
Abstract
Sodium-ion batteries (SIBs) are one of the most promising alternatives to lithium-ion batteries (LIBs), due to the much more abundant resources of Na compared with Li in the world. Developing SIB technology to satisfy the increased demand for energy storage is therefore a significant task . However, one of the biggest bottlenecks is the design of high-performance and low-cost anode materials, since the graphite anode in commercial LIBs is not suitable for SIBs due to thermal dynamic issues. Hard carbon materials have been regarded as having the greatest potential as anodes in commercial SIBs owing to their excellent cost-effectiveness, but their relatively limited performance compared to the graphite in LIBs as well as the dimness of the sodium storage mechanisms still need further investigation. In this review, we summarize the progress of recent research into hard carbons for SIB applications, including the fundamentals of SIBs, sodium storage mechanisms, structures and the electrochemical performances of different types of hard carbons in SIBs and other types of sodium-based energy storage as well as the main challenges in this field. We aim to provide a general insight into hard carbons and their applications in SIBs, opening up future perspectives and possible research directions.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
CO2 emissions caused by burning fossil fuels have significantly increased to a rate which was, in 2017, twice that of the rate of absorption back into the land and ocean [1]. Although researchers have already realized the environmental detriments of using fossil fuels, including oil, coal, natural gas, etc., they continue to play a key role in the current energy scenario. Therefore, the development of renewable and efficient new energy generation, conversion and storage systems to reduce the use of fossil fuels is an urgent challenge today.
Lithium-ion batteries (LIBs), as a representative of secondary batteries, have made a great contribution to energy storage since their first successful commercialization in the 1990s [2, 3]. With the increased usage of electronic devices and electric vehicles (EVs), the demand for LIBs is also increased. In 2018, 11 583 research papers were published in the field of lithium-based batteries. However, they are now facing bottlenecks, especially considering that the average rate of increase of the energy density of commercial LIBs has been no more than 3% in the last 25 years [4]. In addition, lithium resources are not widely distributed in the world and are mainly located in South America, which tremendously restricts the future development of LIBs [5–7].
As a result, the alternative of using less expensive and more abundant metals for rechargeable batteries has attracted research attention. In this respect, sodium, which is the element adjacent to lithium in the same column of the periodic table, became the first option. Sodium is approximately 400 times more abundant than lithium and is widely dispersed in the Earth's crust and sea, so that the cost of sodium is only $150 ton−1, which is much lower than the $5000 ton−1 of lithium [8, 9]. In addition, the use of low-cost metals for the cathode in sodium-ion batteries (SIBs), such as Cu [10], could further reduce the total cost of SIBs compared to the use of Co in LIBs [11].
One of the most significant challenges of SIB technology is designing low-cost and high-performance electrode materials, especially anode materials, due to the unsuitability of graphite. Various cathode materials have also been widely investigated, most of which were inspired by LIBs [8], however, the anode part is still the bottleneck as it dictates the C rates and also plays an important role in the solid electrolyte interphase (SEI) formation and hence the overall Coulombic efficiency. Among all the materials which have been tried as anodes for SIBs [8, 12], carbonaceous materials are regarded as the most promising because of their relatively high structural stability and cost-effectiveness, especially when derived from low-cost precursors like biowaste or plastic waste.
In general, hard carbons cannot be graphitised even at very high carbonisation temperatures due to the high oxygen and disordered structure of the precursors. They are disordered structures with randomly-oriented graphitic domains, higher interlayer spacing and some remaining heteroatoms (mainly oxygenated groups). They also have random closed pores in between the randomly-oriented graphitic crystallites, whose size depends on the size of the crystallites (the higher the crystallites, the larger the pores). Such a mix of crystalline and disordered structures provides more defects, more nanopores and larger interlayer spacing, etc., and is therefore able to afford more sodium diffusion pathways and sodium storage sites. It needs to be clarified that the oxygen content can form cross-linking regions or curved layers in the hard carbon structures during the carbonization process, thus affecting the non-graphitisability of hard carbons. A promising hard carbon precursor, therefore, should possess enough oxygen content to enable the non-graphitisability of hard carbons. Although some other carbonaceous materials such as soft carbons [13–15] or even graphite-based materials [16, 17], etc. could also be used as anodes for SIBs as long as they are properly treated or combined with the hard carbons, hard carbon materials are definitely playing the key role in this field. However, most of the reported hard carbon anode materials are still uncompetitive with graphite in commercial LIBs, and the sodium storage mechanisms of hard carbons are still under debate due to their different structures, which need further consideration.
In this review, the fundamentals of SIBs and hard carbons are introduced. The current sodium storage mechanism models and the electrochemical performance of different types of hard carbons in SIBs are summarized. In addition, a brief introduction to the applications of hard carbons in other sodium-based energy storage systems is also presented. At the end of this review, the main challenges are also addressed, in which we aim to provide researchers with a general insight into hard carbons for SIBs and some possible future directions. A summary of the characteristics and the applications of hard carbons in SIBs and other sodium-based energy storage systems is shown in figure 1.
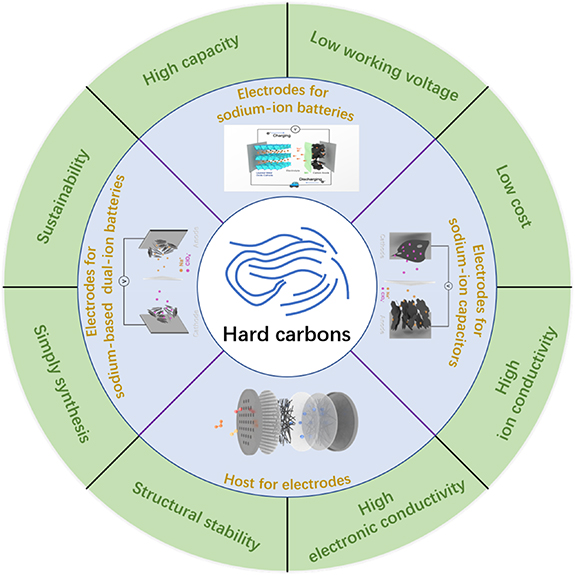
Figure 1. A summary of the characteristics and applications of hard carbons in sodium-based energy storage.
Download figure:
Standard image High-resolution image2. The fundamentals of sodium-ion batteries and hard carbons
2.1. Sodium-ion batteries
2.1.1. Sodium-ion batteries vs. lithium-ion batteries
LIBs, as representatives of rechargeable batteries, have achieved great success and development, being the main portable power source in people's lives over the past decades due to their high energy density. However, with an increasing population and the development of electronic devices and EVs, the demand for LIBs has significantly increased, which will lead to a deficiency of Li resources on the Earth and consequently increasing prices, therefore, LIBs will gradually become unaffordable [18]. Consequently, finding alternative sources to foster new battery technologies is a critical challenge. Due to the similarity of the physical and chemical properties of sodium with lithium (as they are in the same group of elements in the periodic table), SIBs or Na-ion batteries (NIBs) are regarded as one of the most promising candidates to partially replace LIBs.
The working principle of SIBs is same as LIBs, which is called the 'rocking chair' (figure 2): during charge, the cathode desodiates and the Na+ ions 'swim' to the anode through the electrolyte and sodiate into the anode, while electrons move from the cathode to the anode along the external circuit. During discharge, the anode desodiates and Na+ ions return back to the cathode through the electrolyte, while electrons are transported from the anode to the cathode along the external circuit, and the battery provides power to the external circuit [19].

Figure 2. A scheme of the working principles of sodium-ion batteries.
Download figure:
Standard image High-resolution imageThe concept of sodium-ion batteries was first established at almost the same time as lithium-ion batteries (1970s for LIBs and 1980s for SIBs) [19–21]. However, SIBs gradually faded out of sight soon after the successful commercialization of LIBs in the 1990s [8], due to their lower specific capacity, lower energy density and worse rate and cycling performance. The main and simplified reason why SIBs perform worse than LIBs is that sodium is larger, heavier, and has a higher standard potential compared to lithium, which causes SIBs to have a decreased capacity and a reduced range of redox potential. The radius and atomic weight of sodium is 1.06 Å and 23 g mol−1, respectively, while lithium is only 0.76 Å and 6.9 g mol−1 [9] (table 1). This makes for larger volumetric changes when the Na concentration changes, which can end up resulting in a strain build-up upon the insertion or extraction of Na+ ions and a reduce the cycling life. The larger size of the Na+ ions will also affect the kinetic process and cause slower diffusion, which is the main reason for the poorer rate performance [22, 23]. In addition, graphite is usually selected as the anode material in commercial LIBs due to its high cost-effectiveness, and it has a high theoretical specific capacity of 372 mAh g−1 [24] due to the formation of graphite intercalation compounds (GICs). However, the bigger Na+ ions do not intercalate into the graphite layers as do Li+ ions, due to thermal dynamic issues, which further increases the challenges of finding suitable anode materials for commercial SIBs [25].
Table 1. A physical comparison of Li and Na.
| Physical characteristic | Li | Na |
|---|---|---|
| Cation radius (nm) | 0.076 | 0.106 |
| Atomic weight (g mol−1) | 6.9 | 23 |
| Potential (V vs. SHE (standard hydrogen electrode)) | −3.04 | −2.71 |
| Ratio in Earth's crust | 0.0065% | 2.83% |
| Price of carbonates ( | ∼5000 | ∼150 |
| Theoretical capacity of metal (mAh g−1) | 3829 | 1165 |
| Theoretical capacity of GICs in ester-based electrolytes (mAh g−1) | 372 | 35 |
Nevertheless, the assumption that Na+ ions are too large to intercalate into graphite layers (thereby making graphite a poor material for storing sodium) is an oversimplification of a very complex picture. K+ ions with an even larger ion radius (1.38 Å) can happily intercalate into graphite [2, 26]. A graphite lattice can accommodate Li+ ions up to a concentration of LiC6 with a very high theoretical specific capacity of 372 mAh g−1. K+ ions can form K-GICs with a theoretical capacity of 279 mAh g−1 for KC8 [2]. However, in the case of sodium, the concentration is only NaC186 [27] or NaC64 [28] with the extremely low theoretical capacities of 12 and 35 mAh g−1, respectively. Rb+ and Cs+, which have even larger radii, were also investigated for their ease of intercalation into graphite layers [2, 29]. Besides, the desolvation energy for desolving the alkali metal ions from the electrolytes and transferring them to naked ions decreases in most electrolytes from Li+ to K+ ions, meaning that Li+ ionintercalation should 'theoretically' be the most difficult situation compared to Na+ and K+ ions when forming GICs [30, 31]. This inverted phenomenon results in another controversial opinion that it is not the Na+ ions but actually the Li+ ions that suffer from the special intercalation situation. The reason for the inferior intercalation behaviour of Na+ ions into graphite was calculated to be that the attractive interaction between the Na+ ions and the graphite layers is extremely weak, so that there are not enough energetic forces to motivate the intercalation, compared with plating on the graphite surface [32]. Grande et al [33] also found, based on van der Waals density functional theory (DFT) calculations, that the formation enthalpies are positive for NaC6 or NaC8 but negative for LiC6, implying that the formation of Na-rich GICs requires a negative redox potential vs. Na+/Na [34, 35].
On the other hand, the intercalation potentials of Li+ ions (vs. Li+/Li) and Na+ ions (vs. Na+/Na) into host materials are dependent on the materials' structures. Generally, for common hosts, the standard intercalation potentials are lower for sodium compared to lithium due to the higher potential of sodium than lithium (see table 1) [31]. The same trend was also found in cathode materials, so that the operating voltage of LIB and SIB full cells is dependent on the specific combination of both the positive and negative electrode materials. According to the data reported in the literature [31, 36–40], a number of cathode materials have lower sodiation/desodiation potentials (vs. Na+/Na) for sodium than the lithiation/delithiation potentials (vs. Li+/Li) for lithium. The reduced values of the positive electrodes are greater than those of the negative electrodes between Li and Na cells. Therefore, the cell voltage of SIBs is usually lower than LIBs, which means the energy density of SIBs is not as high, compared with LIBs, in most cases [31].
Based on the aforementioned shortcomings of SIBs, the development of SIB technology suffered from a long period of silence before researchers started to realise the problem of Li resources and cost. However, when compared to LIBs, the advantages of SIBs can be summarised as follows: (1) there are much more abundant and widely-distributed resources of sodium than lithium. There is only 0.0065% of lithium in the Earth's crust which is mainly distributed in South America, while sodium makes up 2.83% of the Earth's crust and is plentiful all over the world especially in sea water, and therefore much cheaper [7, 41]; (2) aluminium can be used as the current collector for electrodes in SIBs compared to copper in LIBs since aluminium will form an alloy with lithium but not with sodium, which reduces the total cost and weight [31]; (3) the desolvation energy of sodium ions in various organic solvents is around 30% smaller than for lithium ions, which could decrease the charge transfer resistance and increase the electrode kinetics [31, 42, 43]; (4) in addition to Ni, Co and Mn, which were found to be the only electrochemical active transition metals among the layered oxide cathode materials, more choices are available for SIBs such as Cu, Fe, Ti, V, Cr, etc., showing different electrochemical properties and providing more opportunities for the development of SIBs [44]; (5) the use of hard carbon anodes and the differences in the thermal, chemical and electrochemical properties enable more possibilities for electrolyte optimization, to obtain more favourable sodium-based energy storage devices [45, 46]. In addition, similar technology and equipment to that used for LIBs could be used for SIBs, which makes practical and industrial manufacturing easier. These benefits give SIBs great potential for various markets such as low-speed electrical vehicles, home-use energy storage, electronic devices and especially large-scale energy storage, etc., in the future.
2.1.2. Basic performance parameters for sodium-ion batteries
Generally, the main key parameters for evaluating the performance of SIBanodes include the specific capacity, energy density (gravimetric and volumetric), rate capability, cycling stability, and the initial Coulombic efficiency (ICE), which strongly depends on the electrode materials, electrolytes, and even the additives.
The theoretical specific capacity Qt of an electrode material can be determined by Faraday's law [47]:

where n is the number of transferred electrons, F is the Faraday constant (96 485 C mol−1) and M is the molecular mass of the active material. In a practical situation with a galvanostatic charge/discharge status, the specific capacity is calculated by [48]:

where I (mA) is the charge/discharge current, Δt (h) is the charge/discharge duration and m (g) is the mass of the active material. Taking carbonaceous materials as an example, the capacity is affected by the morphology, pore structures, ordering structures, and defects, etc., which are discussed in the next sections.
The gravimetric energy density of a full SIB cell is the integration of the voltage and the specific capacity. The theoretical energy density is calculated based on the following equation [49]: the Gibbs free energy of a standard chemical reaction is the sum of the formulation energy of the reactants and products:

The maximum electrical work of the reaction equals ΔrG:

where n is the number of transferred electrons of one mole of reactant and E is the thermodynamic equilibrium voltage. Therefore, the theoretical gravimetric energy density can be calculated as:

where ∑M is the sum of the mole weight of all the reactants. In practical situations, the energy density is determined as [48]:

where U represents the voltage window (the volumetric energy density could be calculated in the same way but replacing the mass with the volume). Thus, in addition to increasing the specific capacity of the electrode materials, another way to obtain a high energy density is to elevate the working voltage. That requires a higher voltage of the cathode materials and a lower voltage of the anode materials. The average working voltage of a SIB is the quotient of the integrated area of the charge/discharge profile (the specific energy density) and the total specific capacity. In practice, it usually refers to the voltage at the half of the specific capacity or at the half-integrated area of the galvanostatic charge/discharge profiles. Meanwhile, since a high operating voltage may be beyond the electrochemically stable window of the electrolytes and binders, it is also important to expand the thermodynamically stable potential window of the electrolytes by using proper recipes or adding additives, as well as selecting appropriate binders [50]. Furthermore, it is worth noting that in lab-based experiments, it is usual to calculate the energy density of a full cell based only on the mass of the active materials of both cathode and anode. However, in practical or industrial batteries, the energy density calculation should involve the masses of all the other materials including current collectors, binders, additives, separators and cases, etc. [51]. As a result, controlling the mass of those non-active and packing materials is also very important to improve the energy density.
The rate capability represents the reversible capacity of an electrode material at different current densities. Capacities will typically decrease at higher current rates because the electronic current density is much larger than the ionic current density of the electrolyte and electrodes (and the rate of ion transfer across the electrolyte/electrode interface) [52], meaning that stronger polarisation takes place at high current rates.
The cycling performance relates to the stability of the battery during the long-term charge/discharge process. The lifespan of a battery is defined to be the cycle number at which the capacity retention drops to 80% of its initial capacity [53]. Generally, cycling stability is mainly determined by the interface between the electrode and the electrolyte and by the structural stability of the electrode materials. The electrode/electrolyte interphase is affected by the formation of SEI, which is a passivation layer on the surface of the electrode when the Fermi level of the anode is above the lowest unoccupied molecular orbital (LUMO) of the electrolyte (or the Fermi level of the cathode is lower than the highest occupied molecular orbital (HOMO) of the electrolyte) so that the electrolyte is decomposed to form inorganic insoluble compounds [52, 54]. A stable SEI is beneficial to eliminate further decomposition of the electrolyte during cycling, which ensures the reversible insertion/desertion of Na+ ions. The mechanical properties of the SEI are also important in protecting the electrode materials against volume expansion and maintaining the adsorption capacity on the surface [54]. For some anode materials such as alloys, the huge volume expansion during charge/discharge significantly limits the cycling stability, and some strategies including surface coating, and heteroatom doping, etc., would be useful to overcome this phenomenon and improve the cycling performance [19, 55]. Good interface and structural stability is required to ensure a high enough Coulombic efficiency (CE) during the charge/discharge process, and it is found that a high CE of 99.96% is needed for commercial batteries when cycling to over 500 cycles [4].
ICE is the quotient of the initial discharge capacity divided by the initial charge capacity (for the anode half-cell it is the quotient of the initial charge capacity divided by the initial discharge capacity) which is also a significant parameter for SIB technology but gets less attention compared with other parameters. Since the cathode material usually serves as the Na source in a full battery, it should provide Na+ ions for reversible cycling as well as the consumption of Na+ ions for SEI formation at the first cycle [56]. Therefore, not only does ICE reveal the SEI formation and irreversibility of sodiation/desodiation reactions at the first cycle, but also it affects the energy density of a full battery, as more cathode materials have to be used to cover the initial irreversible Na+ ion consumption which results in an increased weight of the battery and a decreased energy density. Some factors were found to influence the ICE including irreversible decomposition of the electrolyte, an irreversible reaction during sodiation/desodiation, and the defects and porosity, etc. As a result, optimising the electrolytes and tuning the nanostructures and surface area would be helpful to improve the ICE [56].
2.2. Hard carbons
The differences between hard carbons, soft carbons and graphite have been intensively reviewed in the literature. Typically, in addition to the mechanical properties (as suggested, hard carbons are 'harder' compared with the other two forms of carbon), a more specific definition is that these carbon materials cannot be graphitised even at a very high temperature (over 3000 °C), while the soft carbons correspond to the 'graphitizable' carbonaceous materials [57, 58].
On the other hand, some terms or definitions are usually misused in the literature, such as 'amorphous carbons'. It has been reported that 'amorphous carbons' only describes the carbonaceous materials containing localized π-electrons (i.e. diamond-like carbons) [58]. In other words, amorphous carbons belong to the hard carbons, as they cannot be graphitised, but hard carbons are not only the amorphous carbons. For energy storage applications, carbonaceous materials are usually treated at between 600 °C and 2000 °C; within this range both 'hard carbons' and 'soft carbons' have disordered or non-graphitic structures, but 'soft carbons' will be relatively more ordered compared to the 'hard carbons' when carbonized at the same temperature. When the pyrolysis temperature increases to a very high level, the 'hard carbons' are still disordered, while the 'soft carbons' become graphite or pseudo-graphite.
As electrode materials, although hard carbons do not have as high a specific capacity as chalcogen-based materials and alloys, their capacity can approach the commercial graphite anodes of LIBs, and their lower working voltage can be a good point for improving the energy density [9]. In addition, their limited volume change (unlike alloy and conversion anodes) and structural stability ensure that carbonaceous anode materials have a better cycling performance [59]. Hard carbons have more disordered structures, a higher concentration of defects, a higher content of heteroatoms and a larger distance between the graphitic layers as well as more closed pore structures compared with soft carbons, which facilitate more storage sites and diffusion pathways for Na+ ions, and are regarded as the most promising candidates for SIB anode materials. Furthermore, the low cost, sustainability, and simplicity of manufacture also make hard carbon materials the most promising candidates for the practical commercialisation of SIBs [58, 60, 61].
3. Sodium storage mechanisms and interfaces of hard carbons
3.1. Sodium storage models of hard carbons
While extraordinary research achievements have been made in the field of hard carbon anode materials for SIBs, owing to contributions from many research groups, the sodium storage mechanisms in carbon anode materials are still controversial. This is not surprising, given the fact that hard carbons are 'ill-defined' materials with different crystallite sizes, pore structures, heteroatoms, etc. People still debate the sodium storage behaviours at carbon electrodes within the sloping or plateau regions of a typical galvanostatic discharge/charge profile, which can be summarised in three categories: (1) Na+ ions adsorb at the defect sites on the surface; (2) Na+ ions intercalate into graphitic layers; (3) Na+ ions fill the nanopores. Based on these three kinds of sodium storage behaviour, the sodium storage mechanism can be categorised into four models: (1) the 'intercalation-filling' model: Na+ ions intercalate into graphitic layers in the sloping region, and insert into the nanopores between randomly stacked layers at the plateau region [27, 62]; (2) the adsorption-intercalation model: Na+ ions adsorb at the surface or defect sites of the carbon electrodes within the sloping region, while they intercalate into the graphitic layers within the plateau region [63, 64]; (3) the adsorption-filling model: in the sloping region, Na+ ions adsorb at the defect sites, while filling the nanopores in the plateau region [65, 66]; and (4) the 'three-stage' model: defect adsorption of Na+ in the sloping region, but in the plateau region, the Na+ ions first intercalate into the graphitic layers and eventually fill in the nanopores [67]. A schematic of the four types of sodium storage models is shown in figure 3.
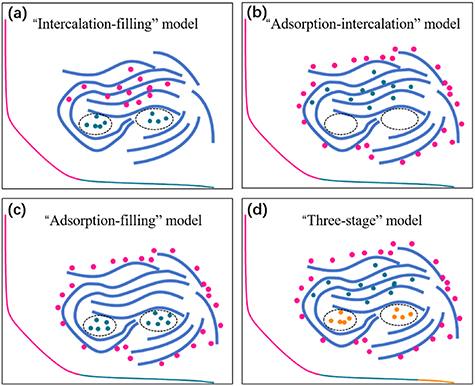
Figure 3. A schematic of the sodium storage models: (a) the 'intercalation-filling' model; (b) the 'adsorption-intercalation' model; (c) the 'adsorption-filling' model and (d) the 'three-stage' model.
Download figure:
Standard image High-resolution imageThe first investigation of sodium storage mechanisms was reported by Stevens and Dahn in 2000 [27]. They thought that the sodium storage mechanism is similar to that of LIBs and proposed the 'intercalation-filling' model. The in situ wide-angle x-ray scattering (WAXS) showed that the Na+ ions intercalate into the graphitic layers within the sloping region based on the (002) peak position shift for soft carbons. Although the (002) peak for hard carbons does not show such an obvious shift, due to the existence of single, double or triple stack layers of hard carbons, the authors think that similar intercalation can still occur for hard carbons. Based on in situ SAXS measurements, the electron density remains unchanged in the high-potential region while decreasing at lower potentials [68], suggesting nanopore filling in the plateau region (figure 4(a)).

Figure 4. (a) The calculated electron-density contrast obtained from in situ small-angle x-ray scattering (SAXS) scans during discharge and charge. Reproduced from [68] © 2000 The Electrochemical Society. All rights reserved. (b) The interlayer spacing changes at different discharge voltages. Reprinted with permission from [64]. Copyright (2013) American Chemical Society. (c) A schematic of the extended 'adsorption-intercalation' model of hard carbons at different stages. [69] John Wiley & Sons. © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (d) Ex situ transmission electron microscopy (TEM) images and (e) ex situ x-ray photoelectron spectroscopy (XPS) Na1s spectra of hard carbon from pristine to 0 V (vs. Na+/Na). [66] John Wiley & Sons. © 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (f) A galvanostatic intermittent titration technique (GITT) profile and the corresponding Na+ ion diffusion coefficients during discharge. Reprinted with permission from [67]. Copyright (2015) American Chemical Society.
Download figure:
Standard image High-resolution imageIn 2012, Cao and Liu et al [63] claimed another adsorption-insertion mechanism, namely that the sloping region is attributed to adsorption of the sodium on the surface of small graphitic clusters, while the plateau region is due to the sodium insertion into the graphitic interlayers. Based on the theoretical simulation, the authors concluded that the minimum interlayer distance for Na+ ionintercalation is 0.37 nm. This model was further supported by Mitlin and Li et al in 2013 [64]. They used carbonised peat moss with (CPM-A) and without activation (CPM) as well as commercial activated carbons (CACs) to investigate sodium storage performance and the sodium storage mechanism. The sloping capacity increased with increasing carbonisation temperature. It was found that CACs with a high surface area and many micro- and mesopores do not show any plateau, suggesting that the low-potential plateau should not be attributed to pore filling. According to the XRD spectra of CPM-1400 and CAC at different discharge voltages, the much more obvious d-spacing changes in the plateau than in the slope indicates that the plateau region is due to the insertion of Na+ ions into the carbon layers (figure 4(b)). Very recently, Xu and co-workers [69] published their research on the sodium storage mechanism of hard carbons and proposed an 'extended adsorption-intercalation model' by using ginkgo leaf (GL)-derived carbons carbonized at various temperatures (GL-T) and correlated the structures with the sodium storage performance (figure 4(c)). High resolution transmission electron microscopy (HRTEM) images show three carbon domains: highly disordered domains, pseudo-graphite domains and graphite-like domains, depending on the carbonisation temperature. Based on the 'volcano-shape' tendency of both sloping and plateau capacities along with the temperature series, the authors correlated the capacities with the structures and proposed a model that can be summarised as follows: (1) the very large interlayer spacing (>0.4 nm) from the highly disordered carbons would be regarded as 'defects' and Na+ could be freely accessed, the same as the defect-adsorption behaviour, which leads to only the sloping region; (2) the smaller interlayer spacing (0.36–0.4 nm) is accessible for intercalation and would cause the plateau region; (3) the graphite-like carbons with a very small interlayer spacing (<0.36 nm) are inaccessible for Na+ ions to intercalate into the graphitic layers, so that defects would predominate in the sodium storage behaviour and only the sloping region behaviour would be exhibited.
Tarascon and co-workers [65] investigated the sodium storage mechanism in 2016 using carbon nanofibres (CNFs) derived from polyacrylonitrile (PAN). In situ XRD was performed and there was no obvious (002) peak shift during the sodiation, suggesting the Na+ ions should not intercalate into the graphitic layers. Also, the sodium metal plating could only happen at around −0.015 V (vs. Na+/Na) and therefore, the plateau region should be attributed neither to intercalation nor to sodium plating. Considering that the low-voltage capacity is prominent only with samples treated above 1000 °C with lots of mesopores, the authors finally claimed that the sloping region was due to defect site adsorption, while the plateau came from pore filling. In the same year, Hu and co-workers [66] reported the same model for the sodium storage mechanism. Based on the correlation of the microstructures and the sloping capacity behaviour observed with the temperature increase, they concluded that the sloping region was attributed to defect adsorption of Na+ ions. Ex situ TEM was used to investigate the microstructures of pristine carbon and the same sample discharged to 0 V (vs. Na+/Na), where the disordered structures did not change and the interlayer spacings were both around 0.404 nm, indicating that no intercalation happened (figure 4(d)). Figure 4(e) shows the ex situ XPS Na 1s spectrum, where the peaks of the carbon samples are approaching the metallic sodium when discharged from 0.12 to 0 V (vs. Na+/Na), which may be due to the lower binding energy of pore surface adsorption. According to the above results, the authors summarized that the Na+ ions are adsorbed at the defect sites in the sloping region, and fill in the nanovoids in the plateau region, which is consistent with some other reports [70, 71].
The last sodium storage model is the 'three-stage' mode proposed by Ji et al [67] The GITT and the corresponding calculated diffusivity (figure 4(f)) indicate that the Na+ ion diffusion in the sloping region is much faster than in the plateau region, suggesting that the sodiation in the sloping region happens at the more easily accessible surfaces and edges. By correlating the defect concentration with the sloping capacity plus the ex situ XRD results showing the position shift of the (002) peaks in the plateau region, an 'adsorption-intercalation' mechanism seems best suited to describe this situation, which is also consistent with the GITT result that the decrease of the diffusion coefficient in the plateau results from the Na+ ions overcoming the repulsive charge from the previously bound defect sites and inserting into the carbon layers. However, the Na+ ions' diffusivity increases again at the end, suggesting that the plateau can not be attributed to only one intercalation mechanism. Therefore, the authors claimed that there is a minor adsorption of Na+ ions on the pore surfaces at the end of the sodiation process, denoting a 'three-stage' model instead.
Although several controversial sodium storage models have been reported so far, they could still guide the design of optimised carbon materials for sodium-ion battery anodes. Making hard carbon materials with more defects, a larger interlayer spacing and more closed pores would be mostly acceptable for achieving high sodium storage performance. However, understanding the exact sodium storage mechanisms is still significant from a fundamental point of view. In our opinion, there may not be only one correct sodium storage model for hard carbon anodes; sodium storage mechanisms could be varied, based on different microstructures. Therefore, generalising the sodium storage behaviours with different microstructures of hard carbons would very important for future research.
3.2. Advanced characterisations for sodium storage mechanism investigations
So as to better understand the sodium storage mechanisms of hard carbons, clear investigations of the structures of hard carbons and the correlation between the structures and electrochemical properties are necessary, which therefore requiresome advanced characterisation techniques. One of the most commonly used methods is in situ or ex situ XRD, which could easily study the intercalation behaviour of Na+ ions within the graphitic layers. Komaba et al [72] have studied the sodiation and desodiation processes of hard carbon electrodes by ex situ XRD and found that the (002) peaks shifted to lower angles and the intensities decreased during sodiation, and could return back to the original position after full oxidation (figure 5(a)), indicating the reversible intercalation/deintercalation of Na+ ions within the graphene layers. On the other hand, in some studies, the (002) peak shifts in the ex situ or in situ XRD only occured at the plateau regions [64, 67], or did not occur at all during the sodiation process [65], based on which, some other sodium storage models have been announced, as illustrated in the last section. However, due to the disordered structures of hard carbons, the (002) peaks are usually weak and broad, and the sample manufacture when using ex situ operations may also influence the accuracy of the results [11]. Therefore, combining the XRD data with other data would be necessary to generally consider the sodium storage mechanism, which needs further studies. Another advanced x-ray analysis method for sodium storage mechanisms is the SAXS, which is able to obtain the pore information. The SAXS patterns of hard carbons usually contain one slope in the low Q region, corresponding to the particles or large pores at the surface, while a shoulder at the higher values of Q is attributed to the nanopores among the turbostratic nanodomains (figure 5(b)). As shown in figure 5(c), it is reported that the slopes of the hard carbons carbonised at various temperatures are quite similar, implying that the surface large pores of hard carbons are almost unchanged with varying pyrolysis temperatures, while the intensities of the shoulder increase with elevated temperatures, indicating larger pore sizes. The pore number was also investigated and found to decrease with increasing temperatures. In situ or ex situ SAXS was also used to observe the pore changes during sodiation [27, 68]; as demonstrated in the above section, the electron density of the nanopores decreases within the plateau regions (figure 4(a)), suggesting the use of the pore filling mechanism, which was further supported by the recent publication of [11] and [77].
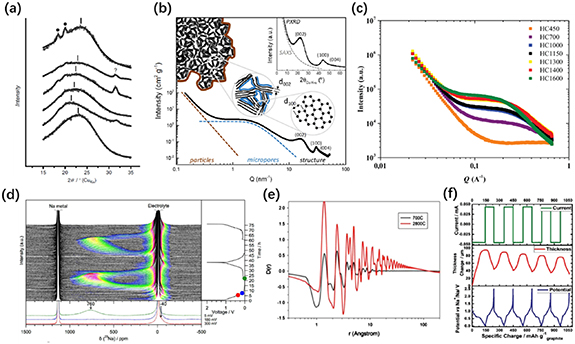
Figure 5. (a) Ex situ XRD patterns of hard carbon electrodes, from top to bottom: pristine, 0.4 V, 0.2 V, 0.1 V, 0 V during sodiation and 2 V upon desodiation. [72] John Wiley & Sons. Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) A schematic illustration of the SAXS patterns with the corresponding nanostructures for each part. Reprinted from [58], Copyright (2019), with permission from Elsevier. (c) The SAXS patterns of hard carbons carbonised at different temperatures. Reprinted from [73] © 2016 Science Press and Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Published by Elsevier B.V. and Science Press. All rights reserved. (d) Operando 23Na NMR spectra for a hard carbon electrode in a half cell. Reproduced from [74]. CC BY 3.0. (e) The neutron PDF spectra of hard carbons carbonised at different temperatures. Reprinted from [75], Copyright (2014), with permission from Elsevier. (f) The in situ electrochemical dilatometry spectra of graphite for the first five cycles. [76] John Wiley & Sons. © 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution imageGotoh and co-workers [78] have further confirmed by in situ and ex situ7Li solid-state nuclear magnetic resonance (NMR) for the case of LIBs that Li metal clustering occurs in the nanopores. In 2016, Grey et al [74] used 23Na solid-state NMR to investigate hard carbon electrodes in SIBs. It was found that during the low-voltage plateau, the 23Na NMR peak shifted from zero ppm which is typical for Na+ ions to around 760 ppm near the metallic Na, suggesting that Na pooling occurred and Na clusters were formed. Within the high-voltage sloping region, the 23Na NMR signal remained at the ionic state, indicating that the Na+ ions formed in this region, which could be due to defects. Another advanced characterisation technique usually applied to the investigation of defects in hard carbon materials is the neutron pair distribution function (PDF). The peak positions in the spectra correspond to the real-space distances between arbitrarily selected pairs of atoms, and the peak integrals suggest the coordination numbers of the atom pairs near a specific distance [79]. Therefore, as shown in figure 5(e), hard carbon carbonised at 2800 °C shows a much higher peak amplitude compared to hard carbon carbonised at 700 °C, indicating the fact that hard carbon materials show more ordered structures and fewer local defects when treated at higher temperatures. Ji et al [67, 79, 80] reported on several studies of the correlation between the neutron PDF results and the electrochemical properties of hard carbons to investigate sodium storage behaviour regarding defects and claimed that the defect adsorption of Na+ ions may be the main reason for the sloping capacity.
Adelhem et al [76] reported on in situ electrochemical dilatometry (ECD) as an effective technique to study the sodium storage behaviours of graphite as a cointercalation electrode. Figure 5(f) shows the ECD results for the graphite electrode. It was found that the thickness of the electrode nearly doubled (from 50 to 95 µm) after the first sodiation, suggesting larger structure rearrangement and particle loosening during the first cycle. During the subsequent cycles, the thickness showed a periodic 'breathing' of about 35–50 µm in each cycle, which could be attributed to the reversible intercalation/deintercalation process. Although this result was for a graphite electrode, the ECD technique can be applied to hard carbon electrodes as well for investigation of the sodium storage mechanism.
Some other advanced characterisation techniques such as x-ray PDF and muon spin, as well as DFT calculations, etc., [81– 83] have also been reported as having been used to investigate the structures and sodium storage mechanisms of hard carbon materials. The utilization of these advanced characterisation techniques for SIB applications would push forward not only the understanding of SIBs but also the techniques themselves. Meanwhile, there are also some limitations when using these methods. For example, ex situ experiments would require the disassembly of cells, which makes for more complexity and may cause sample contamination due to air sensitivity. The neutron and muon analysis methods need a huge number of samples for the measurements, which increases the difficulty of operation. Therefore, the choice of proper techniques and careful combination with other analyses is very important for sodium storage mechanism studies in practical situations.
3.3. The solid electrolyte interphase on hard carbon anodes
During the first cycle of the charge and discharge process, a passivation layer called a solid-electrolyte interphase (SEI) will form on the surface of hard carbon anodes if the Fermi level of the anode material is higher than the lowest unoccupied molecular orbital (LUMO) of the electrolyte [52]. Therefore, the electrolyte is automatically decomposed to form a SEI consisting of both inorganic and organic insoluble compounds so that the voltage window of the system becomes broad enough to allow the redox reaction of energy storage to happen. The SEI layers protect hard carbon anodes from direct contact with the electrolyte but still allow ions to move through the SEI layers, which means the SEI layers are a kind of pure ionic conductor [72].
The formation of an SEI is firstly related to the type of electrolyte used. For ester-based electrolytes, the degraded inorganic and organic components often geometrically form a random mixture, while for ether-based electrolytes, the degraded inorganic particles are intended to be incorporated into a continuous phase consisting of organic components [84]. The use of different types of electrolytes and the different electrode/electrolyte interfaces will affect the sodium storage performance, especially the ICE. It has been reported that the use of ether-based electrolytes is helpful for increasing the ICE, as the LUMO of ether-based electrolytes is calculated to be higher than that of ester-based electrolytes [54]. Even for some carbon anode materials with very high surface areas, the use of ether-based electrolytes can still lead to high ICE [85, 86], implying a great potential. In addition to the electrolytes used, the formation of an SEI is closely related to the properties of hard carbon anodes. Defects and a porous structure can increase the amount of SEI formed during the first cycle, thus decreasing the ICE of hard carbon anodes. For example, although porous hard carbon anodes could provide a high reversible capacity, the ultra-low ICE of those hard carbon anodes requires an excess amount of Na ions from the cathodes to irreversibly form an SEI in the first cycles, which results in a non-negligible waste of the expensive cathode materials. Furthermore, the slope-dominated charge-discharge features will also decrease the working voltages and energy density of full cells, although they could improve the rate performance at high current density. The structures of hard carbons need to be tuned to achieve a balance between several parameters including the ICE, energy and power density. More detailed examples will be discussed in the following sections.
There are several methods that can be employed to characterise the SEI on hard carbon anodes. First of all, the most commonly used two techniques are transmission electron microscopy (TEM) and x-ray photoelectron spectroscopy (XPS). TEM can help characterize the morphology of SEI formation, but the difficulty of sample preparation and the limitation of the resolution of TEM greatly hinder the characterization accuracy of this technique. With the development of TEM techniques, operando TEM characterization and high-resolution cryo-EM appeared, thus significantly improving what we know about the SEI. Cui and co-workers [87] successfully employed cryo-EM to achieve high-resolution images of SEI in different kinds of electrolytes, which opened up the way for future work. XPS can help to partially distinguish the components of SEI. Meng and co-workers [88] applied XPS to characterise the different components of SEI in ester- and ether-based electrolytes, where ether-based electrolytes can provide more organic components in the SEI and thus lead to a good flexibility of the SEI in ether electrolytes. Secondly, modelling methods are also widely employed to help analyse the formation of an SEI on hard carbon anodes [89]. According to the calculation of the energy levels of different electrolyte molecules and active materials, we can determine their effect on the formation of an SEI. Based on molecular dynamic modelling, the effect of electrolyte concentration on the formation of an SEI can also be analysed. Moreover, other methods like 23Na NMR can also help in the characterization of SEI formation.
However, the detailed SEI formation behaviours are quite complex because they are obviously affected by numerous factors, from the properties of the electrode materials to the properties of the electrolytes. More effort is needed to figure out the mechanism of how the SEI is formed during the initial cycles and how the structures of hard carbons affect the formation of an SEI, thus figuring out a suitable solution to achieve a high ICE at the first cycle and a stable SEI during subsequent cycles.
4. Hard carbons for sodium-ion batteries and other sodium-based energy storage
4.1. Biomass-derived hard carbons
Biomass is a class of abundant, sustainable, low-cost and economical precursors, usually containing plenty of C content with some O, H and even some other heteroatoms such as N, S, P, etc. Biomass is a great choice as a renewable and sustainable precursor for producing low-cost and high-performance hard carbon anode materials for SIBs. The conversion of biomass into hard carbons is based on simple strategies such as direct carbonisation [11, 66, 93], hydrothermal carbonisation (HTC) [91, 94, 95], and physical or chemical activation [48, 96], etc.
The first investigation of hard carbon as an anode in SIBs was proposed by Dahn et al [62], using glucose as a precursor. A classical work about hard carbon anode materials with an excellent rate capability achieved by an synthetic hollow nanosphere structure was proposed by Maier and White et al [90]. The hollow carbon nanospheres were made from mixed poly(styrene) and D-glucose dispersion by HTC at 180 °C for 20 h. After washing, the carbon products were carbonised at 1000 °C to remove the polymer template and hollow carbon nanospheres were obtained; their morphology is shown in figure 6(a). The hollow carbon nanospheres were able to deliver a specific capacity of 223 mAh g−1 at 50 mA g−1 with only a sloping region. However, due to a high surface area of 410 m2 g−1, the hollow carbon nanospheres had a very low ICE of 41.5%. More importantly, this hard carbon material showed a very superior rate capability that offered 168, 142, 120, 100, 75 and 50 mAh g−1 at current densities of 0.2, 0.5, 1.2, 5 and 10 A g−1, respectively (figure 6(b)). The authors explained and concluded the reasons for the good rate performance were as follows: (1) the well-connected hollow structures were beneficial for electron transport; (2) a large number of active sites and a good charge transfer reaction were obtained due to the large electrode/electrolyte contact area; (3) the bigger interlayer space was good for sodium storage; (4) the thin shell thickness led to a short Na+ ion diffusion distance. Subsequently, lots of studies about hollow carbon nanospherical materials and their applications for energy storage have been reported [97–100]. Lately, Wan, Cao and Hu et al [71] successfully synthesised multi-shelled hollow carbon nanospheres (MS-HCNs) as high-performance sodium-ion battery anodes. The synthesis strategies were described in the previous report [101]. It was found that with an increased number of shells, the specific capacity would also increase, whereby 4S-HCNs were able to deliver the highest specific capacity and rate capabilities of 360 and 200 mAh g−1 at 30 and 600 mA g−1, respectively.

Figure 6. (a) An SEM image and (b) the rate capability (from 0.1 to 10 A g−1) of a hollow carbon sphere. [90] John Wiley & Sons. Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) The initial discharge/charge curves of hard carbons carbonized at different ramp rates. [91] John Wiley & Sons. © 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (d)–(e) Galvanostatic discharge/charge profiles of various biomass precursors and cellulose pre-treated at different temperatures, respectively. (f) A schematic of the microstructural changes of cellulose due to preheating treatment. Reproduced from [92] with permission of The Royal Society of Chemistry. (g) A schematic diagram of the synthesis of cellulose-derived carbon dots (CCDs) and cellulose-derived HTC carbon spheres (CHTCs) from the supernatant and deposit of a hydrothermal carbonisation (HTC) process. (h)–(i) The initial discharge/charge profiles of CCD and CHTC carbonized at 1000 and 1300 °C, respectively. Reproduced from [77] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageA series of low-defect and low-porosity hard carbon (HC) materials derived from sucrose have been investigated, correlating carbon structures with electrochemical performance at different heating rates (five, two, one and 0.5 °C min−1, denoted as HC-5, HC-2, HC-1 and HC-0.5, respectively). Lower heating rates proved to give rise to relatively more ordered structures and smaller specific surface areas, resulting in specific capacities of 338, 344, 350, and 361 mAh g−1 with an ICE of 78.9%, 81.3%, 85.2% and 86.1%, respectively (figure 6(c)), indicating that the lowest heating rate would also lead to the highest specific capacity and initial Coulombic efficiency. The authors concluded that the lowest defect concentration and nondetectable porosity were also the reasons that the HC-0.5 delivers the highest specific capacity and ICE [91]. Komaba and co-workers [92] have investigated hard carbon anode materials made from various biomass precursors, including glucose, sucrose, maltose, cellulose, glycogen and amylopectin. All the precursors were first pre-heated at 180 °C for 12 h in air and then carbonised to 1300 °C in Ar, while cellulose was also carbonised to 1300 °C with different pre-heating temperatures. The corresponding galvanostatic discharge/charge curves at 25 mA g−1 are shown in figures 6(d)–(e). It is obvious that the cellulose-derived hard carbons delivered the best performance, and a pre-heating temperature of 275 °C further enhanced the specific capacity. It was concluded that a high-temperature pre-heating treatment can lead to dehydration and a highly cross-linked structure and interrupted graphitisation during the high-temperature carbonization process, which results in larger pores and interlayer spacing and provides more sodium storage sites.
HTC is a powerful technology for converting biomass precursors into spherical carbon materials which can be applied for energy storage upon further carbonisation [6, 95, 102]. An HTC pre-treatment plays an important role for the production of biomass-derived hard carbon anode materials in SIBs, especially for sucrose, which would suffer a foaming process under direct carbonisation [67, 103], and HTC-treated hard carbons do show great sodium storage performance according to the literature [61, 104, 105]. Very recently, Titirici et al [77] have reported that not only can the conventional carbon spheres from the precipitate of the HTC process be used as SIB anodes, but the supernatant from the HTC autoclave containing carbon dots also exhibits excellent sodium storage performance upon drying and carbonization (figure 6(g)). In particular, cellulose-derived carbon dots (CCDs) from the liquid phase of HTC can deliver much higher specific capacity and an ICE of 266 mAh g−1 and 82% at 0.1 C, respectively, compared with the cellulose-derived HTC carbon spheres (CHTCs) at 1000 °C figure 6(h). CCDs carbonized at 1300 °C show a comparable reversible capacity of over 300 mAh g−1 but still an ultrahigh ICE of 91% (see figure 6(i)). The same trend for a significantly enhanced ICE also exists in the case of glucose, suggesting the universality of this discovery, which provides a new aspect of the design of hard carbon anode materials for SIBs.
There is another class of hard carbon materials, porous carbons, made through templating strategies such as chemical activation [48, 106], physical activation [96] and salt-templating [107], etc. Many biomass-derived porous carbons have been used as anodes in SIBs, which present excellent performance [108–114]. However, a serious problem with such porous materials is the ultralow ICE caused by the electrolyte decomposition and SEI formation due to the very high specific surface area, which significantly limits their future potential for SIBs. Further consideration of improving the ICE of porous carbons is necessary and important.
In addition to the aforementioned biomass precursors, banana peels [115], peat moss [64], rice husks [93], cotton [66], glucose [116], protein [110], and cellulose nanocrystals [117], etc. have all been utilised as anode materials for SIBs showing good electrochemical properties. A high-temperature-induced hard carbon derived from charcoal (made from wood) was reported to deliver an ultrahigh specific capacity of ∼400 mAh g−1 [118], suggesting the great potential of biomass to facilitate the development of anodes in SIBs. Nevertheless, on the other hand, biomass from Nature usually contains some impurities that need to be removed before it can be applied for SIB anodes. The most-reported biomass-derived hard carbon anodes can deliver reversible capacities of up to 300 mAh g−1 with an ICE below 85%, which still cannot compete with the graphite anode in commercial LIBs. Although biomass has its sustainability and abundance, the general cost is still higher than that of graphite and soft carbons [119]. Therefore, a long journey is still required for biomass-derived hard carbons to be used in commercial high-performance SIBs.
4.2. Heteroatom-doped hard carbons
The introduction of heteroatoms is considered to be an effective approach for enhancing the electrochemical properties, surface wettability, electronic conductivity, and benefitting the charge transfer and electrode/electrolyte interaction for carbonaceous anode materials [61, 120]. The most commonly doped heteroatoms include nitrogen (N), boron (B), sulphur (S), and phosphorus (P), wherein the N- and B-dopants are considered substitutional dopants, whereby these heteroatoms substitute for carbon atoms in the lattice, while S- and P-dopants are interstitial dopants which are introduced into the interlayer spaces [121]. The strategies for the synthesis of heteroatom-doped carbons typically include the carbonization of heteroatom-containing precursors or the mixture of carbon precursors with heteroatom sources (in situ doping), as well as the carbonisation of carbon precursors in the presence of heteroatom-containing gases such as NH3, H2S, etc., which is called postdoping [121].
4.2.1. Nitrogen- and boron-doped hard carbons
N-doped hard carbons are the most widely investigated heteroatom-doped carbonaceous anode materials for SIBs due to their simple synthesis and fabrication. N-doping in hard carbon materials can increase the electrochemical activity, electronic conductivity, surface wettability and interlayer spacing. N is also able to provide more extrinsic defects meaning that more Na+ ions can be adsorbed [122]. Typically, there are three types of doped nitrogen on the carbon lattice, pyridinic-N (N-6), pyrrolic-N (N-5) and graphitic-N or quaternary-N (N-Q), which are shown in figure 7(a). It has been reported that pyridinic-N has a lone pair of electrons which are parallel to the graphene planes, while the long pair of electrons of pyrrolic-N are perpendicular to the graphitic planes, which is beneficial for enlarging the interlayer spacing (figure 7(b)) [123].

Figure 7. (a) A schematic of N-5, N-6 and N-Q configurations. Reproduced from [122] with permission of The Royal Society of Chemistry. (b) A schematic of the impact of pyridinic-N and pyrrolic-N on the enlargement of the interlayer spacing. [123] John Wiley & Sons. © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) The diffusion path of the Na+ ions. (d)–(e) The deformation charge density (DCD) and density of states (DOS) results for S-doped carbon. Reproduced from [124] with permission of The Royal Society of Chemistry. (f) The galvanostatic discharge/charge profiles of HC, P-HC, S-HC and P-HC at 20 mA g−1. [80 John Wiley & Sons. © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (g) The reaction models of the different types of oxygen functional groups with Na+ ions. Reproduced from [125] © 2018 The Electrochemical Society. All rights reserved. (h) The correlation between the ratios of different types of oxygen functional groups and the carbonisation temperatures. Reprinted with permission from [126]. Copyright (2020) American Chemical Society.
Download figure:
Standard image High-resolution imageHuang and co-workers [127] used polypyrrole (PPy) as a N and C precursor and carbonised it at 600 °C under N2 to produce functionalised N-doped carbon nanofibers (FN-CNFs). The weight content of the N was measured to be 13.93% using XPS. The resulting FN-CNFs delivered 172 mAh g−1 at 50 mA g−1 and 87 mAh g−1 at 20 A g−1, with a capacity retention of 88.7% after 200 cycles at 200 mA g−1. The authors claimed that the O- and N-containing functional groups could accelerate the redox reactions on the surface. N-doping could create more topological defects and enlarge the interlayer spacing, which provides more sodium storage sites and a high rate performance. Zhang et al [128] reported the use of bacterial cellulose (BC) together with PPy to synthesise carbon nanofibres within N-doped porous carbon (CNF@NPC) as high-performance anodes for SIBs. Compared with pure CNF, the CNF@NPC shows a much better rate and cycling performance. A PPy and graphene oxide (GO)-derived N-doped carbon nanosheet has also been reported to have the excellent electrochemical properties of 350 mAh g−1 at 50 mA g−1 as an anode in SIBs [129]. Wang and Lou et al [130] have reported a free-standing N-doped nanofiber film, produced by electrospinning and subsequent carbonisation. This material was able to deliver 315 mAh g−1 at 0.5 A g−1 and 154 mAh g−1 at 15 A g−1, indicating a superior rate capability. It also has an excellent cycling stability and the capacity was retained at 210 mAh g−1 after 7000 cycles at 5 A g−1. Guo, Yang and Wang et al [131] reported on a directly carbonized chitin as N-doped amorphous carbon nanofibers (NACFs) for high-performance SIBs. Chitin is the second most abundant biomass in the world and is usually utilised as a nitrogen source for various applications [5, 132, 133]. The NACFs were directly carbonized from 500 °C to 900 °C in Ar. The best sample was carbonized at 700 °C (CC700) showing a high surface area of 369.48 m2 g−1 and a high N content of 7.29%. It was found that with an increasing carbonisation temperature, the N atoms were converted into pyridinic and graphitic N. According to the XPS result, the CC700 also contained some oxygen groups such as C = O (quinone-type and O–I) and C–OH (phenol-type, O–II), which also favoured the sodium storage performance. Furthermore, the nanofibrous structure was able to create fast electron transport channels. As a result, the CC700 delivered reversible capacities of 320.6, and 120.6 mAh g−1 at current densities of 50 and 1000 mA g−1. A capacity retention of 85% was obtained at 1000 mA g−1 after 8000 cycles, indicating an excellent rate and cycling performance.
B-doping is another substitutional doping strategy for enhancing the electrochemical properties of hard carbon anode materials in SIBs by introducing a p-type doping character to reduce the Fermi level [120, 134]. Cao and co-workers [135] found that every boron atom can adsorb three Na atoms due to the hybridized orbitals with carbons. The sodium adsorption energy on B-doped graphene was calculated to be 2.7 and 7.1 times of the intrinsic energy of N-doped graphene, respectively, via the first-principles density functional theory (DFT), indicating much more stable sodium storage on B-doped carbons. Barone et al [136] have also studied the intercalation of Na+ ions into BC3 (NaxBC3) by dispersion-corrected DFT, showing that the most stable intercalation compound for Na is NaBC3 which corresponds to a theoretical capacity of 572 mAh g−1. The sodiation energy of B-doped graphene is calculated to be ∼0.92 V. Boron is a p-type dopant which prefers to accept electrons from the Na+ ion, which means it confers a stronger and stable binding between the doped B and the Na+ ions [80]. However, although the B-doped carbons exhibit great potential as sodium storage electrode materials, there are still not many reports about B-doped hard carbons for SIBs since the fabrication and synthesis methods are relatively harder, compared to other heteroatoms. It was also found that B-doping will introduce high-energy in-plane defects and cause significant irreversibility during the first desodiation process [80], which requires specific consideration in the future.
4.2.2. Sulphur- and phosphorus-doped hard carbons
S- and P-doping have also been proved to have positive effects on sodium storage for hard carbon anode materials. Unlike N and B, S and P have much larger covalent radii and can fit between the graphene layers in most cases, which will significantly increase the interlayer spacing. In addition, the S and P can not only enhance the electronic conductivity, but also reversibly adsorb Na+ ions and provide additional sodium storage capacity [120, 121].
The most commonly used sulphur source is elemental S, which can be easily mixed and carbonized together with hard carbon precursors. An experimental and theoretical study of S-doped carbon nanofibers (S-CNFs) made by simply mixing bacterial cellulose (BC) with elemental S with a mass ratio of 1:2 and carbonising at 500 °C in Ar was reported by Jiang and Wang et al [124]. The S-CNFs showed a high S content of 17.44 wt.% and an excellent rate capability of 355 and 257 mAh g−1 at 0.05 and 8 A g−1, respectively, with reversible electrochemical reactions between the –C–S–C– bonds with the Na+ ions. Theoretical calculations indicated that the interlayer spacing was enlarged and the diffusion barrier was reduced due to the S doping (figure 7(c)). In addition, the deformation charge density (DCD) and density of states (DOS) in figures 7(d)–(e) show that S-doping also improves the electronegativity and electrochemical activity as well as providing more defects, which results in a smaller band gap, which are all beneficial for enhanced sodium storage performance. Biomass-derived S-doped carbon nanosheets (S-CNs) were also synthesised by direct carbonization in Ar upon mixing a biomass precursor (spring onion peel) and elemental S. The resulting S-CNs can deliver ultrahigh reversible capacities of ∼601.2, 545.2, 220.1 and 133.6 mAh g−1 at current densities of 0.05, 0.1, 5 and 10 A g−1, respectively, which are much higher than those of undoped samples. A high capacity of ∼211 mAh g−1 at 5 A g−1 after 2000 cycles was obtained, indicating a great cycling performance [137].
It has been reported that elemental S would lead to unstable configurations in the doped carbon layers, which would make the electrochemical reactions similar to those of Na-S batteries [138]. Other than elemental S, some other S sources have also been investigated for use as a dopant to produce S-doped hard carbons for SIBs. Huang and co-workers [139] pyrolyzed prepared poly(3,4-ethylenedioxythiophene) (PEDOT) at 700 °C in Ar and obtained S-doped carbon (SC) with an enlarged interlayer spacing and a high content of S (15.17 wt.%). Owing to its small surface area of ∼40 m2 g−1, the ICE (73.6%) was relatively higher than for other heteroatom-doped carbons. As an anode in SIBs, SC shows excellent stable reversible capacities of 327.8 and 119.5 mAh g−1 at 0.5 and 5 A g−1 with a retained capacity of 322 mAh g−1 at 0.5 A g−1 after 700 cycles. Hou and co-workers [140] used 2-thiophenemethanol as a S source to synthesise with durian shell-based activated carbon. The resulting S-doped carbon can have a remarkable capacity of 100.02 mAh g−1 at 5 A g−1 after 4500 cycles. CS2, Dimethyl disulphide, Na2S, H2S, and SO2 can also be the S sources for the fabrication of S-doped carbons by either being mixed with the carbon precursors or calcinating the precursors in a S-containing gas atmosphere [141]. S-containing precursors have been found unable to provide sufficient doping, while the major advantage is to increase the d-spacing [138]. S-containing gases cause environmental issues and the doping level is hard to control. In addition, the doped S in the carbon materials is usually located at the defects and edges in a thiophene-type structure [120], therefore selecting proper S sources and synthesising the S-doped hard carbons with a homogenous S distribution would be important aims.
Phosphorus, with low electronegativity and high electron-donating properties can provide more electrochemical active sites and extrinsic defects, and has also attracted enough attention as an alternative doped heteroatom to enhance the sodium storage performance of hard carbons. A comparative study of P-, S- and B-doped hard carbons (P-HC, S-HC and B-HC) was made by using H3PO4, H2SO4 and H3BO4, respectively, with the same doping level of 5 wt.%. Consequently, the P-HC showed more defects and curvatures that may be present because the doped POx is not coplanar with the graphene layers, and it delivered the highest specific capacity of 359 mAh g−1 at 20 mA g−1 compared with other HCs (figure 7(f)) [80]. Ji's group [142] has reported on P-doped large-area carbon nanosheets (P-CNSs) obtained by the conversion of 0D carbon dots (CDs) to 2D nanosheets doped with P. The P-CNSs obtained showed a P content of 1.39% and delivered a high reversible capacity of 328 mAh g−1 at 0.1 A g−1. This material also had an excellent cycling stability of 149 mAh g−1 at 5 A g−1 after 5000 cycles. Recently, Liu, Yang and Lu et al [143] synthesised a 3D well-ordered porous P-doped carbon with phosphoric acid as the P source, which exhibits excellent specific capacities of 270 mAh g−1 at a low current density of 0.2 A g−1 and 140 mAh g−1 at a very high current density of 10 A g−1. The extraordinary sodium storage behaviour was concluded to be due to the larger distance between the graphitic layers, the modified electronic structures and the enhanced Na+ ion adsorption and diffusion kinetics.
On the other hand, it was also reported that phosphorus is an n-type dopant that is reluctant to accept the electrons from Na, which leads to an unstable binding energy between the doped P and the Na [80]. The large radius of phosphorus usually makes the doping more difficult compared to other heteroatoms [120]. Therefore, consideration of both the advantages and disadvantages would be necessary to design proper P-doped hard carbons in the future for high-performance SIBs.
4.2.3. Oxygen- and multiatom-doped hard carbons
Doping with oxygen is usually not regarded as a heteroatom-doping strategy for hard carbon materials because there is always a great amount of intrinsic O contained in the hard carbon precursors. These O-containing functional groups will inherently be retained after carbonisation and in most cases, it is not necessary to dope additional O through other routes. These O-containing groups in hard carbon materials have a strong impact on sodium storage behaviours, which cannot be ignored and require more attention to understand the sodium storage mechanisms and design high-performance hard carbon anodes.
The capacity contributed by the voltage range over 0.1 V (vs. Na+/Na) is believed to correspond to the reactions between the Na and O in most O-doped carbons [144]. Song and Jia et al [125] prepared N-doped carbon nanospheres with different O contents and observed the effect of the O-containing groups on the sodium storage. As shown in figure 7(g), the authors found that the C = O groups are very stable and the sodiation/desodiation is mostly reversible between the Na+ ions and the C = O groups. The C–OH, C–O–C and COOH groups were found to be able to provide more defects and enlarge the interlayer spacing. A higher content of such groups led to higher reaction activity and a higher Na adsorption capacity. Nevertheless, these types of O-containing functional groups also caused strong irreversibility. In a recent study proposed by Huang and co-workers [126], the C = O bond was reported to improve the adsorption capacity of hard carbons in the case of buckwheat husk-derived hard carbons (BPC). BPC carbonised at 1100 °C showed the best performance of 400 mAh g−1 and 116 mAh g−1 at current rates of 0.05 and 2 A g−1, respectively, with a capacity retention of 96% at 2 A g−1 even after 3000 cycles. It was found that with an increase in the carbonisation temperature from 700 °C to 1300 °C, the ICEs of the resulting BPCs also increased from 42% to 72%. Correlated with the ratios of C–O and C = O groups in the BPCs carbonised at different temperatures shown in figure 7(h), the C = O content increased while the C–O increased, suggesting that the C–O groups will cause irreversible loss of capacitiy in the first few cycles.
As opposed to the monoheteroatom doping of carbons, multiatom doping is able to combine the advantages of the single heteroatom with increased electrochemical activity in hard carbon anode materials in SIBs. Jiang and Wang et al [145] synthesised 2D N/B dual-doped carbon nanosheets (NBTs) from gelatin and H3BO3. NBTs show pseudocapacitive-dominated sodium storage properties and the performance was much better, compared with samples containing N doping only. The same group also reported on a N/S co-doped hard carbon using sodium citrate and thiourea. As a comparison, undoped carbon from sodium citrate only and a N-doped carbon from sodium citrate and urea were also produced. It was found that the dual-doping significantly enhanced the electrochemical properties of the hard carbon compared with undoped and single N-doped carbons [146]. Zhou and co-workers [138] have reported on a N/S dual-doped carbon nanosheet for SIBs and studied its performance with and without sulphur doping. With the S doping, the material had a larger interlayer spacing and more defects, presenting a better rate and cycling performance compared to the carbon nanosheet that was only N doped. The sodium storage performance of N/P co-doped carbon microspheres (NPCM) was studied by Li and Peng et al [147]. With heteroatom-doping, the NPCM showed a decreased surface area, more defects and enlarged interlayer spacing. In addition, owing to the spherical morphology, the NPCM delivered a much higher specific capacity and rate capability, compared with undoped carbon.
In general, heteroatom-doping is indeed a popular strategy for enhancing the electrochemical properties of hard carbons by increasing the electronic conductivity and wettability, changing the Fermi level, enlarging the interlayer spacing, and providing more extrinsic defects and functional groups for more sodium adsorption sites. Heteroatom-doped hard carbons usually have highly surface-controlled and capacitive capacities and exhibit only sloping regions in their voltage profiles, ensuring higher safety, fast rate capability and stable cycling performance. However, on the other hand, the introduced defects, heteroatom functional groups and the large surface areas from the low-temperature calcination also cause serious irreversibility during the first cycles and thereby a low ICE. In addition, the synthesis methods of the heteroatom-doped hard carbons are relatively more complicated compared with undoped ones, which may increase the cost and limit the commercialisation process of this kind of material for next-generation SIBs. On the other hand, in some cases, it is still not very clear how each heteroatom specifically contributes to the sodium storage performance and there have been some conflicting reports. A deeper understanding of the exact role of each heteroatom must still be established in further works.
4.3. Other hard carbons
There are still a lot of hard carbon precursors and synthetic strategies that contribute to the preparation of high-performance hard carbon anode materials in SIBs, which will be summarised in this section. For example, Ji and co-workers [148] used acetone and NaOH to synthesise carbon quantum dots (CQDs) and eventually obtained derivative 3D porous carbon frameworks (3D PCFs) upon calcination in an Ar atmosphere. Figures 8(a)–(b) show the scheme of the synthesis strategy and a TEM image of the CQDs, respectively. Owing to the 3D porous frameworks with a large interlayer spacing, more contact surface between the electrode and electrolyte, the short Na+ ion diffusion distance and buffered volume expansion during cycling, the resulting 3D PCFs can deliver an excellent rate capability of 290 and 90 mAh g−1 at 0.2 and 20 A g−1 as well as an ultralong cycle life of 99.8 mAh g−1 retained at 5 A g−1 after 10 000 cycles. Another study showed the use of D-sodium ascorbate as a carbon precursor and Na2CO3 as a salt template to prepare defect-rich porous carbons pyrolyzed at 600 °C–800 °C, from among which the 600 °C sample presented the best electrochemical properties. It had capacities of 280, 229, 130 and 126 mAh g−1 at current densities of 0.05, 0.1, 10 and 20 A g−1, respectively, with a high retained capacity of 115 mAh g−1 after 15 000 cycles at 10 A g−1, indicating superior rate and cycling performance. The b-value calculated, based on the equation i = aυb from the cyclic voltammetry (CV) results was able to achieve 0.82, indicating a high ratio of surface-controlled pseudocapacitive reactions [151].

Figure 8. (a)–(b) A schematic of the synthesis route and an SEM image of carbon quantum dots. [148] John Wiley & Sons. © 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) The galvanostatic discharge/charge curves at 10 mA g−1 of hard carbons carbonised at various temperatures. Reprinted with permission from [149]. Copyright (2019) American Chemical Society. (d) A schematic of the synthesis process of hard carbons using ethanol (EtOH) as a pore-forming agent. (e) The galvanostatic charge/discharge profiles at 0.1 C and 0.2 C of the NaNi1/3Fe1/3Mn1/3O2//HC-21-1400 full cell. Reprinted with permission from [150]. Copyright (2019) American Chemical Society.
Download figure:
Standard image High-resolution imageLately, Komaba et al [149] reported a series of microporous phenolic resin-derived hard carbons with ultrahigh reversible capacities. The hard carbons were synthesised by using a resin as the carbon precursor and an organic polymer as a pore-forming additive. Formalin and maleic acid as crosslinker and catalyst, respectively, were also added for the preparation. After a series of curing, washing and carbonisation processes, hard carbons were obtained. It was found that the surface areas decreased, and the pore sizes increased with increased carbonisation temperatures. Some of the open pores were changed to closed pores by heat treatment above 1500 °C. As shown in figure 8(c), the resulting hard carbons at 1100, 1300 and 1500 °C were able to deliver reversible capacities of 337, 358 and 386 mAh g−1 with the corresponding ICEs of 82%, 81% and 85%, respectively. Hu et al [150] also reported a resin-derived hard carbon by tuning the closed pores for the highest sodium storage capacity. Phenol-formaldehyde resin (PF) was selected as a carbon precursor and ethanol (EtOH) was added as the pore-forming agent. The ratios of PF to EtOH were adjusted for the solvothermal process and the obtained intermediate products were further carbonised at different temperatures to fabricate the final hard carbon materials with closed pores (figure 8(d)). The optimal sample with a PF to EtOH ratio of 2:1 carbonised at 1400 °C had the best performance of 410 mAh g−1 at 0.1 A g−1 with a high ICE of 84%, and the full cell with NaNi1/3Fe1/3Mn1/3O2 as a cathode was able to reach a superior energy density of 300 Wh kg−1.
4.4. Hard-soft carbon composite materials
Hard and soft carbons have both been widely investigated as sodium-ion battery anodes. Soft carbons cannot usually achieve as good a performance as hard carbons (unless properly modified or pre-treated) due to their relatively ordered structures and short interlayer spacing, which is not suitable for Na+ ion intercalation. Hard carbons are usually able to deliver high-performance anodes for SIBs, but their lower carbon yield, larger irreversible capacity (especially when carbonised at low temperatures) and higher cost compared to soft carbon precursors still limits further commercialisation. Table 2 shows the prices and carbon yields of various carbon precursors according to the literature [119, 152]. Therefore, designing hard-soft carbon composite materials would be a smart option to combine the advantages of both and get high-performance and low-cost anode materials for SIBs.
Table 2. The prices and carbon yields of various carbon precursors.
| Precursor | Category | Price ($/ton) | Carbon yield |
|---|---|---|---|
| Graphite | Graphite | 500–2000 | 100% |
| Pitch | Soft carbon | 300 | 56% |
| Anthracite | Soft carbon | 50–200 | 90% |
| Resin | Hard carbon | 2000 | ∼40% |
| Cellulose | Hard carbon | 1000 | <10% |
| Sucrose | Hard carbon | 400 | <10% |
| Starch | Hard carbon | 500 | <10% |
| Lignin | Hard carbon | 450 | 43% |
| Hydrocarbons | Hard carbon | 300 | ∼10% |
Hu and co-workers [94, 153] have reported the production of hard-soft carbon composite materials for SIBs by synthesising pitch with phenolic resin and lignin, respectively. The hard and soft carbon precursors were mixed, and ball milled before carbonisation (figure 9(a)). By adjusting the ratios of pitch to the hard carbon precursors as well as the pyrolysis temperatures, optimal samples were obtained. For the pitch/phenolic resin system, the sample with a ratio of pitch to resin of 3:7 carbonised at 1400 °C (PPAC371400) showed the best performance of 284 mAh g−1 at 0.1 C with a high ICE of 88%. In terms of the pitch/lignin system, the same amount of soft and hard carbon precursors and a carbonisation temperature of 1400 °C (AC111400) were the optimised conditions. When paired with a Na0.9[Cu0.22Fe0.30Mn0.48]O2 cathode [154], the full cell could deliver a high specific capacity of 240 mAh g−1 and a high energy density of 207 Wh kg−1 (figure 9(b)). Another study also reported a hard-soft carbon composite material made from pitch and phenolic resin. With a NaCl template, the resulting sample (3DAC) had a 3D structure. This 3DAC exhibited more capacitive contribution compared to carbon without 3D structures, which is valuable for greater rate performance [155].
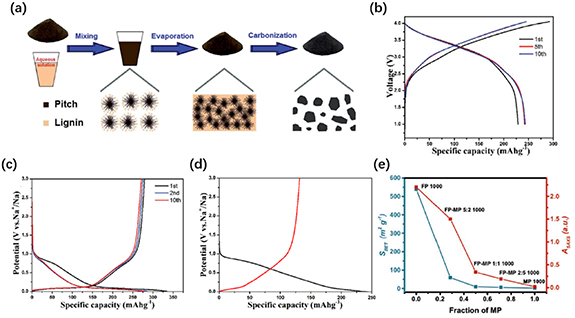
Figure 9. (a) A Schematic of the synthesis route for pitch/lignin-derived hard-soft carbon composites. (b) The galvanostatic charge/discharge curves of a Na0.9[Cu0.22Fe0.30Mn0.48]O2//AC111400 full cell at 0.2C. Reproduced from [152] with permission of The Royal Society of Chemistry. (c)–(d) The galvanostatic discharge/charge curves of hard carbon microspherules with and without a soft carbon coating. Reproduced from [94] with permission of The Royal Society of Chemistry. (e) A comparison between the Brunauer-Emmett-Teller (BET) surface area and the A values fitted from SAXS results. [11] John Wiley & Sons. © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution imageSoft carbons are also usually used for coating the surface of hard carbons and therefore enhancing the sodium storage performance. A series of monodispersed hard carbon spherules (HCS) was produced from sucrose. Interestingly, when carbonised in Ar and a toluene flow at 1000 °C, the HCS1000 delivered a high reversible capacity of 279 mAh g−1 at 0.1 C with a high ICE of 83%, while HCS1000 without a soft carbon coating showed a lower capacity and ICE (figures 9(c)–(d)) [94]. Wang and co-workers [156] also reported the coating effect of soft carbon from coal tar pitch that increased the reversible capacity and the ICE, compared to uncoated hard carbon. Recently, Titirici et al [11] found that a hard-soft carbon composite material made from filter paper (FP) and mesophase pitch (MP) had a synergistic sodium storage performance compared with pure hard and soft carbons prepared in the same conditions. The decrease of the A-values from small-angle x-ray scattering (SAXS) (corresponding to both the open and closed large pores on the surface according to the literature [68]) were not able to scale with the Brunauer-Emmett-Teller (BET) surface areas with the increased ratio of MP (figure 9(e)), suggesting that some pores were inaccessible to N2 adsorption, which means that some open pores were blocked by the addition of MP. Therefore, the blocked open pores reduced the contact area between the electrode and electrolyte and suppressed the SEI formation, which favours the synergistic effect.
4.5. Further applications of hard carbons in sodium-based energy storage
The versatility of hard carbons attracts attention not only for their use as anode materials in SIBs but also extends to many sodium-based electric energy storage (EES) devices, such as sodium-ion capacitors, dual-ion batteries (DIBs) and sodium-ion hybrid capacitors. More functional roles of hard carbons as protective and supportive materials are revealed in traditional SIBs as well as next-generation batteries (e.g. sodium metal batteries, Na-S batteries and sodium-air batteries).
4.5.1. Hard carbons for sodium-ion capacitors
A sodium-ion capacitor (SIC) is an energy storage device consisting of a battery-type anode and a capacitor-type cathode, leading to a balance between high-energy sodium-ion batteries and high-power supercapacitors. This occurs because the diffusion-controlled Faradic reactions in the body phase of the anode and the surface-controlled non-Faradic reaction on the surface of the cathode kinetically constrain each other [159]. In most cases, SICs employ metal oxides as anode materials and activated carbons as cathode materials, which are both usually fabricated using critical materials such as Co, Ni and fossil fuels. Moving towards a more eco-friendly and low-cost energy storage system, hard carbons from biomass have been widely investigated as electrode materials for sodium-ion capacitors [160]. Compared with metal oxides, low-cost hard carbons can also exhibit excellent capacities when they are employed as anodes because of the special storage features of sodium ions which have been mentioned above. When it comes to the cathode, although activated carbons from fossil fuels have been considered to be the default choice, porous hard carbons activated from biomass, in fact, possess more advantages including low-cost, tuneable porosity and surface chemistry. The state-of-the-art rational design strategy of hard carbons for both anode and cathode materials has shown promising application in sodium-ion capacitors.
Titirici and co-workers [48] utilized cellulose as a precursor to rationally synthesize both anode and cathode materials, thus assembling an all-cellulose-based sodium-ion capacitor (figure 10(a)). The anode materials were prepared by the carbonization of cellulose nanocrystals and then formed a non-porous but disordered structure with a reversible sodium storage performance of 256.9 mAh g−1 at 0.1 A g−1, while for the cathode part, carbon materials with hierarchically porous structures (2137 m2 g−1) which were activated from cellulose microfibrils using KHCO3 and Melamine co-activation were able to exhibit a discharge capacitance of 212.4 F g−1 at 0.1 A g−1. This project reasonably took advantage of the inherent properties of the hierarchal building blocks of cellulose to achieve a sustainable sodium-ion capacitor with an optimized electrochemical performance. Utilizing Na+ and ClO4− as charge carriers, a full Na-ion capacitor with two asymmetric carbon electrodes was able to reach an energy density of 181 Wh kg−1 at a power density of 250 W kg−1 (figure 10(b)). In addition, Ding et al [157] prepared carbon materials with specific properties from different parts of peanut shells by corresponding methods, based on their understanding of the natural characteristics of peanut shells. For instance, the inner shells of peanuts, made of lignin with disordered repeating units and three-dimensional structures, can lead to an amorphous carbon whose distance between graphene layers is expanded after carbonization. This specific structure can reduce the reaction barrier to Na ion insertion and achieve an enhanced electrochemical performance of 315 mAh g−1 at 0.1 A g−1. However, the outer shell of the peanut is mainly composed of cellulose microfibrils which can be converted into a graphene-like analogue through hydrothermal treatment and chemical activation. The porous carbons achieved, with a high surface area of 2396 m2 g−1, delivered a specific capacity of 161 mAh g−1 at 0.1 A g−1. The assembled sodium-ion capacitors were able to deliver an energy density of 201 Wh kg−1 when the power density was 285 W kg−1 (figures 10(c)–(d)). Therefore, sodium-ion capacitors become the trade-off between batteries and supercapacitors.
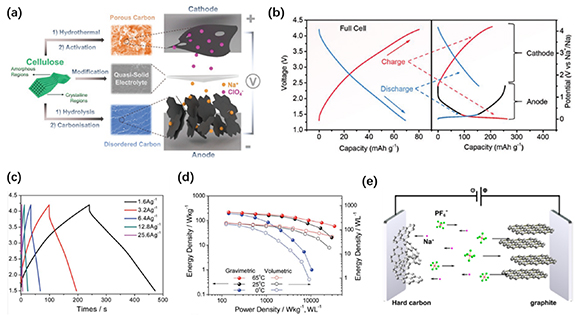
Figure 10. (a) A schematic illustration of an all-cellulose-based synthesis processes for electrodes as well as an asymmetric configuration of SICs. (b) The electrochemical performance of SICs with the corresponding charge–discharge processes in cathodes and anodes. [48] John Wiley & Sons. © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) The galvanostatic profiles of SICs from peanut shells. (d) A Ragone plot of SICs where the calculated energy and power densities are based on the total mass (solid) or volume (hollow) of the active electrodes. Reproduced from [157] with permission of The Royal Society of Chemistry. (e) A schematic illustration of the all-carbon dual-ion battery upon charging. Reprinted with permission from [158]. Copyright (2018) American Chemical Society.
Download figure:
Standard image High-resolution imageAlthough recent research has demonstrated some successful examples of high-performance sodium-ion capacitors, the drawback of fabricating the full cell configuration still needs further consideration because the pre-sodiation of anode materials is usually carried out in half cells against metallic sodium. Without a pre-sodiation process, the lack of charge carriers would cause the loss of reversible capacity and suppress the cycling performance. The most important research direction, therefore, is the in situ pre-sodiation of anode materials during the material preparation step [161]. To sum up, the successful application of hard carbons has explored the road to future commercialization of sodium-ion capacitors, but more scale-up experiments in pouch cells and deep study of their working principles are needed.
4.5.2. Hard carbons for sodium-based dual-ion batteries
Dual-ion batteries (DIBs) are regarded as one of the most promising alternatives to current Li-ion cells due to their comparable energy density. The working principle of DIBs is different from LIBs/SIBs, though. Hu et al [158] reported an all-carbon DIB working by a simple principle of having an electrolyte sandwiched between a hard carbon anode and a graphite cathode. DIBs show the feasibility of designing an all-carbon energy storage system delivering a specific capacity of over 60 mAh g−1 and a good cycling performance. Upon charging, Na+ cations migrate to the anode and are inserted into the hard carbon, while PF6 − anions intercalate into the graphite; upon discharging, Na+ and PF6 − ions move back to the electrolyte (figure 10(e)). The wide range of hard carbon families whose plateau region of both charge and discharge are usually below 0.1 V are suitable for building up a DIB of high energy density, thus hard carbons are categorised as a promising cation hosting material for DIBs. An all-carbon configuration can be achieved in a cell paired with a graphite cathode for anion intercalation (e.g. Hard carbon |1.0 M NaPF6 in a carbonate electrolyte | graphite). Such a carbon sandwich eliminates the use of any layered-metal oxides. The excellent thermal resistance of the carbon electrodes ensures the safety of all-carbon DIBs. The cathode materials in SIBs, typically exhibit a working voltage of ∼3.5 V, while DIBs can deliver an average working voltage of around 4.5 V. The increase in the working voltage may compensate for the limited capacity graphite cathode which has a theoretical capacity of 112 mAh g−1, resulting in a graphite intercalation compound of C20PF6 [162]. This strategy of assembling DIBs has been attracting increasing attention in the battery community in recent years and may also extend to multi-ion batteries in the near future with the hard carbons playing an important role [163].
4.5.3. Hard carbons as functional hosts for other electrode materials
Anode and cathode materials in conventional SIBs can benefit from hard carbon. The NASICON Na3V2(PO4)3 is among the most promising cathode materials for SIBs due to its conductive framework, large channels for transferring Na+ ions and excellent thermal resistance [167, 168]. The rate performance of Na3V2(PO4)3 is not ideal though, so coating hard carbon onto a Na3V2(PO4)3 core via a hydrothermally assisted sol-gel approach is one of the solutions to overcoming this disadvantage, improving the electrochemical performance [169–171]. Zhao and co-workers [170] applied conductive polyaniline (PANI) as a hard carbon coating via a facile sol-gel process onto the NASICON Na3V2(PO4)3 to form a composite. The hard carbon layer consisting of localised graphitised layers was an excellent conductive agent helping charge transfer [61]. After modification, the surface exhibited higher conductivity and protected the Na3V2(PO4)3 core. The double carbon-coating layer enhanced the conductivity and prevented the Na3V2(PO4)3 core from growing over cycling. The protective layer also helped to suppress the volume change during charging and discharging. The modified cathode delivered a discharge capacity of 111.6 mAh g−1 at 1 C, 61.0 mAh g−1 at 50 C and good long cycling performance over 15 000 cycles [170].
Regarding anodes in SIBs, hard carbons coupled with transition metal chalcogenide anodes is a trendy topic. Transition metal chalcogenides (TMCs), usually be denoted as MX2 (M = Ti, V, Nb, Mo, W, Re; X = S, Se, Te), are emerging energy storage technologies, since a weak van der Waals (vdW) force among their interlayers, owing to the layered structure of TMCs, is beneficial for Li+, Na+, and K+ ions to de-/intercalate [172]. TMCs usually have a higher theoretical capacity, for example, MoS2 is a promising anode for SIBs not only because of its high theoretical Na+ storage capability but also its ample space between 2D layers for accommodating more abundant ions [173]. However, TMC materials are not yet commercially possible, as their working principle (conversion reactions), volume expansion and unstable electrochemistry primarily hinder their electrochemical performances for charging and discharging. Therefore, the cycling and rate performance of TMC anodes cannot satisfy the current demands of the market. Hard carbons can be applied to restrict the volume expansion of TMCs, and hydrothermal carbonisation in this regard is an effective way to synthesise hard carbons. Ji and Zhang et al [174] constrained exfoliated MoSe2 inside carbon spheres with a diameter of 100 nm, building a hybrid structure. Polyvinyl pyrrolidone (PVP) was used as a carbon precursor which can easily be dissolved and chemically attached to the surface of MoSe2. Due to such a unique carbon layer, C–O–Mo bonds anchored on the surface, which was reported to activate the Se element generated during cycling and lower adverse reactions, promoting capacity, stability and rate properties. Such beneficial bonds are further confirmed by DFT models. Thanks to the carbon layer, the electrochemical results for modified MoSe2 in SIBs are better than for bulk MoSe2; after 120 cycles, the specific capacity retained was around 441 mAh g−1 at 1 A g−1 and 348 mAh g−1 at 4 A g−1.
Besides acting directly as active materials, hard carbons can also serve as a functional host for other kinds of active materials with different storage mechanisms including alloying and conversion. For example, hard carbons with stable chemical and physical properties can restrict the volume expansion of metal anodes by allowing the homogenous plating and stripping of metallic sodium on the surfaces of hard carbons [175]. Metallic sodium has been considered to be the ultimate anode material for sodium ion storage because of its high theoretical capacity of 1166 mAh g−1. However, during the charge and discharge process, sodium dendrites will be induced because the metallic sodium plating/stripping is a random and uncontrollable process, thus leading to a quick fading of batteries. Luo et al [164] prepared 3D hard carbon hosts with porous channels derived from wood to encapsulate the metallic Na anodes as well as suppress their dendrite formation (figure 11(a)). Pore and defect structures can induce the nucleation of metallic sodium and help it to plate and strip homogenously. Employing Na-hard carbon composite anodes, the cycling performance of symmetric half cells can be greatly improved, up to 500 cycles at 1.0 mA cm−2 (figure 11(b)). Zhu et al [176] also prepared 3D scaffolds from lignin with boron doping for metallic anode protection. Heteroatom-doping can form extrinsic defects on the graphene layers which can act as active sites for the initial nucleation of metallic anodes, thus further improving the homogenous plating and stripping of metallic anodes.
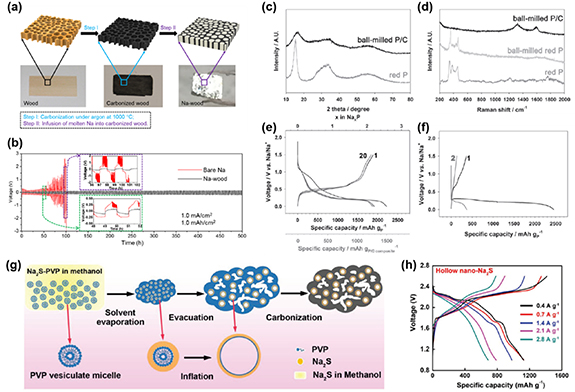
Figure 11. (a) Schematics illustrating the preparation steps for Na-hard carbon composite anodes. (b) The electrochemical performance of symmetric cells using Na − wood electrodes and bare Na metal electrodes at 1 mA cm−2 with a capacity of 1 mAh cm−2. Reprinted with permission from [168]. Copyright (2017) American Chemical Society. (c) The XRD patterns of bare red P and amorphous red P/C composite. (d) The Raman spectra of bare red P, ball-milled bare red P, and amorphous red P/C composite. The voltage profiles of (e) amorphous red P/C composite anodes, and (f) bare red P anodes. [165] John Wiley & Sons. Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (g) A schematic of the preparation procedures for Na2 S nanospheres embedded in a hollow hard carbon matrix. (h) The galvanostatic capacity versus voltage curves of Na2 S-hard carbon composite cathodes at current densities of 0.4, 0.7, 1.4, 2.1, and 2.8 A g−1. [166] John Wiley & Sons. © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution imagePhosphorus (P) is another promising anode with an ultra-high theoretical capacity of 2596 mAh g−1, but its intrinsically low conductivity and huge volume expansion in electrochemical reactions severely hinder its application in sodium battery anodes [177]. Hence, hard carbons with good conductivity can be utilized as a functional host for phosphorous. Oh and Lee et al [165] prepared P/hard carbon composite anodes by ball milling methods (figures 11(c)–(d)). A reversible capacity of 1890 mAh g−1 was obtained at 0.286 A g−1 since hard carbons can not only create conductive channels for composite anodes but also constrain the excess volume expansion of P (figure 11(e)). The composite anode also exhibited an excellent rate performance at a 10-fold current density, where the reversible capacity was able to remain at 1540 mAh g−1. Without hard carbons, bare P anodes only delivered a reversible capacity of around 400 mAh g−1 with a low ICE of below 25%. Furthermore, in the second cycle, the reversible capacity faded to only half of the previous cycle (figure 11(f)). This poor performance further proves that hard carbons play an important role in the improvement of the electrochemical performance of P anodes.
Hard carbons can also contribute to various advanced cathode materials for next-generation sodium batteries. One of the next-generation sodium batteries is sodium-sulphur batteries where S is used as a cathode material, while metallic sodium is used as the anode material as well as the sodium source in full cells. The reactions between the S and Na+ ions can be divided into several steps including the formation of Na2S8, Na2S4, Na2S2 and Na2S. The highest theoretical capacity of S cathodes can be 1675 mAh g−1 when Na2S2 and Na2S form. However, insulated S has a low reaction activity without any conductive agents and a huge volume expansion during the charge and discharge process. The high solubility of sulphur in common electrolytes causes instability in S cathodes and a loss of reversible capacity during cycling [178]. S cathodes, therefore, need to be combined with hard carbon as a functional host to prevent a sulphur shuttle effect and improve the reversibility of electrochemical reactions. Li et al [166] synthesized sodium sulphide (Na2S) nanospheres embedded in hard carbons with a hollow structure and high conductivity, which shortens the ion diffusion pathway (figure 11(g)). As a result, a high initial discharge capacity of 980 mAh g−1 at high current densities of 1.4 A g−1 can be achieved (figure 11(h)). After 100 cycles, the reversible capacity can stabilize at 600 mAh g−1. S-hard carbon composite cathodes can possess enhanced electrochemical reactivity by alleviating the volume change and shuttle effect of sulphur, resulting in excellent rate and cycling performance at various current densities.
Another kind of advanced cathode that can be employed for next-generation sodium batteries is the air cathode. The promising energy density of sodium-air batteries has widely motivated scientists to develop air cathodes. Sodium-air batteries utilize air from outside to participate in the reaction inside the batteries instead of the self-contained battery technologies mentioned above. High overpotential and a limited rate performance are two main restrictions of air cathodes, which result from the sluggish kinetics of oxygen reduction and evolution reactions (ORR/OER) [179]. Hard carbons with specific structures can be used as an effective bifunctional catalyst to boost the kinetics of electrochemical reactions inside the battery at the cathode side. Moreover, hard carbons, as functional hosts, are also able to hold solid discharge products like NaO2 at the same time and provide smooth pathways for mass transport, including air and electrolytes [180]. Zhang et al [181] prepared a co-embedded N-doped hard carbon fabric as a free-standing functional host for an air cathode. A superior electrochemical performance containing a high discharge capacity, a low charge overpotential and a stable cycle life (∼112 cycles) all proved the positive effects of hard carbons in improving ORR/OER reactivity and mass transport. The successful development of next-generation high-performance air cathodes cannot happen without hard carbon functional hosts.
5. Conclusions and prospects
We have reviewed the fundamentals of SIBs as well as the synthesis, structure, sodium storage mechanisms and electrochemical properties of hard carbons. Brief introductions to some other applications of hard carbons in sodium-based energy storage have also been presented, such as their use as electrodes in SICs and sodium-based DIBs, and as functional hosts for other electrode materials.
Although significant achievements have been made in the area of hard carbons for Na-ion batteries, and an impressive amount of papers have been published within the last decade, with their number continuously increasing, many challenges/bottlenecks remain: (1) the sodium storage capacities of most hard carbon materials are still low while the energy density of SIBs with hard carbon as anodes still cannot compete with LIBs; (2) the rate and cycling performance are still not good enough, especially for large-grid energy storage systems; (3) the ICE of hard carbon anodes is usually low and not competitive with the graphite in LIBs, which limits the energy density and total cost; (4) the sodium storage mechanism is still not elucidated. A deeper and more thorough understanding of the sodium storage behaviour is important for better material designs; (5) in most cases, a large portion of the capacity comes from the plateau region below 0.1 V (vs. Na+/Na), which may cause Na deposition and safety issues and needs better understanding.
To address these challenges, numerous strategies have been investigated and some of them have been presented in this review. For instance, heteroatom-doping can enhance the electronic conductivity and interlayer spacing and provide more defects and electrochemical active sites on the surface, leading to more surface-controlled capacitive reactions and better sodium storage performance [120, 121]. A pore-forming strategy is effective for creating and tuning closed pores and obtaining ultrahigh specific capacity [149, 150]. An important concept that has been attracting more and more attention is the 'sloping-type' or 'slope-dominated' carbons. These types of carbon material have a high sloping capacity or even no plateau in the low-potential range [13]. The creation of advanced porous structures [110, 112, 113], the making of defect-rich materials [182, 183] and the aforementioned heteroatom-doping [86, 184], etc., are all able to achieve high sloping capacities. The 'sloping-type' carbons typically have more surface-controlled reactions/adsorptions and have faster electron/ion diffusion kinetics, leading to excellent rate capabilities. Moreover, more capacity obtained from the high-potential sloping region can also reduce the risk of Na deposit, which is safer than the long-plateau situations.
ICE is also a very important parameter for anode materials in SIBs, however, the attention and study devoted to ICE is slightly underestimated in the literature. The ICE can strongly influence the energy density and even the cost of commercial SIBs. Typically, a low ICE and a high irreversible capacity are attributed to the formation of an SEI, due to a large surface area. Hence, reducing the surface area is regarded as the most acceptable way to enhance the ICE. Defects are another important reason for low ICE [91, 185], although they can also provide more specific capacity. Therefore, the shielding of defects is also reported to improve the ICE as well as the cycling stability [121]. Wang and Shao et al [56] reviewed the mechanisms of the ICE and the strategies for its enhancement. In addition to the structural design and modification, optimising the electrolyte and the electrode/electrolyte interface is also important for the Coulombic efficiency and cycling performance. Figure 12 illustrates the specific challenges of hard carbon anode materials for SIBs and some effective methods for corresponding improvements.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 12. A general summary of the challenges of hard carbons for SIBs and the strategies for corresponding improvements.
Download figure:
Standard image High-resolution image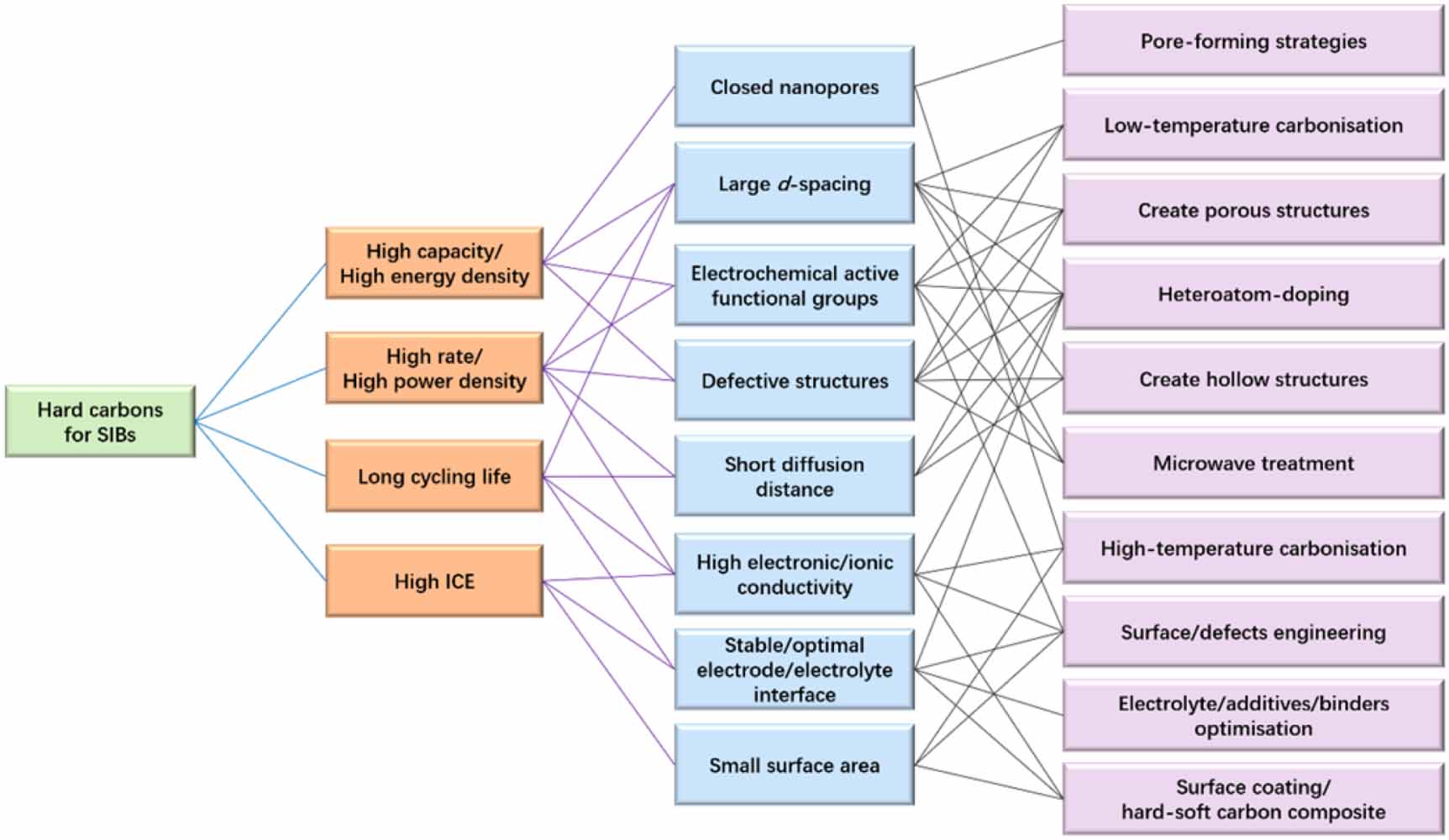{kind=link}
As summarised in figure 3, there are four types of sodium storage mechanism models that are controversial. Several research groups have their own opinions, based on their experimental results, but different carbon materials with different microstructures may have different responses during the sodiation processes, and some characterisation methods such as ex situ XRD have limitations due to the sample manufacture. Na+ ions will choose their most-preferred places for accommodation at each stage, which is hard to generalise and depends on the carbon structure [77]. Therefore, to thoroughly understand the sodium storage behaviour in hard carbon materials, a series of hard carbon materials and some advanced characterisation methods are necessary to carefully grasp and analyse the structures and the electrochemical properties.
Despite all these remaining challenges, hard carbons are the most promising candidates for anodes in commercial SIBs, where they have been showing great potential. We hope that this review article will provide a general insight for researchers to understand hard carbons and SIBs and inspire future works.
Acknowledgments
This work is funded by the Engineering and Physical Sciences Research Council (EP/R021554/1; EP/S018204/1). F X, Z X, and Z G would also like to thank the China Scholarship Council for the PhD scholarship.