testing is performed by the manufacturer, however contract research organisations are often contracted to perform specific testing.
There are few comprehensive standards for specific neural stimulators, however a detailed review of the requirements for safety, functional verification, labelling and reliability reporting for cochlear implants is provided in the ANSI/AAMI Standard CI86:2017 [110]. Briefly, this standard outlines testing for direct current leakage; charge density limits for cochlear implants; thermal levels limit on the surface of the implanted device; risk analysis of potential fault conditions (ISO 14971); hermeticity testing of the assembled device (EN 13185 and EN 1593); MRI compatibility (ISO/TS 10974); testing for immunity to diathermy currents and external defribulators; immunity to stresses of mechanical forces including vibrational and impact stress (e.g. IEC 60068-2-75—Hammer tests); immunity of implant cables to impact, vibration tensile forces and flexural stresses; and performance of implantable connectors. Finally, it is important to note that any upgrade in the implant design requires a review of the previously conducted testing to ascertain if the results may be impacted by the change, with the testing repeated in those instances to ensure ongoing compliance.
8. Conclusions
We are witnessing a rapid expansion in the development of neural stimulators with many devices within the commercialization pipeline. Developing implantable devices for clinical application is a long and expensive process. Because of their complexity they are classified by regulatory bodies into the highest risk category for implantable devices (Class III) and are therefore required to complete a very rigorous regulatory approval process before clinical use. Preclinical studies form an important component of this approval process. The present review describes the design principles associated with a safe neural stimulator and reviews the in vitro, in vivo and human cadaver/ex vivo studies that are available to researchers when considering a suite of preclinical studies to demonstrate device safety.
The major points from this review include the following:
- Preclinical studies form one of several key components in the development pipeline of a neural stimulator from concept to the release of a commercial device.
- This pipeline is long and expensive; careful attention to both device design and its preclinical evaluation will have significant impact on the duration and cost associated with commercialising a new device.
- The successful development of these devices is achieved through close collaboration between many disciplines including engineers, scientists and the surgical/clinical team. It is also important to engage potential end users of the technology.
- The design of a neural stimulator is an iterative process with feedback opportunities impacting on the design occurring at many stages of the preclinical evaluation.
- Preclinical evaluation of a neural stimulator is not typically undertaken on a fully manufactured device; rather prototype components that have a similar form and are manufactured from the same materials as the proposed final product are sufficient. A final reliability and safety study is often performed using a fully assembled device manufactured and sterilized using industry standards.
- Key components of any preclinical evaluation include: in vitro studies that are focussed on materials suitability and device reliability, and include accelerated testing under highly controlled environments; in vivo studies using appropriate animal models of the disease or injury in order to assess safety and, if feasible, the efficacy of the technology; and human cadaver studies designed to ensure the device's form factor conforms to human anatomy, optimize the surgical approach and to develop any specialist surgical tooling.
- There is no specific roadmap outlining the sequence of preclinical studies that must be performed in order to obtain regulatory approval. The studies required will vary from device to device and will be influenced by factors including the novelty of the application and the materials used; the neural target; the surgical approach; and the proposed stimulator.
- Provided the work is relevant to the device under investigation, it is appropriate to cite earlier preclinical studies demonstrating aspects of device safety in order to reduce the burden of the current evaluation.
- Regulatory bodies encourage early dialogue with the development team before initiating preclinical studies. They provide advice and often develop specific guidelines to assist the design of a preclinical study program.
- Many preclinical tests are carefully defined in standards documents. These should be consulted when developing a program of preclinical studies.
- In vitro studies provide relatively rapid feedback on the performance of electrodes, cables and insulation under both passive conditions and in response to repeated electrical stimulation; mechanical failure modes associated with movement; the evaluation of material adhesion and delamination of the electrical insulation; the efficacy of hermetic sealing techniques; the assessment of insertion forces associated with electrode implantation to optimise electrode design; and the in vitro assessment of materials for biological cell–electrode interactions, glial scarring and cytotoxicity.
- In vitro studies under conditions of accelerated aging, including elevated temperatures, harsh electrolyte environments and accelerated mechanical testing, allows the screening of materials prior to in vivo studies and provides estimates of a device's lifetime.
- While small animal models play an important role in in vivo efficacy studies they are not considered suitable for the long-term evaluation of device safety and/or optimizing the surgical approach. Appropriate large animal models more accurately reflect the anatomy, surgical access and the mechanical environment that a neural stimulator would experience clinically. In addition, the use of large animals allows investigators to evaluate electrode arrays and cables that are similar in size to the proposed clinical device.
- A key component of any preclinical chronic active study is the use of the electrode array implanted in the intended anatomical site; and the stimulator, its charge recovery mechanism and the proposed 'worst case' stimulation parameters designed for the specific clinical application. Ideally, these studies would include longitudinal monitoring of electrode impedance and neural function to demonstrate that the electrode array/cable assembly is viable, the stimulator is operating within its voltage compliance and the stimulus levels employed are evoking appropriate neural activity.
- More extensive preclinical testing is typically required for commercial devices undergoing regulatory approval for commercial sale, outside the closely controlled and monitored conditions of a clinical trial. Much of this testing is performed by the manufacturer; however contract research organisations are often employed to perform this specific testing.
Preclinical data associated with neural stimulators is notoriously fragmented, incomplete and highly variable in terms of implant duration, the site of stimulation and stimulation parameters used [12], reflecting the large selection of maladies and neural pathways that have been targeted for neural stimulation. This review will hopefully provide some clarity for investigators new to this space.
Acknowledgments
This work was supported by the following funding agencies for which we are most grateful: NIDCD (R01DC015031), NHMRC (APP1122055, APP1113680 and APP1120664), the Defense Advanced Research Projects Agency (DARPA) BTO under the auspices of Dr Doug Weber and Dr Eric Van Gieson through the Space and Naval Warfare Systems Center (N66001-15-2-4060), and the Garnett Passe and Rodney Williams Memorial Foundation. The Bionics Institute acknowledges the support of the Victorian Government through its Operational Infrastructure Support Program. We thank Jason Leavens, Dr Peter Seligman, Dr Tyler Best, Dr Stuart Cogan and Dr Kip Ludwig for helpful comments on an earlier draft, Mario Huynh and Ceara McGowan for research assistance and Dr Rylie Green, Dr Ulises Aregueta Robels and Dr Laura Poole-Warren for manufacturing the conductive hydrogel electrode used in figure 6.
Conflicts of interest
The authors declare no competing financial interests.