






Abstract
To understand the etiology of moderate-to-severe diarrhea among children in high mortality areas of sub-Saharan Africa and South Asia, we performed a comprehensive case/control study of children aged <5 years at 7 sites. Each site employed an identical case/control study design and each utilized a uniform comprehensive set of microbiological assays to identify the likely bacterial, viral and protozoal etiologies. The selected assays effected a balanced consideration of cost, robustness and performance, and all assays were performed at the study sites. Identification of bacterial pathogens employed streamlined conventional bacteriologic biochemical and serological algorithms. Diarrheagenic Escherichia coli were identified by application of a multiplex polymerase chain reaction assay for enterotoxigenic, enteroaggregative, and enteropathogenic E. coli. Rotavirus, adenovirus, Entamoeba histolytica, Giardia enterica, and Cryptosporidium species were detected by commercially available enzyme immunoassays on stool samples. Samples positive for adenovirus were further evaluated for adenovirus serotypes 40 and 41. We developed a novel multiplex assay to detect norovirus (types 1 and 2), astrovirus, and sapovirus. The portfolio of diagnostic assays used in the GEMS study can be broadly applied in developing countries seeking robust cost-effective methods for enteric pathogen detection.
Diarrheal diseases remain among the leading global causes of death for children <5 years of age. A major shortcoming of diarrheal disease studies conducted prior to The Global Enteric Multicenter Study (GEMS) has been the failure to perform a comprehensive ascertainment of major enteric pathogens, particularly at sites of greatest diarrheal burden. This deficit is understandable, considering that sites with high diarrheal mortality are typically those with the greatest challenges to performing the technically demanding portfolio of assays and protocols required to identify bacterial, viral, and protozoal pathogens. Thus, a goal of GEMS has been to assure accurate and consistent identification of relevant pathogens at all the GEMS study sites.
In order to accomplish the challenging but important task of identifying consistently the key pathogens at all GEMS sites, within the significant internal and external constraints, we established the following requirements for a comprehensive set of diagnostic tests:
Performance: The methods utilized were required to have satisfactory sensitivity and specificity. Although difficult to define, we aspired to achieve performance that equaled the standards necessary for effective clinical management in most settings, and satisfactory to assure sufficiently accurate ascertainment of burden and the generation of reliable data.
Robustness: Although all of the sites introduced stringent quality assurance (QA) structures, the methods needed to be consistent across all the sites, requiring feasible training and oversight, as well as the opportunity for verification and validation using post hoc studies.
Cost-effectiveness: GEMS operated on a generous but limited budget. We were required to introduce assays that could be performed within reasonable financial constraints.
The Delphic perspective: We enlisted respected experts on each pathogen to ensure expert support in method selection, personnel training, and QA programs.
Herein we describe the clinical microbiology laboratory methods and protocols utilized in the GEMS study. Most of these assays were adapted from published methods that had independently been developed, validated and subjected to peer review.
Collection and Processing of Stool Samples
Fecal samples in the GEMS study were delivered to the laboratory in cold containers (see Kotloff et al in this supplement). Either at the point of collection or upon accession in the laboratory, a fecal aliquot was introduced into 2 tubes, one containing Cary-Blair medium [1] and one buffered glycerol saline (BGS) [2]. When no fecal specimen was available, a rectal swab was obtained; these rectal swabs were immediately inserted into tubes containing Cary-Blair and BGS media.
Upon arrival at the laboratory, the lab personnel inspected the sample for temperature and stool volume of at least 3 mL; an accession form was processed. The time between stool collection and inoculation of transport media needed to be not more than 6 hours, and the time between placing the specimen in transport media and accession was not more than 18 hours. Aliquots of stool samples were prepared and frozen for subsequent tests as described below.
Conventional Fecal Microbiology
The GEMS protocol included conventional bacterial culture, primarily so that pure growth of implicated pathogens could be independently validated by central laboratories and characterized further with regard to virulence, serologic, and antimicrobial resistance properties.
Bacteria selected for isolation and identification included gram-negative bacteria of proven or highly suspected pathogenicity and significance in developing world settings, as evidenced by the world's literature. The final list of agents sought was vetted through the investigators and the GEMS Microbiology Steering Committee. The pathogens sought included diarrheagenic (enterotoxigenic [ETEC], enteropathogenic [EPEC], and enteroaggregative [EAEC]) Escherichia coli, serovars of Salmonella enterica, Shigella spp, Campylobacter spp, Vibrio spp, and Aeromonas spp. The algorithm for bacteriologic characterization comprised a differential medium, a moderately selective medium, a highly selective medium, and at least 1 enrichment broth. All protocols were adapted from the Manual of Clinical Microbiology, Eighth Edition [3]. From the Cary-Blair tube, swabs were plated onto MacConkey (MAC), xylose lysine desoxycholate (XLD), thiosulfate citrate bile salts sucrose (TCBS), Aeromonas (Ryan) [4], Campy-BAP [5], and alkaline peptone water media; from the BGS the swab was plated onto MAC and XLD media. Plates were incubated at 37°C with the exception of media for Campylobacter spp (42°C) and Aeromonas spp (10°C–42°C). After incubation, suspicious colonies were selected and subjected to a series of simple biochemical tests that could be performed conveniently in resource-poor settings, minimizing expense, difficulty in procurement of reagents, and need for sophisticated training or equipment. The confirmatory tests utilized are described below.
Enterobacteriaceae
Colonies were inoculated into triple-sugar iron, motility indole ornithine (MIO), and lysine decarboxylase media, as well as citrate and urea biochemical typing media, and incubated at 35°C–37°C overnight. Isolates biochemically suspicious for Salmonella enterica [urea (−) oxidase (−)] were serotyped with polyvalent O and Vi following the manufacturer's instructions (Denka Seiken). All isolates biochemically identified as Shigella spp were serotyped with polyvalent group A, B, C, and D using manufacturer's protocols (Denka Seiken or Reagensia).
Vibrio spp Isolation and Identification
TCBS agar plates were examined for growth on day 2; large yellow and green colonies were subcultured to Trypticase soy agar (TSA) and incubated at 37°C overnight. When there was no growth of colonies resembling Vibrio spp after overnight incubation on the TCBS plates, subculture from TSA was tested for the production of oxidase; if oxidase negative, then no further for testing for Vibrio spp was done. If oxidase positive, the isolates were tested for salt tolerance with different concentration of NaCl supplemented in nutrient broth (0%, 6%, and 8%). If the colony was yellow on TCBS and there was growth in 0% and no growth at 8% NaCl-nutrient broth, then the putative Vibrio isolates were reincubated at 37°C for another 24 hours; at the same time the alkaline peptone water was subcultured to a new TCBS plate and incubated at 37°C. On day 3, each Vibrio cholerae was confirmed serologically using O1 and O139 antisera (Denka Seiken) and V. cholerae O1–positive cultures were typed as Inaba or Ogawa serotypes. If the colony was green on TCBS and there was growth in NaCl concentrations of 6% and 8%, and no growth in 0%, this was considered presumptive for Vibrio parahaemolyticus.
Aeromonas spp Isolation and Identification
On day 2, Ryan agar plates were examined for dark green colonies with darker green centers. Such colonies were subcultured onto TSA plates, tested for salt tolerance with different concentrations of NaCl (0%, 6% and 8%), and incubated aerobically for 24 hours. The next day, oxidase and catalase tests from the TSA plate were performed and tubes read for growth at various NaCl concentrations. Susceptibility to O/129 (2, 4-diamino-6, 7-diisopropyl pteridine) was also assessed [6]. Any isolate that was oxidase (+), catalase (+), grew in 0% NaCl but not in 6% or 8%, and was resistant to O/129, was considered to belong to the species Aeromonas.
Campylobacter spp Isolation and Identification
On day 3 the Campy blood agar plate was observed for growth appearing in one of the following ways: (1) nonhemolytic, gray, yellowish or pinkish tint; (2) flat, spreading, irregular edged colonies; (3) mucoid; (4) thin film; (5) spreading along the streak mark; or (6) round and convex. Oxidase and catalase tests were done and a sodium hippurate tube was inoculated. If isolates were oxidase (+) and catalase (+), smears were prepared for Gram staining. The smear was examined under the light microscope for small gram-negative rods that are slightly curved or “S” shaped. The sodium hippurate hydrolysis test was then performed for confirmation. Hippurate hydrolysis positive isolates were classified as Campylobacter jejuni; if hippurate hydrolysis was negative, strains were classified as Campylobacter coli.
E. coli Isolation and Identification
From 2-day growth on MAC plates, several lactose-fermenting bacterial colonies resembling E. coli were picked and tested using MIO medium. Up to 3 lactose-positive and indole-positive colonies were selected. When there were multiple distinct E. coli–like colony morphologies, each was selected. If there were <3 colonies of lactose-fermenting E. coli–like organisms, then all lactose-positive colonies were picked, and ≥1 lactose-negative colonies were picked to reach the total of 3 colonies per specimen. Indole-positive colonies were saved for further analysis. For indole-negative colonies, a second series of biochemical test, Indole/Methyl Red/Voges Proskauer/Citrate was used to identify E. coli. If any were positive for methyl red, and negative for Voges Proskauer and citrate, they were saved for further analysis. If 3 presumed E. coli were not found (ie, positive for indole or another suggestive biochemical reaction), the microbiologist returned to the original plate and picked up to 3 additional colonies for biochemical testing.
ETEC, EPEC, and EAEC pathotypes were identified using a multiplex polymerase chain reaction (PCR) previously published [7], but adapted for the purpose of GEMS. The targets sought via the PCR reaction included ETEC heat-labile enterotoxin and heat-stable enterotoxin (derived from STh) genes, the EPEC intimin (eae gene) outer membrane protein adhesin; the EPEC plasmid-encoded bundle-forming pilus (BFP); the EAEC plasmid-encoded gene aatA; and the EAEC chromosomally encoded aaiC locus. All of these loci are known virulence determinants of their respective pathogens [8, 9]. Strains positive for eae but not BFP were designated atypical EPEC. Strains positive for either ETEC enterotoxin were considered ETEC and strains positive for either EAEC factor were considered EAEC for the purposes of the GEMS analysis.
The 3 E. coli–like colonies selected from each stool were pooled into a common sample tube and template DNA was prepared from the pooled colonies. Template DNA was prepared by boiling the cultures grown on L-agar for 20 minutes, rapidly cooling on ice, followed by brief centrifugation at 2500g for 10 minutes. This supernatant was used in the PCR assays. Primer nucleotide sequences and the predicted lengths of the resulting amplicons are listed in Table 1.
Primer Sequences and the Expected Amplicon Sizes for the Multiplex Polymerase Chain Reaction Employed in the Detection of Diarrheagenic Escherichia coli
| Pathogen | Primer | Target Gene | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|---|
| ETEC | LT-F | elt | CACACGGAGCTCCTCAGTC | 508 |
| LT-R | CCCCCAGCCTAGCTTAGTTT | |||
| ST-F | est | GCTAAACCAGTAG/AGGTCTTCAAAA | 147 | |
| ST-R | CCCGGTACAG/AGCAGGATTACAACA | |||
| EPEC | BFPA-F | bfpA | GGAAGTCAAATTCATGGGGG | 367 |
| BFPA-R | GGAATCAGACGCAGACTGGT | |||
| EAE-F | eae | CCCGAATTCGGCACAAGCATAAGC | 881 | |
| EAE-R | CCCGGATCCGTCTCGCCAGTATTCG | |||
| EAEC | CVD432F | aatA | CTGGCGAAAGACTGTATCAT | 630 |
| CVD432R | CAATGTATAGAAATCCGCTGTT | |||
| AAIC F | aaiC | ATTGTCCTCAGGCATTTCAC | 215 | |
| AAIC R | ACGACACCCCTGATAAACAA |
| Pathogen | Primer | Target Gene | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|---|
| ETEC | LT-F | elt | CACACGGAGCTCCTCAGTC | 508 |
| LT-R | CCCCCAGCCTAGCTTAGTTT | |||
| ST-F | est | GCTAAACCAGTAG/AGGTCTTCAAAA | 147 | |
| ST-R | CCCGGTACAG/AGCAGGATTACAACA | |||
| EPEC | BFPA-F | bfpA | GGAAGTCAAATTCATGGGGG | 367 |
| BFPA-R | GGAATCAGACGCAGACTGGT | |||
| EAE-F | eae | CCCGAATTCGGCACAAGCATAAGC | 881 | |
| EAE-R | CCCGGATCCGTCTCGCCAGTATTCG | |||
| EAEC | CVD432F | aatA | CTGGCGAAAGACTGTATCAT | 630 |
| CVD432R | CAATGTATAGAAATCCGCTGTT | |||
| AAIC F | aaiC | ATTGTCCTCAGGCATTTCAC | 215 | |
| AAIC R | ACGACACCCCTGATAAACAA |
Abbreviations: EAEC, enteroaggregative Escherichia coli; EPEC, enteropathogenic E. coli; ETEC, enterotoxigenic E. coli.
Primer Sequences and the Expected Amplicon Sizes for the Multiplex Polymerase Chain Reaction Employed in the Detection of Diarrheagenic Escherichia coli
| Pathogen | Primer | Target Gene | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|---|
| ETEC | LT-F | elt | CACACGGAGCTCCTCAGTC | 508 |
| LT-R | CCCCCAGCCTAGCTTAGTTT | |||
| ST-F | est | GCTAAACCAGTAG/AGGTCTTCAAAA | 147 | |
| ST-R | CCCGGTACAG/AGCAGGATTACAACA | |||
| EPEC | BFPA-F | bfpA | GGAAGTCAAATTCATGGGGG | 367 |
| BFPA-R | GGAATCAGACGCAGACTGGT | |||
| EAE-F | eae | CCCGAATTCGGCACAAGCATAAGC | 881 | |
| EAE-R | CCCGGATCCGTCTCGCCAGTATTCG | |||
| EAEC | CVD432F | aatA | CTGGCGAAAGACTGTATCAT | 630 |
| CVD432R | CAATGTATAGAAATCCGCTGTT | |||
| AAIC F | aaiC | ATTGTCCTCAGGCATTTCAC | 215 | |
| AAIC R | ACGACACCCCTGATAAACAA |
| Pathogen | Primer | Target Gene | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|---|
| ETEC | LT-F | elt | CACACGGAGCTCCTCAGTC | 508 |
| LT-R | CCCCCAGCCTAGCTTAGTTT | |||
| ST-F | est | GCTAAACCAGTAG/AGGTCTTCAAAA | 147 | |
| ST-R | CCCGGTACAG/AGCAGGATTACAACA | |||
| EPEC | BFPA-F | bfpA | GGAAGTCAAATTCATGGGGG | 367 |
| BFPA-R | GGAATCAGACGCAGACTGGT | |||
| EAE-F | eae | CCCGAATTCGGCACAAGCATAAGC | 881 | |
| EAE-R | CCCGGATCCGTCTCGCCAGTATTCG | |||
| EAEC | CVD432F | aatA | CTGGCGAAAGACTGTATCAT | 630 |
| CVD432R | CAATGTATAGAAATCCGCTGTT | |||
| AAIC F | aaiC | ATTGTCCTCAGGCATTTCAC | 215 | |
| AAIC R | ACGACACCCCTGATAAACAA |
Abbreviations: EAEC, enteroaggregative Escherichia coli; EPEC, enteropathogenic E. coli; ETEC, enterotoxigenic E. coli.
For the PCR reaction, 3 μL of template DNA was added to the PCR mix containing 2.5 µL of 10× PCR buffer with 2 mM MgCl2 (New England Biolabs), 2.0 µL of 10 mM deoxynucleotide triphosphates (dNTPs) (Fermentas), 0.4 µL of 20 pmol/µL of each primer, 0.25 µL Taq DNA polymerase (5 U/µL, New England Biolabs), and 7.37 µL RNase-free water to a final volume of 20 µL. PCR was performed under the following conditions: preheating at 96°C for 4 minutes, denaturation at 95°C for 20 seconds, annealing at 57°C for 20 seconds, elongation at 72°C for 1 minute. PCR was performed for 35 cycles with final extension at 72°C for 7 minutes in an Eppendorf Mastercycler Gradient thermal cycler. The same model thermal cycler was employed at all sites. The amplification products were separated through a 2% agarose gel and visualized by ultraviolet light transillumination after ethidium bromide staining. The 1-kb plusA 100-bp DNA ladder (New England Biolabs) was used as a molecular size marker in gel. Appearance of the PCR amplicons on agarose gel electrophoresis is shown in Figure 1. Control strains employed in every PCR reaction were ETEC H10407, EAEC 042, and for EPEC strains CVD 28 (eae-positive) and HB101(pMAR7) (bfpA-positive).
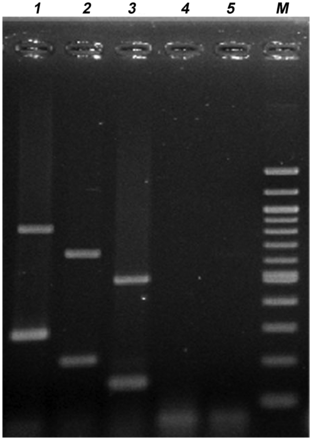
Appearance of diarrheagenic Escherichia coli amplicons separated by agarose gel electrophoresis. Lane 1, enteropathogenic E. coli; lane 2, enteroaggregative E. coli; 3, enterotoxigenic E. coli; lanes 4 and 5, negative control strains; lane 6, 100 bp DNA ladder (New England Biolabs).
Characterization of eae-Positive, bfpA-Negative Strains
As part of a nested study, all E. coli specimens that were negative in the original multiplex PCR for elt, est, bfpA, eae, aatA, and aaiC were investigated at the University of Melbourne, Australia, for eae by using a high-throughput real-time PCR assay. Specimens, consisting of 3 individual isolates, were sent to Melbourne from Baltimore on MAC agar in 96-well flat-bottomed microtiter trays. Upon arrival, the cultures were replica-plated onto MAC agar and grown overnight at 37°C. To generate template DNA for use in the real-time PCR, a sterile pipette tip was used to transfer a portion of a culture sample from the MAC replica plate into a single well of a 96-well PCR tray (Bio-Rad) containing 100 µL DNase-free water. This procedure was repeated for the remaining 2 samples of the specimen, so that each well contained 1 specimen comprising 3 separate isolates. The plate was sealed with Microseal “A” adhesive (Bio-Rad). To lyse the bacterial cells, the samples were heated to 99°C for 10 minutes in a C1000 PCR machine (Bio-Rad) followed by cooling at 12°C. Before use the plate was centrifuged for 1 minute at 3000g and the supernatant was used as the template DNA in the real-time PCR assay.
For the real-time PCR, 8 µL of a master mix was added to individual wells of a 96-well PCR tray (Bio-Rad) followed by 2 μL of template DNA. The real-time master mix, for one reaction, comprised 5.0 µL of 2 × SSoFast EvaGreen Supermix (Bio-Rad), 1.4 µL DNase-free water, and 0.8 µL of 5 µM of each primer. The plate was sealed with Microseal “B” adhesive (Bio-Rad) and centrifuged for 30 seconds at 3000g. Real-time PCR was performed using a CFX96 real-time PCR machine (Bio-Rad) using the following protocol: 95°C for 2 minutes, followed by 35 cycles of 95°C for 1 second, and 60°C for 5 seconds. The duration of one complete reaction was 24 minutes and upon completion the results were analyzed using the CFX Manager Software (Bio-Rad). Binding of the SSoFast EvaGreen dye to double-stranded DNA PCR products causes the dye to fluoresce. The cycle threshold is the number of cycles at which the fluorescence exceeds the background level (Figure 2). In our study, specimens with a cycle threshold of ≤30 were analyzed further. Control strains employed in every PCR included EPEC strains E2348/69, E128010, W1056, and TR952, which carry intimin alpha, beta, gamma, and epsilon, respectively (positive controls); and ETEC strain H10407 and E. coli K-12 strain MC4100 (negative controls). Three “no DNA template” controls were also included. Each individual isolate within an eae-positive specimen was analyzed by using a multiplex PCR to confirm the presence of eae, and also to test for the presence of genetic markers of typical EPEC (bfpA), Shiga toxin–producing E. coli, and/or enterohemorrhagic E. coli (EHEC) (stx1, stx2, ehxA). Template DNA for use in this PCR was prepared by resuspending a loopful of the individual culture samples from the MAC replica plate in 500 μL of DNase-free water and then boiling the suspension for 10 minutes. The boiled bacterial lysate was rapidly cooled on ice for 5 minutes followed by centrifugation for 5 minutes at 16 000g. The supernatant containing the DNA was transferred to a fresh microfuge tube and placed on ice or at 4°C until used in the PCR.
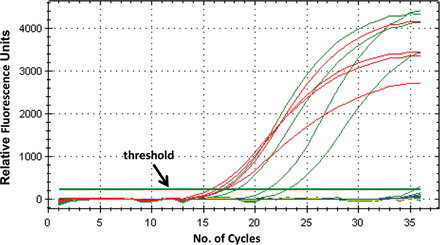
An example of the graphical results of real-time polymerase chain reaction performed on 4 eae-positive specimens (red), 4 unknown specimens (green), and negative controls (yellow and blue). A threshold for detection of DNA-based fluorescence is set slightly above background fluorescence levels.
For this PCR a GoTaq Green Master Mix (Promega), which contained Taq DNA polymerase, dNTPs, MgCl2, reaction buffers, and loading dye, was used. The PCR was performed in a C1000 PCR machine (Bio-Rad) using the following protocol: 95°C for 5 minutes, followed by 35 cycles of 95°C for 20 seconds, 55°C for 45 seconds, and 72°C for 30 seconds, followed by 1 cycle of 72°C for 7 minutes. The amplification products were separated through a 2% Tris-acetate-EDTA agarose gel and visualized by ultraviolet light transillumination. A 100-bp DNA ladder (New England Biolabs) was used as a molecular size marker. Examples of the results of this PCR are shown in Figure 3. Control strains included in every PCR reaction were EPEC strain E2348/69 for eae and bfpA and EHEC strain EH48 for stx1, stx2, and ehxA. Primers that were used in the eae real-time PCR are listed in Table 2; reaction conditions are listed in Table 3; primer nucleotide sequences and the predicted lengths of the resulting amplicons are listed in Table 4.
Primer Sequences and Expected Amplicon Size for Real-time Polymerase Chain Reaction
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae83-F | CAGGCTTCGTCACAGTTG | eae | 83 |
| eae83-R | CCGTCAAAGTTATTACCACTCTG |
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae83-F | CAGGCTTCGTCACAGTTG | eae | 83 |
| eae83-R | CCGTCAAAGTTATTACCACTCTG |
Primer Sequences and Expected Amplicon Size for Real-time Polymerase Chain Reaction
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae83-F | CAGGCTTCGTCACAGTTG | eae | 83 |
| eae83-R | CCGTCAAAGTTATTACCACTCTG |
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae83-F | CAGGCTTCGTCACAGTTG | eae | 83 |
| eae83-R | CCGTCAAAGTTATTACCACTCTG |
Components of the Multiplex Polymerase Chain Reaction
| 1 × polymerase chain reaction | |
| 2 × GoTaq Green Master Mix | 15.0 μL |
| 20 μM bfpA-F | 1.0 μL |
| 20 μM bfpA-R | 1.0 μL |
| 20 μM ehxA-F | 1.0 μL |
| 20 μM ehxA-R | 1.0 μL |
| 20 μM eae-F | 1.0 μL |
| 20 μM eae-R | 1.0 μL |
| 20 μM stx1-F | 0.5 μL |
| 20 μM stx1-R | 0.5 μL |
| 20 μM stx2-F | 0.5 μL |
| 20 μM stx2-R | 0.5 μL |
| DNA template | 2.0 μL |
| Total volume | 25.0 μL |
| 1 × polymerase chain reaction | |
| 2 × GoTaq Green Master Mix | 15.0 μL |
| 20 μM bfpA-F | 1.0 μL |
| 20 μM bfpA-R | 1.0 μL |
| 20 μM ehxA-F | 1.0 μL |
| 20 μM ehxA-R | 1.0 μL |
| 20 μM eae-F | 1.0 μL |
| 20 μM eae-R | 1.0 μL |
| 20 μM stx1-F | 0.5 μL |
| 20 μM stx1-R | 0.5 μL |
| 20 μM stx2-F | 0.5 μL |
| 20 μM stx2-R | 0.5 μL |
| DNA template | 2.0 μL |
| Total volume | 25.0 μL |
Components of the Multiplex Polymerase Chain Reaction
| 1 × polymerase chain reaction | |
| 2 × GoTaq Green Master Mix | 15.0 μL |
| 20 μM bfpA-F | 1.0 μL |
| 20 μM bfpA-R | 1.0 μL |
| 20 μM ehxA-F | 1.0 μL |
| 20 μM ehxA-R | 1.0 μL |
| 20 μM eae-F | 1.0 μL |
| 20 μM eae-R | 1.0 μL |
| 20 μM stx1-F | 0.5 μL |
| 20 μM stx1-R | 0.5 μL |
| 20 μM stx2-F | 0.5 μL |
| 20 μM stx2-R | 0.5 μL |
| DNA template | 2.0 μL |
| Total volume | 25.0 μL |
| 1 × polymerase chain reaction | |
| 2 × GoTaq Green Master Mix | 15.0 μL |
| 20 μM bfpA-F | 1.0 μL |
| 20 μM bfpA-R | 1.0 μL |
| 20 μM ehxA-F | 1.0 μL |
| 20 μM ehxA-R | 1.0 μL |
| 20 μM eae-F | 1.0 μL |
| 20 μM eae-R | 1.0 μL |
| 20 μM stx1-F | 0.5 μL |
| 20 μM stx1-R | 0.5 μL |
| 20 μM stx2-F | 0.5 μL |
| 20 μM stx2-R | 0.5 μL |
| DNA template | 2.0 μL |
| Total volume | 25.0 μL |
Primer Sequences and the Expected Amplicon Sizes for the Multiplex Polymerase Chain Reaction
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae-F | GACCCGGCACAAGCATAAGC | eae | 384 |
| eae-R | CCACCTGCAGCAACAAGAGG | ||
| ehxA-F | GCATCATCAAGCGTACGTTCC | ehxA | 534 |
| ehxA-R | AATGAGCCAAGCTGGTTAAGCT | ||
| stx1-F | ATAAATCGCCATTCGTTGACTAC | stx1 | 180 |
| stx1-R | AGAACGCCCACTGAGATCATC | ||
| stx2-F | GGCACTGTCTGAAACTGCTCC | stx2 | 255 |
| stx2-R | TCGCCAGTTATCTGACATTCTG | ||
| bfpA-F | GGAAGTCAAATTCATGGGGG | bfpA | 300 |
| bfpA-R | GGAATCAGACGCAGACTGGT |
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae-F | GACCCGGCACAAGCATAAGC | eae | 384 |
| eae-R | CCACCTGCAGCAACAAGAGG | ||
| ehxA-F | GCATCATCAAGCGTACGTTCC | ehxA | 534 |
| ehxA-R | AATGAGCCAAGCTGGTTAAGCT | ||
| stx1-F | ATAAATCGCCATTCGTTGACTAC | stx1 | 180 |
| stx1-R | AGAACGCCCACTGAGATCATC | ||
| stx2-F | GGCACTGTCTGAAACTGCTCC | stx2 | 255 |
| stx2-R | TCGCCAGTTATCTGACATTCTG | ||
| bfpA-F | GGAAGTCAAATTCATGGGGG | bfpA | 300 |
| bfpA-R | GGAATCAGACGCAGACTGGT |
Primer Sequences and the Expected Amplicon Sizes for the Multiplex Polymerase Chain Reaction
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae-F | GACCCGGCACAAGCATAAGC | eae | 384 |
| eae-R | CCACCTGCAGCAACAAGAGG | ||
| ehxA-F | GCATCATCAAGCGTACGTTCC | ehxA | 534 |
| ehxA-R | AATGAGCCAAGCTGGTTAAGCT | ||
| stx1-F | ATAAATCGCCATTCGTTGACTAC | stx1 | 180 |
| stx1-R | AGAACGCCCACTGAGATCATC | ||
| stx2-F | GGCACTGTCTGAAACTGCTCC | stx2 | 255 |
| stx2-R | TCGCCAGTTATCTGACATTCTG | ||
| bfpA-F | GGAAGTCAAATTCATGGGGG | bfpA | 300 |
| bfpA-R | GGAATCAGACGCAGACTGGT |
| Primer | Sequence (5′-3′) | TargetGene | Amplicon(bp) |
|---|---|---|---|
| eae-F | GACCCGGCACAAGCATAAGC | eae | 384 |
| eae-R | CCACCTGCAGCAACAAGAGG | ||
| ehxA-F | GCATCATCAAGCGTACGTTCC | ehxA | 534 |
| ehxA-R | AATGAGCCAAGCTGGTTAAGCT | ||
| stx1-F | ATAAATCGCCATTCGTTGACTAC | stx1 | 180 |
| stx1-R | AGAACGCCCACTGAGATCATC | ||
| stx2-F | GGCACTGTCTGAAACTGCTCC | stx2 | 255 |
| stx2-R | TCGCCAGTTATCTGACATTCTG | ||
| bfpA-F | GGAAGTCAAATTCATGGGGG | bfpA | 300 |
| bfpA-R | GGAATCAGACGCAGACTGGT |

Gels showing the results of a multiplex polymerase chain reaction (PCR) assay for enteropathogenic Escherichia coli (EPEC), Shiga toxin–producing E. coli, and enterohemorrhagic E. coli (EHEC). Individual isolates from 34 specimens were subjected to a multiplex PCR as described in the text. Each specimen, separated by yellow vertical lines, consists of 3 individual isolates. The yellow values indicate the cycle threshold obtained for each specimen in the real-time PCR used in the initial screening for eae. The amplicons produced by the positive controls, EPEC E2348/69 (eae and bfpA) and EHEC EH48 (stx1, stx2, and ehxA) are also shown. 100 bp DNA ladder was used as a molecular size marker. Abbreviation: NTC, no template control.
Virus Immunoassays
Enzyme immunoassays are rapid, robust, sensitive, and specific diagnostic assays for some viral pathogens. We used well-validated commercial immunoassays for rotavirus and adenovirus according to established protocols.
Rotavirus
Rotavirus VP6 antigen was detected in stools by the ProSpecT ELISA Rotavirus kit following the manufacturer's instructions (Oxoid).
Adenovirus
General adenovirus hexon protein was detected using ProSpecT Adenovirus Microplate assays according to the manufacturer's instructions (Oxoid). This test utilizes a genus-specific monoclonal antibody to detect epitopes common to all human adenovirus serotypes.
Samples for adenovirus by the ProSpecT assay were further tested for the presence of enteric adenovirus serotypes 40/41 using Premier Adenoclone kit (Meridian Bioscience) following the manufacturer's instructions.
Multiplex PCR for Detection of RNA Viruses
Stool specimens were diluted to 10% (w/v or v/v) suspensions in Vertrel XF (Miller Stephenson) and centrifuged at 1000g for 10 minutes. The supernatant was collected and stored at 4°C prior to RNA extraction.
Viral RNA was extracted from stool supernatant using Nuclisens (bioMérieux) as per the manufacturer's instructions. In brief, 900 μL of lysis buffer was added to 200 µL of supernatant, vortexed and incubated for 10 minutes, then 50 μL of silica suspension was added, vortexed and centrifuged at 10 000g for 30 seconds. Washing was done by adding 1 mL of wash buffer twice followed by washing with 1 mL of 70% ethanol twice. Finally 1 mL of acetone was added to the pellet. At the end of each washing step, tubes were vortexed and centrifuged at room temperature for 30 seconds at 10 000g; supernatant was carefully discarded without disturbing silica pellet. The silica pellet was dried at 56°C for 10 minutes and the pellet was reconstituted by adding 50 μL of elution buffer. Samples were vortexed and incubated at 56°C for 5 minutes, the incubation step was repeated, and the specimen was centrifuged for 2 minutes at 10 000g. RNA containing supernatant was collected containing RNA and stored at −70°C until use.
RNA was reverse transcribed in a total volume of 15 µL containing 1× First strand buffer (Invitrogen), 0.5 mM dNTPs (Roche), 0.5 mM dithiothreitol (Invitrogen), 0.5 µg of random primers (TaKaRa), 20 units of RNase Inhibitor (Roche), and 150 units of Superscript II Reverse Transcriptase (RT; Invitrogen). The mixture was incubated at 42°C for 1 hour and then heated at 99°C for 5 minutes.
A multiplex PCR reaction was designed to amplify norovirus, astrovirus, and sapovirus complementary DNA (cDNA) present in the reverse transcription reactions described above. The method was adapted from a published protocol [10]. After cDNA synthesis, multiplex PCR was performed using specific primers (Table 5). PCR master mix contained 0.5 µM concentration of specific primers, 0.2 mM dNTPs (Roche), 1× AmpliTaq buffer I, and 1.25 U of AmpliTaq DNA polymerase (Applied Biosystems) for a 25-µL reaction. Master Mix was distributed to 0.2-mL PCR tubes, and 5 µL of template cDNA was added. The assay was confirmed using positive and negative controls cDNA from confirmed prior reactions. PCR reactions were conducted in a Eppendorf Mastercycler Gradient thermal cycler starting with a denaturing step of 3 minutes at 94°C, followed by 35 cycles of 30 seconds at 94°C, 30 seconds at 55°C, and 30 seconds at 72°C, followed by an extension of 72°C for 7 minutes. After the thermocycling step, all PCR products were electrophoresed on a 2.0% agarose gel and sized with a 100-bp ladder (Promega) (Figure 4).
Primer Sequences and the Expected Amplicon Sizes for the Multiplex Polymerase Chain Reaction Used in the Detection of RNA Viruses
| Pathogen | Primer | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|
| Norovirus GI | G1SKR | CCAACCCARCCATTRTACA | 330 |
| G1SKF | CTGCCCGAATTYGTAAATGA | ||
| Norovirus GII | G2SKR | CCRCCNGCATRHCCRTTRTACAT | 387 |
| COG2F | CARGARBCNATGTTYAGRTGGATGAG | ||
| Sapovirus | SLV5749 | CGGRCYTCAAAVSTACCBCCCCA | 434 |
| SLV5317 | CTCGCCACCTACRAWGCBTGGTT | ||
| Astrovirus | 82b | GTGAGCCACCAGCCATCCCT | 719 |
| PreCAP1 | GGACTGCAAAGCAGCTTCGTG | ||
| Cog2R | TCGACGCCATCTTCATTCACA |
| Pathogen | Primer | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|
| Norovirus GI | G1SKR | CCAACCCARCCATTRTACA | 330 |
| G1SKF | CTGCCCGAATTYGTAAATGA | ||
| Norovirus GII | G2SKR | CCRCCNGCATRHCCRTTRTACAT | 387 |
| COG2F | CARGARBCNATGTTYAGRTGGATGAG | ||
| Sapovirus | SLV5749 | CGGRCYTCAAAVSTACCBCCCCA | 434 |
| SLV5317 | CTCGCCACCTACRAWGCBTGGTT | ||
| Astrovirus | 82b | GTGAGCCACCAGCCATCCCT | 719 |
| PreCAP1 | GGACTGCAAAGCAGCTTCGTG | ||
| Cog2R | TCGACGCCATCTTCATTCACA |
Primer Sequences and the Expected Amplicon Sizes for the Multiplex Polymerase Chain Reaction Used in the Detection of RNA Viruses
| Pathogen | Primer | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|
| Norovirus GI | G1SKR | CCAACCCARCCATTRTACA | 330 |
| G1SKF | CTGCCCGAATTYGTAAATGA | ||
| Norovirus GII | G2SKR | CCRCCNGCATRHCCRTTRTACAT | 387 |
| COG2F | CARGARBCNATGTTYAGRTGGATGAG | ||
| Sapovirus | SLV5749 | CGGRCYTCAAAVSTACCBCCCCA | 434 |
| SLV5317 | CTCGCCACCTACRAWGCBTGGTT | ||
| Astrovirus | 82b | GTGAGCCACCAGCCATCCCT | 719 |
| PreCAP1 | GGACTGCAAAGCAGCTTCGTG | ||
| Cog2R | TCGACGCCATCTTCATTCACA |
| Pathogen | Primer | Primer Sequence (5′-3′) | Amplicon (bp) |
|---|---|---|---|
| Norovirus GI | G1SKR | CCAACCCARCCATTRTACA | 330 |
| G1SKF | CTGCCCGAATTYGTAAATGA | ||
| Norovirus GII | G2SKR | CCRCCNGCATRHCCRTTRTACAT | 387 |
| COG2F | CARGARBCNATGTTYAGRTGGATGAG | ||
| Sapovirus | SLV5749 | CGGRCYTCAAAVSTACCBCCCCA | 434 |
| SLV5317 | CTCGCCACCTACRAWGCBTGGTT | ||
| Astrovirus | 82b | GTGAGCCACCAGCCATCCCT | 719 |
| PreCAP1 | GGACTGCAAAGCAGCTTCGTG | ||
| Cog2R | TCGACGCCATCTTCATTCACA |
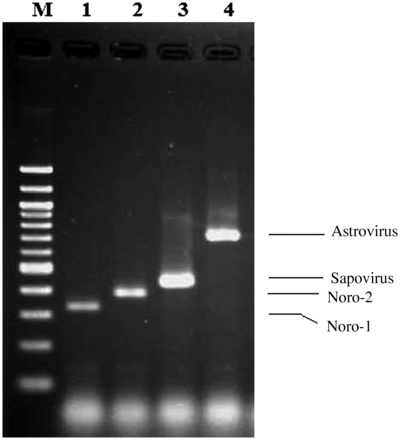
Appearance of enteric viral amplicons separated by agarose gel electrophoresis. Lane M, 100 bp DNA ladder (New England Biolabs); lane 1, Norovirus GI (330 bp); lane 2, Norovirus GII positive (387 bp); lane 3, sapovirus (434 bp); lane 4, astrovirus (719 bp).
Detection of Protozoal Pathogens
Giardia enterica [11, 12], Entamoeba histolytica [13], and Cryptosporidium spp [12] were detected using immunoassays available commercially from TechLab, Inc and according to manufacturer's protocols. Studies have demonstrated excellent performance of these assays, superior to microscopic detection [14–17].
Quality Control Methods
Initial Training
An investigators’ meeting was held at the start of the study at the Center for Vaccine Development (CVD) in Baltimore, to review the procedures to be used. All the laboratory heads from the field sites and some technicians attended the meeting. CVD Quality Control (QC)/QA staff reviewed the execution of each standard operating procedure (SOP) during site visits and provided retraining if necessary.
Standard Operating Procedures
In order to streamline processes at each site, SOPs were generated to ensure that all procedures were executed in consistent fashion at each site. SOPs clearly defined the purpose, the required materials and equipment, safety guidelines, responsibility, procedures, and documentation and provided related documents.
By introducing controlled forms for each SOP, a “quality checklist” was created that would ensure that each step in the SOP was carried out as directed, and that the materials used were as stipulated in the SOP and prior to their expiration dates. These forms were also reviewed by the laboratory supervisor or designee to ensure adherence to the SOP and that quality deliverables were generated. Forms also included, for some tests, negative, positive, and cutoff values. Samples that were not valid, or test runs in which control values were not valid, were repeated. The rate of sample reworking was tracked as a quality metric.
Quality Assurance
Quality incidents and deviations from the SOP were reported and documented on designated forms and reviewed by the supervisor on site and by QC/QA personnel during regular site visits. Corrective and preventive actions were executed on site, by the laboratory supervisor or designee. Very few or no quality incidents or deviations occurred for each protocol. All forms were reviewed by the QC/QA CVD staff during routine site visits.
All case report forms were also reviewed by the data coordinating center (DCC) for completeness. Missing data and/or missing forms were communicated to the sites via email. Other information, such as ranges of time, was also calculated by the DCC.
Biannual proficiency testing was conducted at each site. Sites were expected to score 80% on identification of “unknown” samples sent from Baltimore. All sites attained this score. Any incorrect results were investigated and any errors corrected and retraining provided if necessary on site by the laboratory managers.
Post Hoc Studies and Validation Studies
The GEMS study has generated a cornucopia of bacterial strains, fecal nucleic acid, and frozen stool strains that will yield priceless information regarding the agents associated with diarrhea in infants and young children in developing countries and their genomic and serologic diversity. The analyses proposed in the GEMS protocol include typing of the major ETEC adhesins, the colonization factors. In addition, Shigella dysenteriae isolates were tested to detect S. dysenteriae 1 (the Shiga bacillus), all Shigella sonnei were serologically confirmed, and all Shigella flexneri isolates were typed and subtyped. These 2 analyses will profile the antigenic diversity of these 2 important pathogens and inform future vaccine development priorities and will be reported elsewhere.
The availability of GEMS clinical samples also provides the opportunity for diagnostic method development and validation. For example, a rigorous comparison of the multiplex RT-PCR assay with real-time PCR for detection of norovirus has been completed and will be described elsewhere. In addition, the sample archive provides the platform for the development of new, high-throughput and highly multiplexed diagnostic technologies, comparing their performance with gold standard methodologies.
DISCUSSION
The GEMS study employed a portfolio of diagnostic tests that balanced practicality and economy, as well as good sensitivity and specificity. A number of important issues warrant elaboration.
We decided to employ conventional bacteriologic methods for isolation of putative bacterial pathogens, followed by molecular and/or phenotypic characterization. The derivation of pure bacterial stocks permitted not only downstream characterization of genetic and surface markers of relevance to epidemiology and vaccine development, but also allowed us to revisit the diagnostic performance of the selected assays on archived strain collections. Escherichia coli colonies, for example, were isolated, archived, and tested for the presence of virulence-related genes that define diarrheagenic pathotypes; Shigella strains were serotyped at reference laboratories in order to inform future vaccine development strategies. As noted, validation of both EPEC and ETEC primers sets was performed on the E. coli archive using high-throughput PCR analysis. For EAEC, which was not associated with diarrhea overall, the availability of archived bacterial cultures permitted extensive genomic characterization of isolates, thereby identifying potentially pathogenic genotypes [18].
Agarose gel–based detection of PCR amplicons was the preferred in the GEMS diagnostic set for the following reasons. At the time the GEMS protocol was developed, there was little expertise in real-time PCR at any of the sites in the GEMS network, and the added complexity of real-time was beyond what the training programs could realistically accomplish. Additional advantages of the gel-based method include substantially lower cost, the availability of gel images that could be shared across sites for validation and quality control purposes, and greater availability of supplies at the sites.
We decided to employ immunoassays for detection of protozoal pathogens for many of the same reasons. Direct microscopic detection of protozoal pathogens requires significant expertise and is not readily amenable to downstream validation. Immunoassays, also employed for detection of some viral agents, followed a simple, highly standardized, and centrally validated method that was easily deployed at the study sites. An additional advantage to enzyme immunoassay methods was the availability of product support from the kit manufacturers.
GEMS investigators applied multiple criteria by which to select agents for detection. These criteria included published citation as a significant agent of childhood diarrhea at multiple sites in the developing world, and practical detection methodology. Toxigenic Bacteroides fragilis, for example, could have been included in the portfolio but would have required either anaerobic bacteriology or use of tests that could not be validated post hoc on pure cultures. The availability of the GEMS specimen archive permits post hoc detection of additional agents using molecular and other technologies, and these efforts are under way.
All primers employed in PCR reactions were selected from published studies, thereby conferring both validation by an independent laboratory and peer review, and were also validated in the laboratories of the GEMS investigators in Baltimore. Post hoc validation was nevertheless carried out employing nested studies of individual block PCR reactions and/or the use of alternative primer sets.
The GEMS study offers a quantum leap in our understanding of the burden and etiology of diarrhea afflicting infants and young children in developing countries. The GEMS etiology data and specimen collections will be grist for further advances far into the future.
Notes
Financial support. This work was supported by the Bill & Melinda Gates Foundation (grant number 38874).
Supplement sponsorship. This article was published as part of the supplement entitled “The Global Enteric Multicenter Study (GEMS),” sponsored by the Bill & Melinda Gates Foundation.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments