





Abstract
The organization and maturation of ventricular cardiomyocytes from the embryonic to the adult form is crucial for normal cardiac function. We have shown that a polarity protein, Scrib, may be involved in regulating the early stages of this process. Our goal was to establish whether Scrib plays a cell autonomous role in the ventricular myocardium, and whether this involves well-known polarity pathways.
Deletion of Scrib in cardiac precursors utilizing Scribflox mice together with the Nkx2.5-Cre driver resulted in disruption of the cytoarchitecture of the forming trabeculae and ventricular septal defects. Although the majority of mice lacking Scrib in the myocardium survived to adulthood, they developed marked cardiac fibrosis. Scrib did not physically interact with the planar cell polarity (PCP) protein, Vangl2, in early cardiomyocytes as it does in other tissues, suggesting that the anomalies did not result from disruption of PCP signalling. However, Scrib interacted with Rac1 physically in embryonic cardiomyocytes and genetically to result in ventricular abnormalities, suggesting that this interaction is crucial for the development of the early myocardium.
The Scrib–Rac1 interaction plays a crucial role in the organization of developing cardiomyocytes and formation of the ventricular myocardium. Thus, we have identified a novel signalling pathway in the early, functioning, heart muscle. These data also show that the foetus can recover from relatively severe abnormalities in prenatal ventricular development, although cardiac fibrosis can be a long-term consequence.
1. Introduction
Maturation of ventricular cardiomyocytes is poorly understood.1 Abnormalities in the arrangement of cardiomyocytes within the myocardium are associated with a range of adult diseases, including cardiomyopathy, hypertrophy, and heart failure.2 Anything more than minor abnormalities in the formation of the ventricular myocardium during embryonic life are likely to result in death of the embryo. Nevertheless, minor defects may be compatible with survival in the postnatal period. The relationship between abnormal development of the myocardium and adult heart disease, however, remains unclear.
Scrib is a large scaffolding protein that forms complexes with other proteins at the cell junctions of expressing cells.3,4 In Drosophila, scribble plays roles in maintaining apical–basal polarity and its loss is associated with tissue overgrowth suggesting roles in cell proliferation and/or neoplasia.4 Related roles in neoplasia have been identified in mammals,5 with mammalian Scrib shown to play roles in a variety of cellular processes including the establishment of apical–basal and planar cell polarity, migration, proliferation, differentiation, and vesicle trafficking.3,4 Mutations in Scrib are found in the mouse mutant circletail (Crc).6Crc mutants develop a range of developmental defects that affect the neural tube, body wall, and branching organs such as the lung.6,7 These abnormalities have been ascribed to disruption of planar cell polarity (PCP) signalling, at least in part via the interaction of Scrib with the core PCP protein, Vangl2.6,8
Crc mutants also develop congenital heart defects that include abnormalities of the ventricular myocardium, as well as malalignment defects affecting the outflow region and the atrioventricular canal. It was previously assumed that all of these defects result directly from loss of Scrib in the myocardium.9 In this study, we have deleted Scrib solely in the myocardium and shown that this recapitulates the ventricular septal defects and thinned myocardial wall observed in Crc mutants. Interestingly, the mice largely recover from these defects and survive to adulthood, although they develop cardiac fibrosis. Unlike elsewhere in the embryo, interactions between Scrib and Vangl2 do not appear to be crucial for the development of the ventricular myocardium. However, we show that Scrib interacts, both physically and genetically, with Rac1 during formation of the ventricular myocardium.
2. Methods
2.1 Mouse strains and histological analysis
Scribflox,5Rac1flox,10 and Nkx2.5-Cre11 mice were used extensively in these studies. Other Cre drivers, including PGK-Cre,12Isl1-Cre,13Mlc2v-Cre,14Wnt1-Cre,15 and Gata5-Cre,16 were also used to conditionally delete Scrib or Rac1 in the required cell type. Timed matings were carried out overnight and the presence of a copulation plug was designated embryonic day (E) 0.5. All animals were sacrificed by cervical dislocation. All mice were maintained on a C57Bl/6 background, backcrossed for three generations, and then maintained by brother–sister matings. Mice were maintained according to the Animals (Scientific Procedures) Act 1986, UK, under project license PPL 30/3876. All experiments were approved by the Newcastle University Ethical Review Panel and conformed to Directive 2010/63/EU of the European Parliament. Littermate controls were used in all experiments.
Embryos were harvested at different developmental stages, rinsed in ice-cold phosphate-buffered saline, and either fixed overnight in 4% paraformaldehyde (PFA) before paraffin embedding, processing for cryoembedding, or western blotting and co-immunoprecipitation. For basic histological analysis, paraffin-embedded embryos or isolated hearts were sectioned and stained with H&E or alizarin red to detect fibrosis, following standard protocols. A minimum of three mutants and controls were used for each analysis. For analysis of the ventricular wall thickness, ImageJ was used to measure the cross section of the myocardium in three fixed positions (four controls and six mutants), averaged for three measurements in each region. For proliferation and cell death analyses, alternate sections through the E10.5 ventricular myocardium were stained and counted for positive cells, as a percentage of the total number of nuclei. In each case, a minimum of three animals of each genotype were analysed. For quantification of cardiac hypertrophy, adult hearts were dissected at 6 months of age, fixed in 4% PFA then sectioned and stained with H&E. Cardiomyocyte cell nuclei were counted per fixed unit area using the ImageJ analysis software. Five different areas were chosen within the myocardium of each mouse and the average numbers were compared. Due to the relatively small numbers of samples in each data set, the Kruskal–Wallis ANOVA test for non-parametric data was used for statistical analysis.
2.2 Cell culture
Cardiac H9C2 cells17 (undifferentiated neonatal rat cardiomyoblasts) were grown in Dulbecco's modified Eagle's medium, 10% foetal bovine serum, and 5% antibiotics (streptomycin + penicillin) under standard conditions.
2.3 Immunofluorescence
Samples for immunohistochemistry were either fixed in PFA and paraffin-embedded or equilibrated through a sucrose series (to 15%) and subsequently mounted and frozen in OTC (Tissue-tek). In the latter case, air-dried sections were fixed with methanol or 4% PFA for immunostaining. Primary antibodies utilized were Scribble (Santa Cruz); Vangl2 (gift from Dr Charlotte Dean, London, UK), MF20 (DSHB), phospho-histone H3 (Millipore), cleaved caspase-3 (Cell Signalling), sarcomeric α actinin (Abcam), alpha-smooth muscle actin (α-SMA) (Sigma), cardiac troponin I (Hytest Ltd), Rac1 (Millipore), β-PIX (Millipore), β-catenin (BD Transduction Laboratories), N-cadherin (BD Transduction Laboratories), and connexin-43 (Chemicon). Alexa fluor 488 and 596-conjugated secondary antibodies (Invitrogen) were used to detect the primary antibody. Phalloidin (Sigma) was used to stain the actin cytoskeleton and wheat germ agglutinin (Alexa fluor 647; Invitrogen) was used to stain cell membranes. Cell nuclei were identified using DAPI. Immunofluorescence images were collected with using a Zeiss Axioimager Z1 fluorescence microscope equipped with Zeiss Apotome 2 (Zeiss, Germany). Acquired images were processed with the AxioVision Rel 4.9 software.
2.4 Western blotting
H9C2 cells and mouse hearts were stored at −80°C until use for western blotting.18 Cells were lysed and then the extracts were cleared by centrifugation and stored at −80°C until use. The extracts were boiled in 2× Laemmli sample buffer. Samples were than subjected to SDS–polyacrylamide gel electrophoresis followed by western blot analysis using specific antibodies raised against Scrib (Santa Cruz), Vangl2 (Santa Cruz), Rac1 (Millipore), β-PIX (Millipore), Git1 (Novus Biologicals), or β-tubulin (Abcam). Horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were used for detection using the enhanced chemiluminescence method (GE Healthcare BioSciences). Quantification of protein levels was determined by densitometry using the ImageJ software. Band intensities were normalized to β-tubulin. The Kruskal–Wallis ANOVA test for non-parametric data was used for statistical analysis.
2.5 Co-immunoprecipitation
Embryonic hearts (E10.5) were homogenized in ice-cold NP-40 lysis buffer, and the lysates pre-cleared with protein G beads (Pierce, Thermo Scientific) and then incubated with the primary antibody [Scrib from Santa Cruz; Rac1 (Millipore); β-PIX (Millipore), β-catenin (BD Transduction Laboratories) at 4°C, overnight]. Lysates were incubated with protein G beads to precipitate complexes for 2 h at 4°C. The immunocomplexes were recovered by centrifugation, washed, precipitated, and denatured. Proteins were size separated by SDS–polyacrylamide gel electrophoresis and western blot analysis using antibodies as above.
3. Results
3.1 Scribf/f;PGK-Cre recapitulate the cardiac phenotype of Crc mice
To understand the tissue-specific requirement for Scrib function during cardiovascular development, we utilized Scribflox mice (Scribf;5). Crossing these mice to PGK-Cre mice (Figure 1A), deleting Scrib throughout the embryo (Figure 1B), recapitulated the Crc/Crc external phenotype.6 Neural tube defects and gastroschisis were observed in all Scribf/f;PGK-Cre mutants (Table 1 and Figure 1C–E). Sectioning showed that the Scribf/f;PGK-Cre embryos developed heart malformations including abnormalities in the ventricular myocardium, alignment of the outflow vessels with the ventricular chambers, and abnormal positioning of the heart in the chest, recapitulating those seen in Crc mutants9 (Figure 1F–K).
Analysis of phenotypes of Scrib-depleted embryos at E14.5–E15.5
| Genotype | Tissue specificity | Percentage of embryos with external phenotype | External abnormalities observed | Percentage of embryos with heart defect | Types of cardiac malformation |
|---|---|---|---|---|---|
| Scribf/f;PGK-Cre | Somatic and germ cells | 100 | Gastroschisis Neural tube defects | 100 | VSD, DORV Malpositioning in chest |
| Scribf/f;Wnt1-Cre | Neural crest cells | 25 | Exencephaly | 25 | VSD |
| Scribf/f;GATA5-Cre | Epicardium and left ventricular myocardium | 20 | Axial rotation of the lower spine Gastroschisis | 20 | VSD, DORV Malpositioning in chest |
| Scribf/f;Isl1-Cre | Second heart field | 0 | None | 0 | None |
| Scribf/f;Mlc2v-Cre | Myocardium | 0 | None | 40 | Thin ventricular wall |
| Scribf/f;Nkx2.5-Cre | Cardiac progenitors | 0 | None | 100 | Thin ventricular wall, VSD |
| Genotype | Tissue specificity | Percentage of embryos with external phenotype | External abnormalities observed | Percentage of embryos with heart defect | Types of cardiac malformation |
|---|---|---|---|---|---|
| Scribf/f;PGK-Cre | Somatic and germ cells | 100 | Gastroschisis Neural tube defects | 100 | VSD, DORV Malpositioning in chest |
| Scribf/f;Wnt1-Cre | Neural crest cells | 25 | Exencephaly | 25 | VSD |
| Scribf/f;GATA5-Cre | Epicardium and left ventricular myocardium | 20 | Axial rotation of the lower spine Gastroschisis | 20 | VSD, DORV Malpositioning in chest |
| Scribf/f;Isl1-Cre | Second heart field | 0 | None | 0 | None |
| Scribf/f;Mlc2v-Cre | Myocardium | 0 | None | 40 | Thin ventricular wall |
| Scribf/f;Nkx2.5-Cre | Cardiac progenitors | 0 | None | 100 | Thin ventricular wall, VSD |
VSD, ventricular septal defect; DORV, double outlet right ventricle.
Analysis of phenotypes of Scrib-depleted embryos at E14.5–E15.5
| Genotype | Tissue specificity | Percentage of embryos with external phenotype | External abnormalities observed | Percentage of embryos with heart defect | Types of cardiac malformation |
|---|---|---|---|---|---|
| Scribf/f;PGK-Cre | Somatic and germ cells | 100 | Gastroschisis Neural tube defects | 100 | VSD, DORV Malpositioning in chest |
| Scribf/f;Wnt1-Cre | Neural crest cells | 25 | Exencephaly | 25 | VSD |
| Scribf/f;GATA5-Cre | Epicardium and left ventricular myocardium | 20 | Axial rotation of the lower spine Gastroschisis | 20 | VSD, DORV Malpositioning in chest |
| Scribf/f;Isl1-Cre | Second heart field | 0 | None | 0 | None |
| Scribf/f;Mlc2v-Cre | Myocardium | 0 | None | 40 | Thin ventricular wall |
| Scribf/f;Nkx2.5-Cre | Cardiac progenitors | 0 | None | 100 | Thin ventricular wall, VSD |
| Genotype | Tissue specificity | Percentage of embryos with external phenotype | External abnormalities observed | Percentage of embryos with heart defect | Types of cardiac malformation |
|---|---|---|---|---|---|
| Scribf/f;PGK-Cre | Somatic and germ cells | 100 | Gastroschisis Neural tube defects | 100 | VSD, DORV Malpositioning in chest |
| Scribf/f;Wnt1-Cre | Neural crest cells | 25 | Exencephaly | 25 | VSD |
| Scribf/f;GATA5-Cre | Epicardium and left ventricular myocardium | 20 | Axial rotation of the lower spine Gastroschisis | 20 | VSD, DORV Malpositioning in chest |
| Scribf/f;Isl1-Cre | Second heart field | 0 | None | 0 | None |
| Scribf/f;Mlc2v-Cre | Myocardium | 0 | None | 40 | Thin ventricular wall |
| Scribf/f;Nkx2.5-Cre | Cardiac progenitors | 0 | None | 100 | Thin ventricular wall, VSD |
VSD, ventricular septal defect; DORV, double outlet right ventricle.
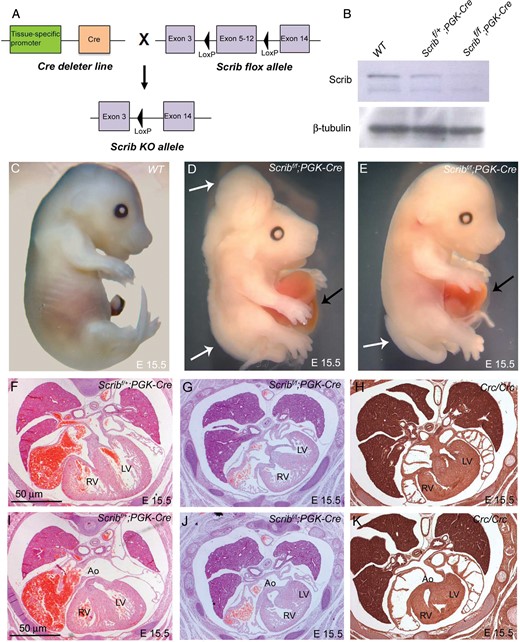
Cardiac anomalies in Scribf/f;PGK-Cre embryos. (A) Schematic representation of breeding strategy to obtain Scrib-deleted mice. (B) Western blotting (n = 3) showed that Scrib protein levels were markedly reduced in Scribf/f;PGK-Cre at E15.5. β-Tubulin was used as a loading control. (C–E) Scribf/f;PGK-Cre embryos display neural tube defects (white arrows) and gastroschisis (black arrows) at E14.5. (F and I) Scribf/+;PGK-Cre have normal hearts. (G and J) Scribf/f;PGK-Cre exhibit double outlet right ventricle, ventricular septal defects, and septal hyperplasia that closely resemble those seen in Crc/Crc (H and K). See Table 1 for numbers of animals analysed. Ao, aorta; LV, left ventricle; RV, right ventricle. Scale bar = 50 μm.
3.2 Scrib is required in myocardial progenitors for normal development of the ventricular myocardium
As the global knockout of Scrib encompassed the Crc cardiac phenotype, we set out to investigate the role of different cell lineages in creating this phenotype. Tissue-specific deletion of Scrib using Nkx2.5-Cre (myocardial progenitors) had no effect on the external phenotype of the embryos (Figure 2A,C). However, ∼25% of the mutants died in utero by E14.5 (2 of 7; Table 1). Isolated hearts appeared less mature than control littermates at E14.5 (Figure 2B,D), suggesting that there might be an abnormality in the development of the heart muscle. Defects in the ventricular myocardium were observed as early as E11.5 in the mutant embryos, manifested as stunted trabeculation and poor formation of the interventricular septum (Figure 2E–J). Closer analysis using wheat germ agglutinin staining to reveal cell boundaries showed that the cellular architecture of the trabeculae was disrupted in Scribf/f;Nkx2.5-Cre compared with control littermates (Figure 2G,J). Sectioning of E14.5 hearts revealed peri-membranous and muscular ventricular septal defects, often in combination with thinned ventricular wall (4 of 7; Figure 2K–O). Abnormalities in the formation of the muscular ventricular septum were also found in around half of the Scribf/f;Mlc2v-Cre embryos (in which Scrib was deleted in ventricular cardiomyocytes) examined at E14.5 (Table 1 and see Supplementary material online, Figure S1). Strikingly, there were no outflow malalignment defects or abnormalities in heart positioning in any of the Scribf/f;Nkx2.5-Cre or Scribf/f;Mlc2v-Cre embryos examined, indicating that these phenotypes did not result from direct roles of Scrib in the myocardium.
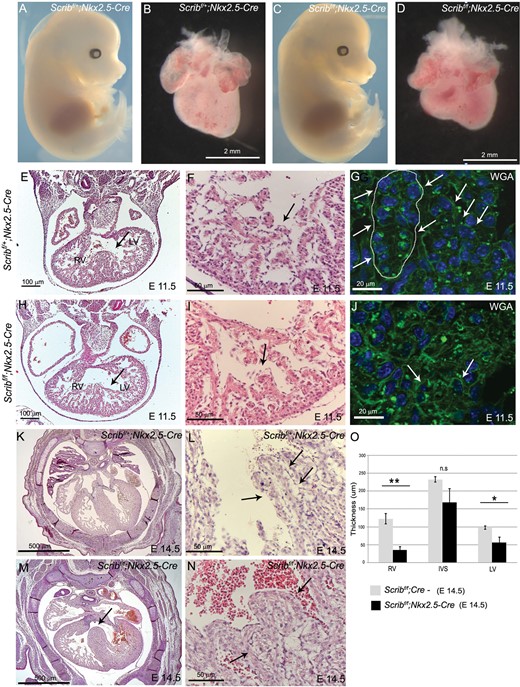
Defects in the ventricular myocardium in Scribf/f;Nkx2.5-Cre. (A–D) Scribf/f;Nkx2.5-Cre mouse mutants have a normal external phenotype at E14.5, whereas the heart appears smaller and immature when compared with control littermates. See Table 1 for numbers of animals analysed. (E–J) Transverse sectioning of E11.5 Scribf/f;Nkx2.5-Cre hearts (n = 5) revealed abnormalities in the trabeculae and in the formation of the interventricular septum (arrows). High-power views of the trabeculae show that the trabecular network (arrow) is less well developed in the mutant hearts than in control littermates (compare I with F). Labelling of the cell membranes with wheat germ agglutinin reveals that the cellular architecture of the trabeculae (arrows) appears more disorganized in the mutant (J) than in the control (G) (n = 3). (K–N) Transverse sections of Scribf/f;Nkx2.5-Cre hearts at E14.5 reveal peri-membranous septal defects (arrow). High-power views show that the trabeculae (arrows) appear thickened in the mutants (N) compared with control littermates (L). (O) Measurements (n = 6) revealed significant differences in the thickness of the right and left ventricular walls, but not the septum. Error bars represent standard deviation, *P < 0.05. LV, left ventricle; RV, right ventricle; WGA, wheat germ agglutinin. Scale bar: B,D = 2 mm. E,H = 100 µm, F,I,L,N = 50 µm, G,J = 20 µm, K,M = 500 µm.
To investigate the cause of the abnormalities in the formation of the trabeculae within the ventricle, we carried out proliferation and cell death analyses at E10.5, before an obvious abnormality in the ventricular myocardium was apparent in the Scribf/f;Nkx2.5-Cre hearts. However, there were no significant differences in either parameter between mutant and control littermates (Figure 3A,B). We carried out immunohistochemsitry for a range of proteins, including filamentous actin, myosin heavy chain, α-SMA, α-actinin, and cardiac troponin-I, which label developing cardiomyocytes. These markers showed that there was abnormal expression of some differentiated cardiomyocyte markers in the mutant hearts with a delay in the formation of Z-lines as shown by α-SMA and α-actinin (Figure 3G–J). Moreover, there was reduced expression of cardiac troponin I, particularly in the trabeculae (Figure 3K,L). These markers were, however, indistinguishable from controls by E14.5 (data not shown).
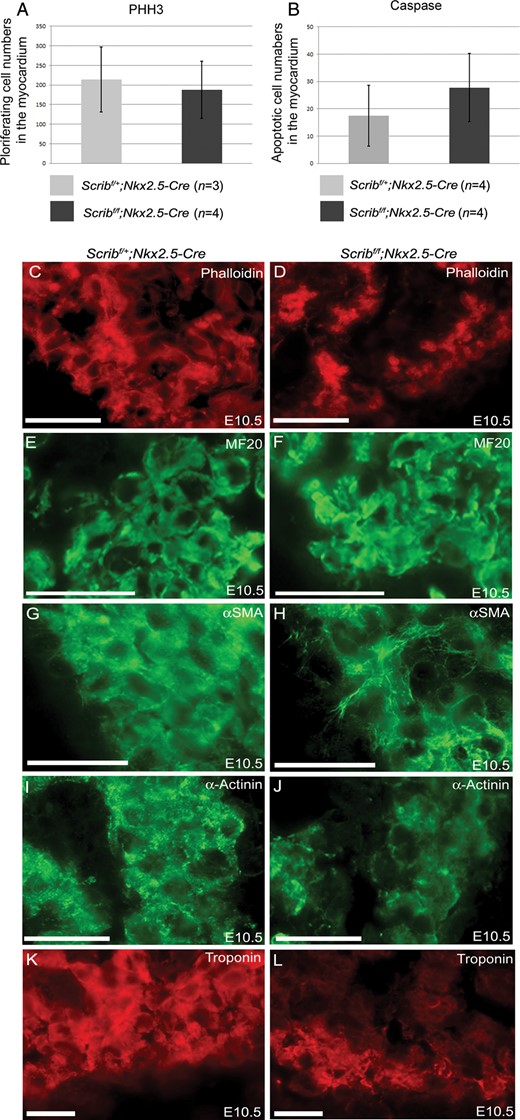
Abnormal expression of cardiomyocyte markers in Scribf/f;Nkx2.5-Cre. (A and B) Neither proliferation (pHH3) nor cell death (cleaved caspase-3) was altered in myocardium from Scribf/f;Nkx2.5-Cre ventricles compared with control littermates at E10.5. (C–L) While phalloidin and MF20 staining did not show any reproducible differences between control and mutant cardiomyocytes in the E10.5 ventricle (C–F), analysis of other markers suggested that the Scribf/f;Nkx2.5-Cre ventricles were immature in comparison with control littermates; whereas striations suggesting the presence of sarcomeres (labelled by α-SMA and α-actinin) were readily apparent in control cells, these were absent in the mutant cells (H and J). In addition, there was markedly reduced expression of cardiac troponin I (K and L) in Scribf/f;Nkx2.5-Cre trabeculae. n = 3 for all experiments. Scale bar = 20 µm.
To investigate whether the ventricular maturation abnormalities we observed had long-term consequences, we allowed Scribf/f;Nkx2.5-Cre litters to be born. Four of five Scribf/f;Nkx2.5-Cre animals survived to 6 months of age. Heart weight to body weight ratios (mg/g) were not significantly different between Scribf/f;Nkx2.5-Cre and control littermates (Figure 4 and see Supplementary material online, Table S1). Cardiomyocytes appeared normal in the Scribf/f;Nkx2.5-Cre hearts with no evidence of cardiac hypertrophy; however, there was evidence of increased cardiac fibrosis within the interstitium of the ventricular myocardium as shown by staining for Sirius Red (Figure 4C–H). Fibrosis was also seen in Scribf/f;Mlc2v-Cre animals (data not shown). Thus, these data suggest that the mutant hearts largely recover from the abnormalities in the ventricular myocardium seen earlier in gestation, although cardiac fibrosis develops as a consequence.
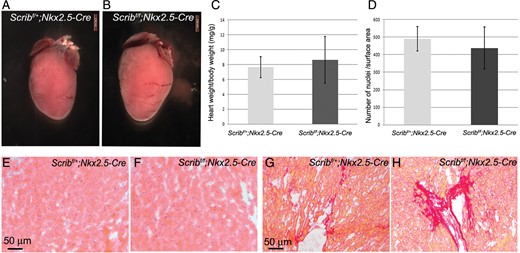
Surviving Scribf/f;Nkx2.5-Cre hearts develop cardiac fibrosis. (A–C) Six-month-old Scribf/f;Nkx2.5-Cre hearts appeared slightly larger than control littermates, although heart weight:body weight ratios showed that this was variable and not statistically significant. Error bars in C represent standard deviation. (D–F) H&E staining of myocardium shows no obvious signs of abnormalities in Scribf/f Nkx2.5-Cre hearts. Quantification of cardiomyocyte nuclei/unit surface area showed no significant difference compared with control littermates (D). (G and H) Sirius Red staining revealed increased fibrosis of the ventricular myocardium of Scribf/f;Nkx2.5-Cre mice compared with control littermates.
3.3 Cardiac malalignment defects are secondary to other defects
As deletion of Scrib solely in cardiomyocytes did not explain the cardiac malalignment defects observed in Crc mutants, we wanted to establish which other cell type might require Scrib function for normal heart development. We utilized a number of other mouse Cre driver lines that direct expression to specific cell types during heart development, including Wnt1-Cre [neural crest cells (NCC)], Isl1-Cre (second heart field), and Gata5-Cre (epicardium and left ventricular myocardium). There was no external phenotype associated with deletion of Scrib from these cell types, except in two individual embryos (out of a total of 18) where exencephaly and gastroschisis were seen (Table 1). In each case, the conditionally null mutants with a normal external phenotype had normal hearts. In contrast, a ventricular septal defect was observed in the single Scribf/f;Wnt1-Cre embryo that developed exencephaly, and a double outlet right ventricle and ventricular septal defect were found in the single Scribf/f;Gata5-Cre embryo that manifested gastroschisis (Table 1 and see Supplementary material online, Figure S2). As all Scribf/f;PGK-Cre and Crc/Crc embryos displayed neural tube defects and/or gastroschisis, these data suggest that the cardiac malalignment and positioning defects are secondary to the gross abnormalities in body patterning also observed in these mutants.
3.4 Scrib:Vangl2 interactions are not crucial for ventricular development
Our previous studies suggested that Scrib might interact with the PCP protein, Vangl2, in the heart.9 To begin to uncover the roles these two proteins might play in developing cardiomyocytes, we co-localized each with junctional proteins at E8.5 and E10.5. Whereas Scrib did not co-localize with markers of tight junctions [Zona occludins 1 (ZO-1)], desmosomes (desmoplakin), or focal adhesions (vinculin) (data not shown), it did co-localize with both β-catenin and N-cadherin in E8.5 and E10.5 myocardium (Figure 5A,B and see Supplementary material online, Figure S3). Moreover, Scrib also co-localized with the gap junction protein, connexin-43, in developing cardiomyocytes (Figure 5C,D). In contrast, Vangl2 was mostly cytoplasmic in developing cardiomyocytes, with only minimal localization to cell membranes (Figure 5E–H and data not shown). This contrasted with its expression in epithelial tissues such as the pharyngeal endoderm, where it was closely associated with the cell membrane (see Supplementary material online, Figure S3). Thus, while Vangl2 and Scrib were both associated with the cell membrane, although to differing extents, they did not obviously co-localize in ventricular cardiomyocytes (Figure 5I–K). In support of this, Scrib and Vangl2 failed to co-immunoprecipitate from a protein extract from H9C2 cardiomyocytes (Figure 5L). Thus, we conclude that although Vangl2 and Scrib genetically and physically interact in some tissues, there is no direct interaction in the developing myocardium, at least before the phenotype becomes apparent in the Scribf/f;Nkx2.5Cre embryos.
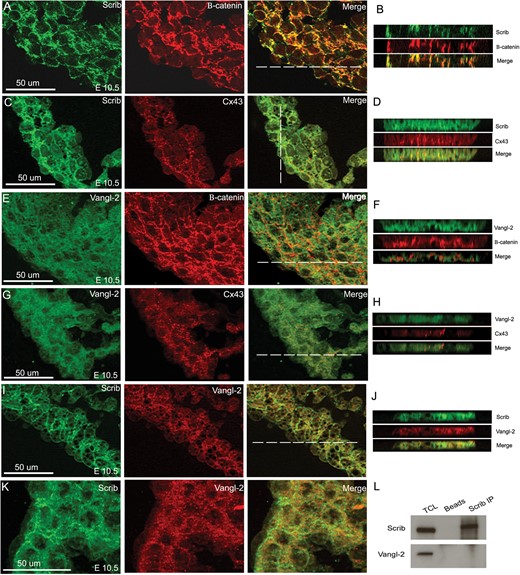
Scrib co-localizes with adherens junctions and gap junctions in the developing myocardium. (A–D) Scrib localizes to cardiomyocyte cell membranes at E10.5, co-localizing with β-catenin (A and B) and connexin-43 (C and D). (E–H) In contrast, Vangl2 is found in the cytoplasm of the ventricular cardiomyocytes, with reduced staining in the cell membrane where there is no evidence of co-localization with β-catenin (E and F) or connexin-43 (G and H). (I–K) Although both Scrib and Vangl2 are found in cardiomyocytes, they do not appear to co-localize at E10.5. The position of the acquired z-axis images (B, D, F, H, and J) are indicated by the horizontal white line on the composite images. (L) Co-immunoprecipitation with Scrib antibody in H9C2 cell lysate confirmed no physical interaction with Vangl2. n = 3 for all experiments. Scale bar = 50 µm.
3.5 Scrib interacts with β-PIX and Rac1 in the developing myocardium
We sought to identify potential Scrib-interacting proteins that might be important in the developing myocardium. Scrib interacts with β-PIX, a guanosine exchange factor for the RhoGTPAse Rac1, in epithelial and neuronal cells.19,20 As RhoGTPases, including Rac1, are known to play crucial roles in processes such as cell adhesion and cell shape changes,21 we reasoned that Scrib might also be interacting with these factors in the developing myocardium.
We first carried out fluorescent immunohistochemistry for Scrib, β-PIX, and Rac1 in E8.5 and E10.5 hearts, revealing that β-PIX and Rac1 co-localize with Scrib in the developing myocardium (Figure 6A–H and data not shown). We next carried out co-immunoprecipitation reactions for endogenous Scrib, β-PIX, and Rac1 in H9C2 cells. Both Rac1 and β-PIX were immunoprecipitated by Scrib from the H9C2 cell lysate (Figure 6I). In contrast, β-catenin was not, despite also co-localizing with Scrib at the cell membrane. The experiment was repeated using crude lysates from isolated hearts from E10.5 embryos. Again, both β-PIX and Rac1, but not β-catenin, were pulled down with the Scrib antibody (Figure 6I). These results clearly demonstrate that both β-PIX and Rac1 are present in a protein complex with Scrib not only in H9C2 cells, but also in embryonic cardiomyocytes.
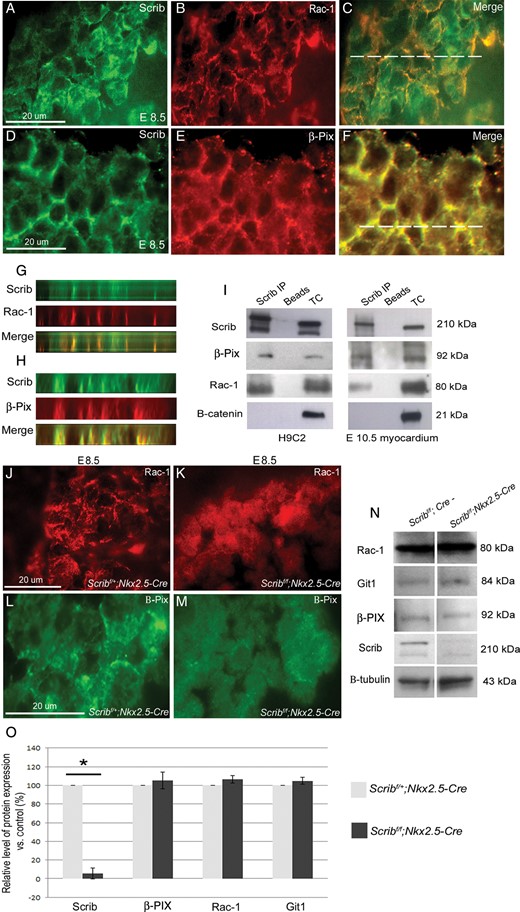
Scrib, β-PIX, and Rac1 interact in the developing myocardium. (A–H) Immunostaining for Scrib (A and D), Rac1 (B), and β-PIX (E) in the E8.5 myocardium reveals overlapping localization at the cardiomyocyte membrane (C and F). The position of the acquired z-axis images (G and H) is indicated by the horizontal white line on the composite images (C and F). Co-localization (yellow) of Scrib/Rac1/β-PIX can be seen (G and H). (I) Co-immunoprecipitation was performed in lysates from H9C2 cells and in crude E10.5 heart homogenate (n = 3). Scrib forms protein complexes with both β-PIX and Rac1. No precipitation of β-catenin with Scrib was observed. (J–M) Rac1 and β-PIX are lost from the cell membrane in the Scrib-depleted myocardium when compared with control sections. (N and O) Western blot analysis was performed for Scrib, Rac1, Git-1, β-PIX, and β-tubulin (as a loading control), in homogenates from E10.5 control and Scribf/f;Nkx2.5-Cre hearts (n = 3 for each). The graphs display the percentage of protein expression in the mutant relative to the control. Only Scrib was significantly reduced in the Scribf/f;Nkx2.5-Cre relative to the control, *P < 0.05. Error bars represent standard deviation. Scale bar = 20 µm (A–F,J–M).
3.6 Scrib is required for membrane association of β-PIX and Rac1 in cardiomyocytes
To investigate the relevance of the Scrib:Rac1:β-PIX interaction, we examined the expression of Rac1 and β-PIX in Scribf/f;Nkx2.5-Cre mutant and control littermate hearts at E8.5 and E10.5. Both were depleted from the cardiomyocyte membrane in the Scrib-deficient myocardium (Figure 6J–M and data not shown). Western blotting was carried out using protein extracts from isolated E10.5 Scribf/f;Nkx2.5-Cre hearts and from control littermates. These analyses showed that the levels of Rac1 and β-PIX were similar in the Scribf/f;Nkx2.5-Cre mutant hearts, compared with control littermates. Moreover, the levels of GIT1, another component of the β-PIX/Rac1 complex, were also similar in control and mutant hearts (Figure 6N,O). Thus, these data support the idea that Scrib, as a scaffolding protein, may stabilize the Rac1/β-PIX complex at the membrane of cardiomyocytes, rather than regulate the level of its expression.
3.7 Rac1 plays essential roles in the development of the myocardium
We next wanted to test the in vivo importance of the Scrib/β-PIX/Rac1 interaction in myocardial development. Although β-PIXflox mice are not available, Rac1flox (Rac1f) mice have been described.9 To test the idea that Rac1 plays crucial roles in the developing myocardium, we intercrossed Rac1f mice with Nkx2.5-Cre mice. At E10.5–E11.5, the Rac1f/f;Nkx2.5-Cre embryos (n = 4) were well formed and of normal size (data not shown). At E12.5, Rac1f/f;Nkx2.5-Cre embryos showed marked cardiac oedema (5/5; Figure 7A,B) and were dead by E13.5 of gestation. Sectioning of the E12.5 embryos revealed significant cardiac abnormalities, including small, underdeveloped ventricles with a thinned ventricular wall (Figure 7C,D). Thus, Rac1 is essential for the normal development of the ventricular myocardium.
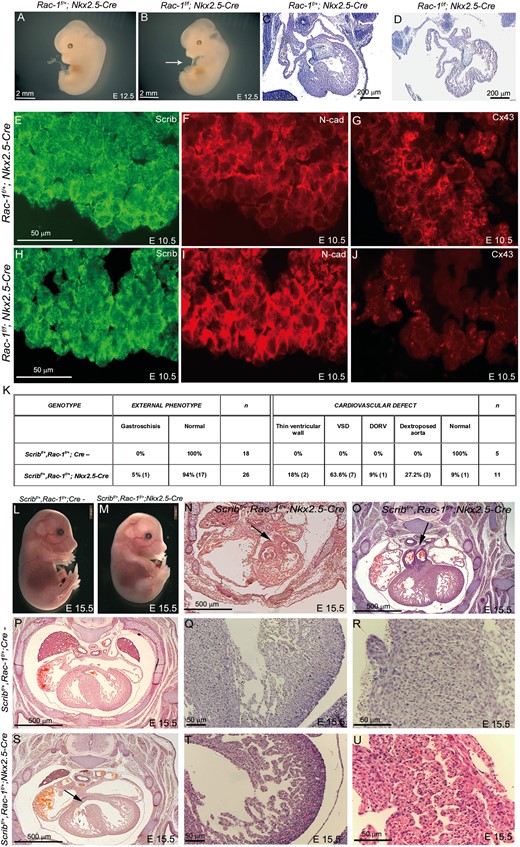
Rac1 is required for normal development of the myocardium. (A and B) Rac1f/f;Nkx2.5-Cre embryos develop cardiac oedema by E12.5 (white arrow in B). (C–D) Sectioning of Rac-1f/f;Nkx2.5-Cre at E12.5 reveals an immature heart with thin ventricular walls and a poorly developed interventricular septum. (E–J) Fluorescent immunostaining for Scrib, N-cadherin, and connexin-43 at E10.5 reveals mislocalization of Scrib and N-cadherin in the Rac1f/f;Nkx2.5-Cre, particularly in the outer region of the ventricular wall, and loss of connexin-43. (K) Table showing numbers of Scribf/+;Rac-1f/+;Nkx2.5-Cre embryos with an external or cardiac phenotype, compared with littermate controls. (L and M) The majority (16/18) of Scribf/+;Rac1f/+;Nkx2.5-Cre had no external phenotype at E15.5. (N–U) Transverse sectioning of control and Scribf/+;Rac1f/+;Nkx2.5-Cre embryos reveals double outlet right ventricle (N,O; black arrows) and ventricular septal defects in the latter (S; black arrow). The ventricular myocardium is immature in the double heterozygotes (T), compared with control littermates (Q), and the interventricular septum is poorly compacted with channels running through it (compare U with R). Scale bar: A,B,L,M = 2 mm; C,D = 200 µm, E–J,O R,T,U = 50 µm; P,S,N,O = 500 µm.
At E10.5, Rac1f/f;Nkx2.5-Cre and control littermates were analysed for the expression of Scrib. Scrib staining was much more diffuse in these hearts, particularly in the ventricular wall where Rac1 and Scrib are found at a highest level (Figure 7E,H). Moreover, N-cadherin and β-catenin were lost from the cell membrane in Rac1f/f;Nkx2.5-Cre ventricular cardiomyocytes, with increased cytoplasmic staining (Figure 7F,I and data not shown). More obviously still, expression of connexin-43 was almost completely lost from cardiomyocytes in Rac1f/f;Nkx2.5-Cre embryos (Figure 7G,J). Taken together, these data support the idea that Scrib and Rac1 act together to regulate junctional complexes in cardiomyocytes.
3.8 Scrib and Rac1 genetically interact in the development of the ventricular myocardium
Finally, we wanted to test directly the idea that Scrib and Rac1 act together in the ventricular myocardium. To do this, Rac1f, Scribf, and Nkx2.5-Cre mice were intercrossed. Of 18 Scribf/+;Rac1f/+;Nkx2.5-Cre embryos examined at E15.5, one was dead at the time of collection and one had gastroschisis (Figure 7K). The remaining 16 embryos were externally normal (Figure 7L,M). Serial sectioning revealed cardiac malformations in 11/11 of the Scribf/+;Rac1f/+;Nkx2.5-Cre embryos examined at E15.5. The most common malformation observed was ventricular septal defect, with thinned ventricular wall, dextraoposed aorta, and double outlet right ventricle also observed (Figure 7K,N–U). No cardiac malformations were observed in any of the control littermates (0/5 sectioned; Figure 7K). These data support the idea that the Scrib–Rac1 interaction is playing an essential role in the development of the ventricular myocardium.
4. Discussion
We show here that a protein complex that forms at the cell membrane, composed of Scrib, β-PIX, and Rac1, plays an essential role in the early ventricular myocardium. Disruption of this complex leads to loss of key proteins from the cardiomyocyte cell membrane, disturbed cellular architecture, and results in structural abnormalities of the ventricular myocardium including ventricular septal defects. Deletion of Scrib in either cardiomyocyte progenitors (with Nkx2.5-Cre) or differentiating ventricular myocardium (with Mlc2v-Cre) results in ventricular septal defects. There are several possible explanations for why these defects might arise, including abnormalities in cell proliferation or death, cell migration into the heart, and interactions between cardiomyocytes and maturation of the ventricle. Cell proliferation and cell death analyses, carried out before the appearance of the ventricular anomalies, showed there were no differences in these parameters between control and mutants at stages immediately before abnormalities become apparent. It seems highly unlikely that abnormalities in cell migration into or within the heart cause the defects, as Scribf/f;Wnt1-Cre and Scribf/f;Isl1-Cre, in which Scrib is deleted in migratory NCC and second heart field cells, respectively, are phenotypically normal. Moreover, the migration of epicardially derived cells into the myocardium, which is known to be crucial for ventricular development, occurs after the appearance of the defects in the Scribf/f;Nkx2.5-Cre mutants. We have previously shown that the distribution of junctional proteins, including N-cadherin and ZO-1, is abnormal in cardiomyocytes from the Scrib mutant, Crc, and that the cardiomyocytes appear disorganized in the early heart.9 To complement this, we show here that there is an abnormality in the cellular architecture of the trabeculae. Moreover, the mutant myocardium appears immature and markers of differentiated cardiomyocytes show delayed expression. Scrib is known to be associated with junctional complexes between cells and thus, abnormalities in the relationships between adjacent cardiomyocytes could lead to delayed and abnormal formation of trabeculae, which could in itself impact on the development of the ventricular septum. These defects in myocardial architecture, although subtle, persist through foetal life, with abnormal patterning of the trabeculae and growth of the ventricular septum. Analysis of these mice at 6 months of age showed that their heart was well formed with no obvious signs of hypertrophy, although there was marked fibrosis of the ventricular myocardium. Thus, the mice largely recover from, or at least tolerate, the early anomalies in the ventricular myocardium. This is analogous to the situation in humans, where muscular ventricular septal defects frequently close spontaneously during infancy.22 Moreover, fibrosis is a common finding in adults with unrepaired ventricular septal defects.23 Fibrosis is initially a reparative process acting to maintain the functional integrity of the myocardium, but can have adverse consequences for ventricular mobility if it becomes widespread. Thus, it is possible that the Scribf/f;Nkx2.5-Cre mice might progress to heart failure in the longer term, although this analysis was outside the scope of this study.
Mice lacking functional Scrib, in the Crc mutant, have a range of classical PCP defects that include shortened body axis and neural tube defects.6,24 A similar pattern of anomalies are observed in other PCP mutants, including the Vangl2 mutant loop-tail (Lp), and the genetic link between these abnormalities and the PCP pathway has been confirmed.6,9 Although our previous studies suggested that Crc and Lp mutants interact genetically to regulate cardiac development,9 and Lp mutants have significant myocardial disorganization25 similar to that seen when Scrib is lost from the myocardium, the current study suggests that this is unlikely to be a consequence of a physical interaction between the two proteins. Whilst Scrib and Vangl2 are known to interact in some tissues,8 we cannot show this in cardiomyocytes. Thus, Scrib appears to act outside the Vangl2-associated PCP pathway in the early ventricular myocardium.
Although we were able to confirm crucial roles for Scrib in the development of the myocardium, we show here that cardiac malalignment defects are likely secondary to the neural tube defects and gastroschisis observed when Scrib is lost throughout the whole embryo. Cardiac malalignment was only observed in the presence of these severe embryonic patterning defects. Gastroschisis grossly distorts the abdominal wall and thus may well directly affect positioning of the heart in the chest, mechanically disrupting normal heart remodelling. Thus, caution must be observed when interpreting cardiac phenotypes and determining causality in the presence of other embryonic patterning abnormalities. Indeed, the high incidence of cardiovascular defects found in babies with either neural tube or body wall defects, reported to be as high as 40%,26,27 may reflect the possibility that the heart malformations are a secondary consequence of the other structural anomalies.
Although our data rule out direct interactions with Vangl2 in developing cardiomyocytes, Scrib has been shown to interact with a number of other proteins. For example, Scrib interacts with β-PIX in a number of cell types. Scrib localization to the cell membrane has been shown to be required for Rac1/β-PIX exocytosis in neuroendocrine cells,20 whereas Scrib function has been shown to be required for recruitment of Rac1 to the lamellipodium in a mammary cell line28 and in migrating fibroblasts.29 Our study shows that Scrib, β-PIX, and Rac1 are found in a complex in native protein extracts from cardiomyocyte cell lines and hearts from early mouse embryos. Moreover, loss of either Scrib or Rac1 results in a reduction in the membrane association of the other component, although not a reduction in the overall levels of the protein. The overall phenotype of Scribf/f;Nkx2.5Cre and Rac1f/f;Nkx2.5Cre embryos is similar at E12.5, although the latter is more severe and the mice die by E13.5, before ventricular septation and alignment with the outflow vessels is complete. Scribf/+;Rac1f/+;Nkx2.5-Cre (double heterozygote) embryos also have a similar phenotype, with abnormalities in the formation of the ventricular myocardium, although in this case there are additional abnormalities in the ventriculo-arterial connections that are not apparent in Scrib mutants. It is possible though that they might have been observed in Rac1f/f;Nkx2.5Cre embryos, if these had survived long enough for the abnormalities to become apparent. Although Rac1 has not been previously implicated in myocardial development, as Rac1 null mice die early in development before the heart is well formed,30 it has been implicated in adult cardiovascular pathologies. For example, loss of Rac1 from cardiomyocytes is protective against angiotensin II-induced hypertrophy.31 In contrast, expression of a constitutively active form of Rac1 in the mature myocardium leads to severe dilated cardiomyopathy and early death,32 a result that correlates with the finding of elevated levels of RAC1 in human patients with cardiac hypertrophy and a range of other cardiovascular pathologies.33 Our data suggest that Rac1 also plays a crucial role in the developing myocardium. The observations that Rac1 is required for cardiomyocyte alignment in response to mechanical stress, at least in vitro,34 and that N-cadherin signals via Rac1 to localize connexin-43 in cardiomyocytes,35 support the idea that Scrib–Rac1 signalling may be required for the organization and maturation of the developing cardiomyocytes. The loss of β-catenin, N-cadherin, and connexin-43 from the membrane of Rac1f/f;Nkx2.5-Cre myocardial cells implies a role in cardiomyocyte–cardiomyocyte contacts and suggests that this pathway may be crucial not only during development, but also in cardiac disease.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by the British Heart Foundation (PG/07/086 and PG/11/76/29108). Funding to pay the Open Access publication charges for this article was provided by the British Heart Foundation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}