
Abstract
Polyaromatic carbon is widely held to be strongly diamagnetic and hydrophobic, with textbook van der Waals and 'π-stacked' binding of hydrocarbons, which disrupt their self-assembled supramolecular structures. The NMR of organic molecules sequestered by polyaromatic carbon is expected to be dominated by shielding from the orbital diamagnetism of π electrons. We report the first evidence of very different polar and magnetic behavior in water, wherein graphene remained well-dispersed after extensive dialysis and behaved as a 1H-NMR-silent ghost. Magnetic effects dominated the NMR of organic structures which interacted with graphene, with changes in spin–spin coupling, vast increase in relaxation, line broadening and decrease in NMR peak heights when bound to graphene. However, the interactions were weak, reversible and did not disrupt organic self-assemblies reliant on hydrophobic 'π-stacking', even when substantially sequestered on the surface of graphene by the high surface area available. Interacting assemblies of aromatic molecules retained their strongly-shielded NMR signals and remained within self-assembled structures, with slower rates of diffusion from association with graphene, but with no further shielding from graphene. Binding to graphene was selective for positively-charged organic assemblies, weaker for non-aromatic and negligible for strongly-negatively-charged molecules, presumably repelled by a negative zeta potential of graphene in water. Stronger binders, or considerable excess of weaker binders readily reversed physisorption, with no evidence of structural changes from chemisorption. The fundamental nature of these different electronic interactions between organic and polyaromatic carbon is considered with relevance to electronics, charge storage, sensor, medical, pharmaceutical and environmental research.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The non-covalent interactions between polyaromatic carbon and the hydrocarbon-based structure of organic molecules are both of fundamental interest and a promising means of modifying graphene-based electronics [1–7], spintronics [8–10], optoelectronics and sensors [11–14]. The high relative surface area of graphene is attractive for the sequestration of xenobiotics in environmental [15–17] and drugs in pharmaceutical [18–21] applications. Organic molecular interactions with graphene are visible in microelectronic devices [22] and in Raman studies in the dried state [23–25]. However, the structural and electronic nature of the interactions of organic compounds with graphene is unclear and misunderstood in polar solvents such as water.
Computational studies often model the polyaromatic carbon of graphene as a finite polyaromatic hydrocarbon (e.g. coronene), when saturation of its dangling bonds with hydrogen atoms introduces a significant quadrupole potential, which tends to zero in flat graphene [26]. Such models contribute a widely-held view that graphene is hydrophobic, like coronenes, where van der Waals dispersive forces together with 'π–π stacking' are responsible for interactions with organic molecules [26, 27]. This may be the case when polar solvents are not used or removed prior to analysis in the dried or crystallized state [28–30]. Comparison of the differences in behavior in polar solvents is less considered. Notably, the 'π–π interactions' of hydrophobic aromatic amino acids with graphene can also be modeled as strongest in the absence of water. However, when water was included in simulations, polar and positively-charged amino acids (e.g. arginine) interacted more strongly with graphene than hydrophobic amino acids [31]. Computational studies of graphene interactions have mainly considered monomeric molecules [26–31], whose self-assembly is also often avoided or overlooked [32]. As self-assembly of organic molecules into supra-molecular structures include weak hydrophobic interactions, which may be overcome by polyaromatic carbon in the absence of water, interaction with graphene has been assumed to disrupt natural self-assemblies of organic molecules, including lipid bilayers, proteins and DNA [33–35].
Nuclear magnetic resonance (NMR) spectroscopy is established as the tool to understand organic structures at the atomic level of their electronic interactions [36]. Even though the structural nature of electronic interactions of polyaromatic carbon and hydrocarbons are of fundamental interest [37–39], and lie at the heart of a huge range of research and potential applications [18], NMR has been little considered nor understood.
The electronic structure of monolayer graphene is dominated by its conduction and valence bands touching at the Dirac point with zero band gap and linear energy dipersion [37]. This singularity in energy dispersion lies at the origin of its diamagnetic susceptibility [37–39]. Although few-layer graphene (FLG) is more complicated, the zero band gap is retained [39]. Consequently, FLG also exhibits strong orbital diamagnetism which, unlike the case of a metal, overcomes its paramagnetic spin–spin coupling [38, 39], and has been predicted to circulate at the edges of larger FLG flakes at room temperature considered here [40].
NMR models of the nuclear shielding, spin–spin coupling and the T1 spin-lattice relaxation times of graphene are important in quantum computing and spintronics [41, 42] and have progressed from coronene hydrocarbon [43] to bilayer graphene [44]. However, the direct 1H-NMR observation of unmodified pristine graphene is infeasible due to the absence of the hydrogen atoms in its structure. Experimental 13C-NMR study of natural graphene dispersed in solvents is challenging because of the low level (~1%) of magnetically-active 13C nuclei with net spin of 1/2 (~1 per (16 Å)2 area graphene layer) in natural graphite and their low gyromagnetic ratio (6.7283 × 107 rad s−1 T−1). Solid-state NMR [45–47] and/or highly 13C-enriched samples are required [41, 42]. Solid-state NMR revealed a metallic-like anisotropy of the inner nanotubes of double-walled 13C-enriched carbon nanotubes (CNTs), where the Knight shift from the spins of conduction electrons contributed hyperfine coupling [48, 49]. A zero-field NMR signal has been characterized in 13C-enriched 'ferromagnetic graphene' as providing strong direct evidence of the hyperfine magnetic field created around defects in graphene, due to the coupling between nuclear and ordered electron spins [50].
In the absence of chemical modification, natural pristine graphene dispersions may be silent in 1H-NMR, when their ghost-like effects studied here may arise in the NMR spectra of organic compounds interacting with graphene from: (a) nuclear-independent shielding from the high orbital diamagnetism of graphene [37–40], larger than [46, 47] the shielding effects of aromatic hydrocarbon ring currents; and (b) any significant intrinsic magnetism [38, 39] accelerating nuclear spin relaxation [36, 46, 47] and signal loss, resulting in a fall in peak height. Doping of graphene by organic compounds [1–7, 37] may also cause chemical shift changes in their NMR spectra.
Solid-state NMR signals of molecules trapped between graphene layers and in porous activated carbons in supercapacitors are shifted upfield to lower frequencies by the nuclear-independent shielding from orbital diamagnetism [46, 47, 51–54], which may dominate and mask any other interactions present. Similarly, the ring currents of CNTs are also predicted to be diamagnetic and greater at their outer surfaces, possibly due to the localized current at each wall opposing the inner delocalized current [55]. However, although ring currents are expected to shield the NMR signals of organic compounds adsorbed to CNTs [56], the expected upfield chemical shift changes appear to be rather small [57] or even downfield [58].
Herein, when the above shielding effects were at a minimum, graphene behaved as a 1H-NMR-silent ghost, which was only visible to proton NMR through its magnetic effects on nearby protons in interacting molecular structures. In this contribution, we report the magnetic-like effects of molecular engagement with graphene in water, which allow the structural nature of interactions with different aromatic and aliphatic hydrocarbons to be characterized for the first time. Even though high concentrations of organic compounds were fully sequestered by graphene, we find the interactions involved in water to be weak, reversible and benign, with no evidence of disruption of organic self-assembly, assumed for graphene from computational and surface science studies [34, 35].
2. Methods
Details of all materials, equipment and methods are described in 'Experimental details' in the first section of supplementary material (stacks.iop.org/TDM/5/015003/mmedia).
2.1. Materials
Organic compounds were selected to include the non-aromatic organic solvents DMF (dimethylformamide, Sigma-Aldrich) and NMP (N-methyl-2-pyrrolidone, Sigma-Aldrich) commonly used to exfoliate graphene; and polyaromatic and heteronuclear dyes and drugs: chlorpromazine HCl (CPZ, Fluka), 3,6-diaminoacridine (DAA, Sigma-Aldrich), Pyronin Y (PyY, Fluka), pyrenemethylamine HCl (PN1, Sigma-Aldrich), 1-pyrene sulfonic acid sodium salt (PS1, Sigma-Aldrich), 6,8-dihydroxy-pyrene 1,3-disulfonic acid di-sodium salt (PS2, Sigma-Aldrich), 8-hydroxy-1,3,6-pyrene tri-sulfonic acid tri-sodium salt (PS3, Sigma-Aldrich), (1,3,8,6-pyrene tetrasulfonic acid tetra-sodium salt (PS4, Sigma-Aldrich).
The graphene dispersion was the same batch previously characterized in organic solvents (NMP) and in water (D2O) [33]. The graphene dispersion was exfoliated from graphite in NMP and dialyzed against 3 successive 10-fold volumes of H2O, followed by additional dialysis against 6 successive 3-fold volumes of D2O to reduce the undesirable H2O NMR signal and any residual contamination to negligible levels. Graphene concentrations were estimated by optical absorption at 660 nm (4632 ml mg−1 m−1 absorption coefficient, estimated from >5 different preparations using gravimetric analysis) [33]. UV–visible spectra were recorded at 25 °C using a Varian Cary 4000 spectrophotometer with wavelength range between 200 and 800 nm. Unless specified otherwise, the concentration of graphene in the key NMR experiments was either 0 µg ml−1 (for (−G) samples) or 62.3 µg ml−1 (for (+G) samples), whereas the concentration of the studied compounds was 0.5 mM.
2.2. NMR
NMR spectroscopy was used to characterize the interactions between graphene dispersed in D2O and the selected organic compounds, and to measure the changes in their diffusion coefficients and T1 spin-lattice relaxation times. 1H-NMR spectra were recorded using a Bruker AVANCE II + 400 Ultra Shield NMR spectrometer (with a field of 9.4 Tesla) operating at proton frequencies of 400 MHz using a 5 mm BBI 1H/D-BB Z-GRD Z8202/0347 probe.
3. Results and discussion
3.1. Few-layer graphene
Graphene was exfoliated from graphite into organic solvent (NMP), which was later removed by extensive dialysis against H2O and then exchanged by further dialysis with D2O to reduce H2O to trace levels for 1H-NMR studies. This exhaustive dialysis diluted neat exfoliating solvent to negligible levels (<0.0002%), with no visible changes in the dispersion (figure S1), which was stable for many months in D2O [33]. Trimethyl phosphate (TMP; 0.1 mM) was used as an internal NMR standard to estimate the residual NMP signal from the –N(CH3) group after dialysis. Only residual NMP (0.017 mM) and water, together with the internal standard were visible in the 1H NMR spectrum of graphene dispersions used in this work (figure 1(d)).
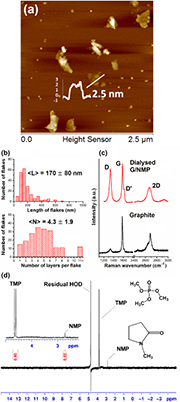
Figure 1. Characterization of graphene exfoliated in NMP and dialyzed into water (Dialyzed G/NMP). (a) AFM height scans and (b) histograms of the flake length distribution and number of layers per flake. (c) Raman bands compared to the starting graphite. (d) 1H-NMR spectrum (400 MHz; Bruker AVANCE II+) of NMP-exfoliated graphene dialyzed into D2O. Water suppression was achieved using pre-saturation pulse program zgcppr. The concentration of graphene in the NMR experiment was 62.3 µg ml−1, and the concentration of TMP (an internal NMR standard) was 0.1 mM.
Download figure:
Standard image High-resolution imageThe graphene was the same batch as previously characterized as few layer (mean 4.3 ± 1.9 layers) with a 170 ± 80 nm mean flake length (figure 1(b)) and about 200 nm hydrodynamic diameter with negative zeta potential in polar solvents (NMP, DMF) and aqueous media [33]. The mean flake length  and mean number of layers per flake
and mean number of layers per flake  for the dialyzed dispersions (figure 1(b)) were similar to the starting undialyzed dispersion in NMP [33].
for the dialyzed dispersions (figure 1(b)) were similar to the starting undialyzed dispersion in NMP [33].
The proportion of few layer graphene was  , with monolayer content of
, with monolayer content of  . The dialyzed graphene showed typical Raman bands: a sharp G band at ~1580 cm−1, a pronounced D band at ~1330 cm−1, a broad and symmetric 2D band at ~2660 cm−1 and shoulder peak D' band at 1617 cm−1. The shape of the 2D band and peak ratios (
. The dialyzed graphene showed typical Raman bands: a sharp G band at ~1580 cm−1, a pronounced D band at ~1330 cm−1, a broad and symmetric 2D band at ~2660 cm−1 and shoulder peak D' band at 1617 cm−1. The shape of the 2D band and peak ratios ( ) of the dialyzed dispersions also suggest graphene with few layers (<5). The
) of the dialyzed dispersions also suggest graphene with few layers (<5). The  suggests edge or boundary-type defects [25].
suggests edge or boundary-type defects [25].
As expected, the 1H-NMR spectrum of the dialyzed graphene was flat and featureless (figure 1(d)), with no detectable protons from any possible chemical modification of dangling bonds [46, 50]. Neither the residual H2O signal nor the TMP doublet signal showed any signal broadening (figure 1(d)), which would be usually indicative of magnetic contamination, such as from electrolytes in graphite [51], metals in CNTs [59] or ferromagnetic organic interactions [46, 9, 10, 60].
3.2. Molecular self-assembly
In order to explore the electronic nature of molecular interactions with graphene, we selected 10 structurally-diverse organic compounds, carrying various functional groups and/or net charges (see figures 2 and 3 for structures). None of the compounds were paramagnetic organic radicals [60, 61]. To investigate the possibility of the studied compounds to form aggregates in aqueous solutions, we compared NMR spectra of these compounds (see figure S2) recorded at relatively low concentration (0.5 mM) with those acquired at 20-fold increased concentration (10 mM). Most of the dye and drug compounds studied here, except for the highly-sulfonated PS3 and PS4, stacked into multimeric assemblies at the concentrations considered here (0.1–10 mM) and useful in their applications. Upfield shifts in 1H-NMR signals (figure S2) and changes in fluorescence spectra (figure S3), induced by increased concentration of these compounds, reported the structural nature of their self-assembly. Dimerization and multimerization by 'π–π interactions' are often assumed to be face-centered 'π stacking', but which is only favored in special cases between electron-deficient and electron-rich rings, due to the presence of charge-donating and charge-withdrawing groups (respectively) [61, 62]. Otherwise, off-centered parallel (in line) or edge-to-face oblique to perpendicular stacking is favored by the quadrupole moment of the π electron density on most aromatic hydrocarbon rings, due to repulsion between the negative charge above and below the ring and the positive charge around its periphery [62, 63].
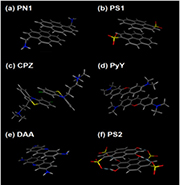
Figure 2. NMR-informed models of dimerization. Dimers of the organic compounds showing self-assembly by NMR were modeled by DFT methodology informed by the interactions identified by NMR (see supporting material for details).
Download figure:
Standard image High-resolution image
Figure 3. Effect of graphene on 1H-NMR spectra. Chemical shifts of 1H protons of aromatic compounds (0.5 mM) in the absence (red) and presence (black) of graphene (G, 62.3 µg ml−1). Spectra were referenced against TMP (d; 3,82 ppm).
Download figure:
Standard image High-resolution imageThe diatropic ring currents of the cyclically-delocalized (4n + 2) π electrons of aromatic rings induce a magnetic field, which shields protons assembling within the influence of aromatic rings or deshields those falling outside; whereas, the opposite arises for the paratropic ring current of the 4n π electron system of anti-aromatic rings [64, 65]. Large upfield shifts of the 1H NMR signals (up to 0.758 ppm in extreme cases) from the shielding effects of aromatic ring currents on self-assembly were clear here (figure S2 and table S1) for those of the aromatic compounds (PN1, PS1, CPZ, DAA, PyY and PS2), which were also shown to associate with graphene (see section 3.3 below). From our detailed 2D NMR assignments (COSY, DQF COSY and NOESY [36]; figure S4, table S1), and from analysis of the relative changes in chemical shifts arising from increased concentration, in-line and intermediate stacking arrangements were involved in the self-assembly of these compounds. Stacking arrangements are illustrated by our NMR-informed 3D models of the self-assembly of PN1, PS1, CPZ, DAA, PyY and PS2 (figure 2).
PN1 and PS1 showed extensive interaction of their aromatic rings, such that the charged substituting groups (–CH2– and –
and – , respectively) seem to locate on opposite sides of the dimeric structure, to ensure the most distant orientation from each other. CPZ showed a 'vertical' stacking of the molecules in an offset manner: with the positively-charged alkyl side-chains located on the opposite sides of the stack and with the maximum overlap of the chlorinated rings; in agreement with earlier work, recognizing the V-shaped phenothiazine ring system of CPZ [66]. DAA and PyY seemed to form partly-overlapping aggregates, interacting mainly in the hydrophobic regions of their structures, free of polar heteroatoms (N and O, respectively) and substituting groups (–NH2 and –N–(CH3)2, respectively). PS2 formed partly-overlapped, anti-parallel, in-line aggregates with major involvement of the OH-bearing rings and exclusion of the SO3-bearing rings from the stack, stabilized by 4 hydrogen bonds between the oxygens of the SO3-groups and the hydrogens of the OH groups. PS3 and PS4 showed no tendency to dimerize, due to repulsion from negatively-charged sulfonate groups (figure S2, table S1).
, respectively) seem to locate on opposite sides of the dimeric structure, to ensure the most distant orientation from each other. CPZ showed a 'vertical' stacking of the molecules in an offset manner: with the positively-charged alkyl side-chains located on the opposite sides of the stack and with the maximum overlap of the chlorinated rings; in agreement with earlier work, recognizing the V-shaped phenothiazine ring system of CPZ [66]. DAA and PyY seemed to form partly-overlapping aggregates, interacting mainly in the hydrophobic regions of their structures, free of polar heteroatoms (N and O, respectively) and substituting groups (–NH2 and –N–(CH3)2, respectively). PS2 formed partly-overlapped, anti-parallel, in-line aggregates with major involvement of the OH-bearing rings and exclusion of the SO3-bearing rings from the stack, stabilized by 4 hydrogen bonds between the oxygens of the SO3-groups and the hydrogens of the OH groups. PS3 and PS4 showed no tendency to dimerize, due to repulsion from negatively-charged sulfonate groups (figure S2, table S1).
Fluorescence spectra were less informative, but similarly provided no indication of assembly into face-centered parallel stacks [65]. Parallel or H-type assembly results in oscillating dipole moments, which are in phase, higher energy and so result in blue-shifted excitation and poor fluorescence. The small blue shift of CPZ (figure S3) reflects a partial face-centered stacking of the overlapping chlorinated rings (figure 2). The reverse arises for in-line or J- type assembly, with out-of-phase dipole moments, with sharp red-shifted excitation, which excites fluorescence emission with a small Stokes shift. Intermediate or more oblique assembly results in both blue- and red-splitting of excitation [67, 68]. Both pyrenes forming excimers (PN1 and PS1) and the highly-sulfonated pyrenes (PS3 and PS4) showed blue and red shifts at the concentrations used here, as did others (DAA and PS2) indicating intermediate conformations (figure S3), but weak (e.g. collisional) interactions were not distinguished from self-assembly.
Self-assembly of PN1, PS1, CPZ, DAA, PyY and PS2 compounds in water was demonstrated here over a wide range of concentrations from 0.1 mM to 10 mM (e.g. PN1 in figure S5). Self-assembly was also evident from the distinctive decrease in diffusion coefficients of these compounds upon 10-fold increase of their concentrations from 1 mM to 10 mM (figure S6), as demonstrated by diffusion-ordered spectroscopy (DOSY) [36]. However, the negatively-charged, highly-sulfonated pyrenes showed little (PS3) or no (PS4) change in diffusion coefficients (viz. (g) and (h) in figure S6). The absence of ring current shielding for PS3 and PS4 over a wide range of concentrations (from 0.5 mM to 10 mM) was also indicative of a lack of self-assembly (viz. (g) and (h) figures S2 and S6).
3.3. Molecular interactions with graphene
Comparison of the 1H-NMR spectra of organic compounds exposed to a higher concentration of graphene with those of free organic assemblies identified structures, which were able to interact with graphene, and to rank them in terms of their affinity towards graphene (from (a) to (f) figure 3). Spectral changes associated with the concentration-dependent variations (see section 3.2) in the level of self-assembly were avoided by maintaining exactly the same concentration of organic compounds (0.5 mM), in order to observe the direct effect of graphene on 1H-NMR signals.
On exposure to graphene, 5 out of 8 studied aromatic compounds (i.e. PN1, PS1, CPZ, DAA and PyY) showed considerable 1H-NMR signal broadening, accompanied by decreases in the peak heights of 1H-NMR signals (figures 3(a)–(e)). For these 5 compounds, we also detected a considerable decrease in the overall peak area, measured as an area under the signal curve (the 'integral') against TMP, an internal NMR reference, which was present in each NMR sample at exactly the same concentration (0.05 mM). This decrease in the overall peak area reached ∼60% for PN1 and was in the range of 30%–40% for PS1, CPZ, DAA and PyY (figure 3). These interactions seem to be highly discriminative in terms of the chemical structure and declined in the order: PN1 > PS1 > CPZ > DAA ⩾ PyY, which strongly followed the degrees of change in spin-lattice relaxation times (T1) for these compounds on addition of graphene (table S2). For example, PN1 showed 7.7- fold decrease in T1, followed by CPZ and PS1 (6.0- and 1.7-fold, respectively, decrease in the T1 of aromatic protons). In the case of DAA and PyY, only a few protons showed a statistically-significant (p < 0.05) decrease in T1 in response to graphene, thus indicating less interaction with graphene.
A typical example of the gradual signal broadening accompanied by the decrease of the NMR peak heights in response to stepwise increase of graphene concentration is given for CPZ (figure 4), which showed a greater level of response from aromatic than aliphatic functionalities. Notably also, the signal of the internal reference (TMP; 3.2 ppm) present in each NMR sample remained unaffected by graphene (figure 4), thus confirming that the observed effects can be attributed to the selective interactions with graphene, rather than to some non-specific effects in bulk solution.
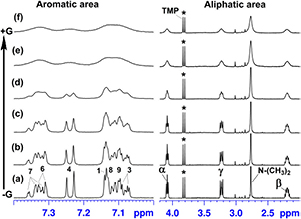
Figure 4. Gradual response of CPZ 1H-NMR signals to stepwise increase of graphene concentration. Graphene concentrations (µg ml−1) were 0 (a), 8.2 (b), 10.9 (c), 27.3 (d), 54.7 (e), and 62.3 µg ml−1 (f) in CPZ (0.5 mM). Signal assignments for CPZ protons are shown on (a). Asterisk indicates TMP (0.05 mM), internal NMR reference (d; 3.82 ppm).
Download figure:
Standard image High-resolution imageThese selective interactions of graphene with the polyaromatic compounds seem to be largely influenced by the effective charge of the substituting functional group(s) in their aromatic rings. Unlike the hydrophobic nature of polyaromatic hydrocarbon widely assumed for graphene, polyaromatic carbon appears to be mildly hydrophilic with a net negative zeta potential [33, 72–75]. Presumably, consequent of the repulsion between graphene and their negative charge, the highly-sulfonated pyrene derivatives (PS2, PS3 and especially PS4) showed from weak to negligible interaction with graphene (PS2 > PS3 > PS4), evident from the absence of signal broadening and negligible decrease in the 1H-NMR peak heights (figure 3).
Statistically-significant (p < 0.05) decrease in T1 was detected only for a few specific protons of these compounds, presumably indicating the localized nature of collisional contacts with graphene seen for PS2, PS3 and especially PS4 (table S2). In contrast, all aromatic compounds, which were fully or partly positively-charged at neutral pH, were identified by 1H-NMR as good binders to graphene (PN1 > CPZ > DAA ⩾ PyY; figure 3).
The large upfield shielding, which had been gained by the studied aromatic compounds through self-assembly (figure S2), remained unchanged upon addition of graphene, even at the higher levels of graphene, which caused considerable line broadening and major loss of the 1H-NMR peak heights (figures 3 and 4). Graphene thus interacted with intact multimeric assemblies, without changing their stacking and associated shielding, and did not add to the shielding. The stacking interactions between molecules within the assemblies appeared to be stronger than any similar interactions between the molecules in the assemblies with graphene, which would have otherwise disrupted the assemblies. Indeed, any change in the stacking of the organic assemblies or shift in the equilibrium and redistribution of different free and bound assemblies would have caused large spectral changes [32, 64–68]. Disruption of the assemblies on interaction with graphene would have resulted in large downfield chemical shift changes, with the lessened shielding from aromatic ring currents [64, 65]. No such changes were evident from the 1H-NMR spectra (figure 3), thus implying that graphene engaged mainly with molecular assemblies when present, rather than with individual molecules. Moreover, those polyaromatic compounds that did not form multimeric structures (e.g. PS3 and PS4) showed either very weak or negligible interaction with graphene.
In order to investigate the interaction of non-aromatic hydrocarbons with graphene, we selected NMP and DMF, which are normally used as solvents for graphene exfoliation and thus widely assumed to interact strongly with graphene. However, our study of the impact of graphene on the 1H-NMR spectra of NMP and DMF (figures 5(a) and (b), respectively) showed that these interactions are fairly weak and readily reversible in water.
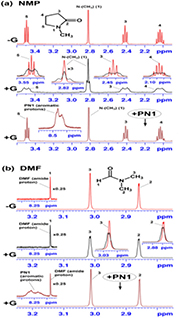
Figure 5. Changes in multiplet 1H-NMR signals of NMP (a) and DMF (b) (both present at 0.5 mM concentration) induced by interaction with graphene (G, 62.3 µg ml−1).
Download figure:
Standard image High-resolution imageIn the absence of graphene, the 1H-NMR spectrum of NMP (figure 5(a) top) is fairly straightforward to interpret, since the geminal protons at positions 1, 3, 4 and 5 display both chemical shift equivalence and magnetic equivalence due to a rapid interconversion of ring pucker in solution. Consequently, these protons are not normally involved in geminal proton-proton couplings and, as expected, appeared as well-resolved singlet (1), triplet (3), pentet (4) and triplet (5). On exposure to graphene, NMP showed extensive signal broadening of all aliphatic protons (figure 5(a) middle), which is symptomatic of interaction with graphene. However, this was accompanied by a very distinctive change in the signal splitting patterns seen for both the N-CH3 and methylene protons upon addition of graphene (figure 5(a) and table S1). It looks like the interactions with graphene led to a complete loss of the magnetic equivalence for the above geminal protons, presumably due to a restrained conformational freedom of the NMP ring and/or induced magnetic fields. This resulted in appearance of additional (i.e. geminal) proton–proton coupling (ranging from −12.4 to −14.7 Hz for methylene protons) leading to a higher level of signal multiplicity (table S1). Consequently, the signal from the N–CH3 group is now seen as a triplet (J = 5.0 Hz), whereas the methylene protons at positions 3, 4 and 5 are seen as broad, poorly-resolved multiplets.
Although DMF showed considerably weaker interactions with graphene (evident from very modest line broadening on addition of graphene), the overall trend seemed to be similar to that seen for the NMP-graphene interactions (figure 5(b)). Indeed, the originally sharp singlets from the two (non-equivalent) methyl groups (2) and (3) (figure 5(b), top spectrum) now show some evidence of additional geminal proton–proton coupling and indicate a tendency to form poorly-resolved broad triplets (figure 5(b) middle spectrum). In contrast, the signal from the amide proton (1), which is not involved in any spin-spin coupling, remained intact.
Interestingly, neither NMP nor DMF showed decrease in the overall peak area, and displayed only decreased 1H-NMR peak heights coupled with line broadening (figure 5), thus suggesting a different nature of their interactions with graphene, as compared to those of the aromatic hydrocarbons. More importantly here, both NMP and DMF showed complete and effortless signal recovery (in terms of both peak height and shape) after being displaced from graphene by addition of an equimolar concentration (0.5 mM) of aromatic compounds (e.g. PN1 in figures 5(a) and (b) bottom). Although the interaction of aromatic hydrocarbons with graphene seems to be also reversible (e.g. PN1 and CPZ in figure S7), their displacement from the graphene surface required a large excess of NMP (1000-fold and 2000-fold, respectively) to initiate the recovery process for the aromatic signals, previously lost on interaction with graphene.
Notably, the spin–spin coupling patterns of other aliphatic protons associated with aromatic compounds were not changed on graphene interaction (e.g. CPZ in figure 4(b)), although the usual signal broadening was also observed on addition of graphene. Presumably, the interactions involved in the self-assembly of these aromatic compounds dominated over those with graphene, which were also dominated by aromatic rather than aliphatic interactions with graphene (e.g. CPZ in figure 4). This seems to distance the aliphatic groups from the graphene surface, thus preventing these geminal protons from losing their magnetic equivalence. Moreover, the 2.3-fold increase in T1 of the CPZ aliphatic chain protons (table S2) suggested both a reduced conformational freedom and their distancing from magnetic interaction (which otherwise caused shortening of relaxation times of aromatic protons interacting with graphene).
The above observations provide qualitative indication of the different nature and affinity of chemically diverse structures towards graphene, which is further quantified in section 3.5.
3.4. Role of magnetism in interactions with graphene
NMR line broadening, which seemed to be the common indicator of interaction with graphene, can be the result of many contributing factors (e.g. induced conformational restriction, loss of magnetic field homogeneity, exchange between different conformational states) [36]. However, the significant decrease in the overall 1H-NMR peak area (figure 3), often coupled with considerable decrease in relaxation times (table S2), is typically observed upon magnetic interactions additional to the applied magnetic field, which cause rapid relaxation of the RF-induced precession in nuclear spins back to their thermodynamic states [36]. Selective magnetic effects were seen here, similar to the selective interaction of organo-metallic compounds, also detected by 1H-NMR through their signal disappearance [69]. Indeed, spin-lattice relaxation times T1 were most shortened for those compounds (e.g. PN1, CPZ and PS1; table S2) which showed decline in the peak area, upon interaction with graphene (figure 3).
If molecular interactions with graphene were within the field of the comparatively large orbital diamagnetism of graphene [45–47], additional shielding in their 1H-NMR spectra would be obvious (as would be deshielding for any physisorption outside ring currents). The absence of considerable extra shielding effects on exposure to graphene (figure 3) suggests that orbital diamagnetism was at a minimum. Diamagnetic alignment of graphene flakes parallel to the applied magnetic field may be expected in a 9.4 Tesla NMR magnet to minimize diamagnetism [55, 70, 71], because the field component penetrating their graphene planes at any oblique or orthogonal angle raises the total energy due to diamagnetism [40–43]. Albeit with rotational and translational diffusion disorder in other vectors, in-plane alignment of the applied field reduces ring currents and their shielding effects to a minimum. Ring currents from single-walled CNTs aligned parallel to a magnetic field are also predicted to be less intense [55], which may have similarly affected NMR studies of physisorption of compounds to CNTs [56–58]. Further, the orbital diamagnetism of graphene is predicted to decline sharply with the opening of a band gap [72]. Water (and other polar solvents) may n-dope graphene, contributing to its negative zeta potential, including at edge-defect locations [33, 73–76].
Although 'π–π stacked' aromatic hydrocarbon interactions with graphene may arise in non-polar solvents or in the dry state [28–30], this does not appear to be the case here in water. Interactions with the polyaromatic carbon of graphene in aqueous (i.e. D2O) solutions (figure 3) were clearly of a different nature from those seen for the polyaromatic hydrocarbon interactions involved in their self-assembly, where there was large upfield shielding (up to 0.758 ppm) from ring currents and no decrease in the overall 1H-NMR peak area (table S1 and figure S2). Unlike FLG flakes, aromatic assemblies are not large enough to align parallel to the applied magnetic field [40–43]. However, any parallel face-face physisorption to the larger FLG flakes of face-centered or in-line stacks of organic compounds would have resulted in a large shielding loss, with major downfield change in the chemical shifts of the assemblies. Indeed, when so-associated with graphene, the aromatic rings within the assemblies would also be aligned parallel to the applied magnetic field, thus minimizing their ring current shielding. No such alignment had effect here, as the upfield shielding of assemblies remained unchanged.
3.5. Association–dissociation characteristics
Diffusion-ordered spectroscopy (DOSY) clearly evidenced the slower diffusion of the molecular assemblies associated with graphene, as compared with the free assemblies alone (figure 6, black and red traces, respectively; and table S2).
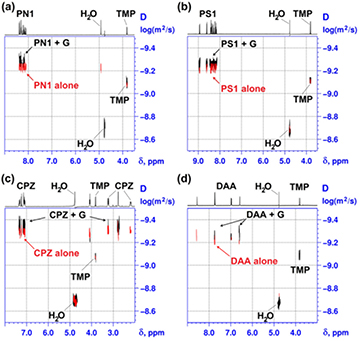
Figure 6. Change in diffusion coefficient on physisorption of molecular assemblies to graphene. The overlaid 2D DOSY spectra of (a) PN1, (b) PS1, (c) CPZ and (d) DAA in the absence (red) and presence (black) of graphene (G, 62.3 µg ml−1). The concentration of each dye in (a)–(d) was 0.5 mM.
Download figure:
Standard image High-resolution imageThe larger size of graphene flakes (~200 nm hydrodynamic diameter) [33] shifted the overall diffusion coefficient of the much (>100×) smaller assemblies according to the proportion of time spent associated with the FLG flakes during the DOSY measurements. DOSY spectra (figure 6) provided qualitative indication of molecular mobilities through the 2D-correlations of diffusion characteristics with the 1H-NMR chemical shifts.
More precise values of diffusion coefficients (D) of each studied compound in the presence and absence of graphene were estimated using the T1/T2 relaxation module of Bruker TopSpin 2.1 software to obtain numerical values for the diffusion constant at a given chemical shift (see supplementary 'experimental details'). A small, but significant (p < 0.05), change in molecular mobility was seen for six compounds (i.e. PN1, PS1, CPZ, DAA, PyY and PS2) with up to ~18% decrease in their diffusion coefficients (table S2), thus reflecting weak associations with graphene with significant dissociation rates. The representative DOSY spectra of four-studied compounds, which were earlier identified by NMR as the strongest binders, show a clear change in their molecular mobility on addition of graphene, thus reflecting the contribution of the slower diffusion of their graphene-associated fraction (figure 6 and table S2). Other compounds (PS3, PS4, NMP and DMF) showed no significant (p > 0.05) change in their diffusion coefficients, indicative of the dynamic, collisional nature of their interactions with graphene (table S2).
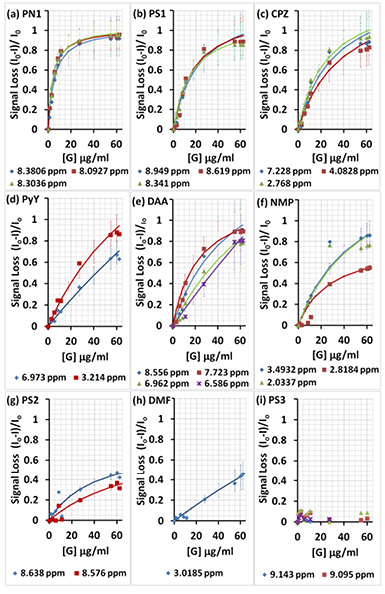
Given the clear link with graphene association, decrease in proton NMR signal peak heights was considered as a quantitative measure of physisorption. As expected [73], the relative decrease in the 1H-NMR peak heights followed a hyperbolic relationship (figure 7). The relative decrease in the NMR peak heights approached unity (100% loss of the 1H NMR peak height) for assemblies with greater association with graphene (PN1, PS1 and CPZ; figures 7(a)–(c)). Higher graphene concentrations did sequester the assemblies of compounds with weaker association (PyY, DAA and NMP; figures 7(d)–(f)). However, other compounds showed only partial (PS2 and DMF in figures 7(g) and (h)) or negligible association when highly-sulfonated (e.g. PS3 in figure 7(i)), which is in a full agreement with the qualitative NMR data presented above.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Relative decrease in the 1H-NMR signal heights for aromatic and non-aromatic protons, as the ratio of the difference between selected signal heights in the presence (I) and absence (Io) of graphene, in the order from high (a) to low (h) affinity. Continuous traces are the least squares estimates of the hyperbolic regression model, with 95% confidence intervals. The concentration of the studied compounds in (a)–(i) was maintained constant (0.5 mM), whereas the concentration of graphene in each experiment varied from 0 to 1.1, 2.7, 5.5, 8.2, 10.9, 27.3, 54.7, 60.1 and 62.3 µg ml−1.
Download figure:
Standard image High-resolution image{kind=link}
In parallel, we studied the interaction of graphene with the above aromatic compounds under identical conditions (0.5 mM) by following their natural fluorescence (figure S8) upon gradual increase of the graphene concentration (0.0–62.3 µg ml−1). In contrast to the data obtained from NMR, the binding profiles from quenching of fluorescence by graphene appeared to be significantly less discriminative towards chemical structures and followed the same trend for all compounds. Indeed, highly-sulfonated pyrenes (PS3 and PS4), which consistently demonstrated a lack of engagement with graphene by NMR, showed rather similar loss of fluorescence. According to the results obtained from quenching profiles, the difference ratios of all compounds approached unity (100% fluorescence signal loss; figure S8), thus implying that all studied compounds were able to reach 100% binding with graphene. Signal loss from quenching of fluorescence has been widely considered as a convenient measure of physisorption to graphene dispersions. However, absorption of light by graphene is often ignored [77–81].
Various methods are used [82, 83] to correct fluorescence quenching for light absorption by the sample. Such corrections, when applied to our fluorescence quenching profiles, had greater effect here on the more sulfonated compounds, which showed less (PS2 and PS3) or no (PS4) association with graphene by NMR (figure 7). Indeed, in the case of PS2, PS3 and especially PS4, much of the loss of fluorescence appeared to be due to light absorption, rather than fluorescence quenching induced by interaction with graphene (figure S8). Some apparent quenching remained though after correction in the case of these compounds (PS2, PS3 and PS4), particularly at higher graphene concentrations. However, correction of fluorescence quenching for the large extent of light absorption by higher graphene concentrations cannot be relied upon [82, 83]. When corrections were applied more reliably at the lower levels of graphene, the estimated dissociation characteristics of physisorption of the compounds by graphene varied 20 fold across the range of weak association, thus reflecting the expected diversity in binding affinities of these compounds to graphene, as earlier predicted by NMR. In fact, association characteristics estimated from quenching profiles were much closer to those estimated by NMR (figures 7 and S9) when loss of fluorescence was corrected for light absorption by graphene (figure S10). Indeed, when the ground-state association characteristics estimated by NMR were subtracted from those estimated by fluorescence, little remained (PN1, PS1, CPZ, PyG, DAA in figure S10) to be considered as possible dynamic quenching from collisions in the excited state. In comparison with many other analytical methods, NMR seems to offer the advantage of being more precise, direct and reliable description of molecular interactions with graphene. Further, with regard to proton NMR spectroscopy, graphene remained 1H-NMR-silent, as an 'invisible ghost', thus allowing direct measurement of responses from interacting partners without undesirable interferences and corrections.
Fluorescence quenching is generally considered as the sum of static quenching from binding in the ground state and dynamic quenching from collisional interactions in the excited state. The contribution of dynamic quenching is often estimated from changes in the fluorescence lifetime on quenching [77–81]. However, graphene interactions here resulted in negligible change in fluorescence decay characteristics of the assemblies (data not presented), further suggesting predominant association with graphene in the ground state, as also estimated by NMR. Charge transfer from the photoexcited state of the assemblies associated in the ground state cannot be eliminated [78, 81]. Neither the fluorescence excitation nor emission maxima changed upon varying graphene levels across all studied aromatic compounds. Similar to 1H-NMR peak height loss, only the fluorescence peak heights declined on increased interaction with graphene without any detectable shifts in excitation or emission bands (figure S11). Little change in the ratios of the vibronic and excimer bands of pyrene [80] also indicated a ground-state quenching with no loss of self-assembly, upon fluorescence quenching by graphene (figure S12).
Freundlich and Langmuir models of adsorption are considered when the bound and free fractions can be separated and determined [76]. Physisorption here was assumed to result in loss of signal of the graphene-associated fraction, when the remaining measured signal was assumed to represent the free dissociated fraction of the total concentration (0.5 mM) at equilibrium. On this basis, the approximated maximum association was around 2000 wt% for PN1 and around 1000 wt% for the PS1 assemblies, with the maximum association of other assemblies a few fold less (figure S13). However, relatively weak organic affinities for graphene were indicated (Ka 0.04–1.3 mM; figure S13). The largest signal loss arose at the highest graphene concentrations, where the density of physisorption of assemblies to graphene was least (e.g. PN1 figure S13). Sequestration of assemblies by graphene (almost complete loss of signal) relied on the higher frequency of interaction with the high surface area available at greater graphene concentrations. However, the surface density of bound assemblies was then much less (<50×) than the maximum density associated with graphene when organic compounds were in excess, at lower levels of graphene.
Graphene is widely assumed to be hydrophobic, with the strength of physisorption of organic molecular structures to disrupt biomolecular assemblies and their biology, notably: denaturation of proteins, disruption of lipid bilayers in biomembranes by hydrophobic bonding, and strong binding of single stranded DNA via melting double-stranded DNA base pairing [34, 35]. However, the evidence seems to be based on assumptions or convenience, such as: computational and surface studies in the absence of water, and from assuming the behavior of graphene from studies with graphite or graphene oxide [34, 35]. In contrast, up to a few layers in thickness, graphene is rather transparent, and mildly hydrophilic with negative zeta potential in water [33, 72–75], which we proffer herein results in benign organic interactions, without disruption of self-assembled organic structures. We found no disruption of aromatic assemblies (similar or weaker in strength than in DNA and proteins), even weaker aliphatic interactions (inadequate to disrupt lipid bilayers) [33], and repulsion of negative charge (see DNA).
4. Conclusions
NMR detects room temperature magnetic-like effects in graphene-associated organic molecular assemblies, when shielding from the orbital diamagnetism of the conduction electrons of graphene is at a minimum, which will be considered in a subsequent paper.
The magnetic-like effects upon hydrocarbon protons are selective for the type of association with graphene. Non-aromatic solvent interactions result in additional signal splitting with little relaxation change and insignificant decrease in 1H-NMR peak heights, whereas the relaxation of spin precessions of the aromatic proton nuclei back to their thermodynamic states results in vast decrease in the 1H-NMR peak heights for interacting assemblies, accompanied by considerable line broadening.
The interactions of water-dispersed graphene with aromatic hydrocarbons are weak but selective for positively-charged organic assemblies with affinities declining up to 2 orders of magnitude for strongly negatively-charged molecules. Despite significant dissociation rates, the large surface area of excess graphene allows almost complete physisorption of interacting organic assemblies.
The attractive forces within aromatic hydrocarbon assemblies are greater than those interacting with graphene, as there was no disruption and loss of shielding from the aromatic ring currents within organic self-assemblies and, similarly, no change in their fluorescence excitation and emission maxima upon addition of graphene.
Computational and surface science studies of the interactions of organic molecules and their supra-molecular structures should include the effects of water on graphene, and NMR studies to be meaningful in the life sciences and medicine.
Acknowledgments
APAR is grateful for the support of the Dean's Scholarship and the Graphene Environment, Health and Safety Committee, University of Manchester. IAK acknowledges the EU Graphene Flagship grant agreement no. 604391 and the EPSRC (EP/I023879/1). The authors acknowledge financial support from the Graphene Bioscience Interdisciplinary Grand Challenges, University of Manchester. The authors acknowledge the assistance given by Dr R Bryce and by IT Services in the use of the Computational Shared Facility at The University of Manchester, and the assistance given by Prof RJ Young, Dr P Gorgojo, Dr C Vallés and Dr A Vijayraghavan in graphene characterization.
Author information
Conflicts of interest
The authors declare no competing financial interests.
Author contributions
Graphene materials were prepared by APAR under the supervision of IAK and DJC under the direction of KSN. KKB undertook computational modeling. NMR experiments were performed by EVB and fluorescent studies by DJC. EVB and DJC analyzed the data and prepared the paper. All authors commented and saw the final version of the manuscript.