




Abstract
The Ras–Erk pathway is frequently overactivated in human tumors. Neurofibromatosis types 1 and 2 (NF1, NF2) are characterized by multiple tumors of Schwann cell origin. The NF1 tumor suppressor neurofibromin is a principal Ras-GAP accelerating Ras inactivation, whereas the NF2 tumor suppressor merlin is a scaffold protein coordinating multiple signaling pathways. We have previously reported that merlin interacts with Ras and p120RasGAP. Here, we show that merlin can also interact with the neurofibromin/Spred1 complex via merlin-binding sites present on both proteins. Further, merlin can directly bind to the Ras-binding domain (RBD) and the kinase domain (KiD) of Raf1. As the third component of the neurofibromin/Spred1 complex, merlin cannot increase the Ras-GAP activity; rather, it blocks Ras binding to Raf1 by functioning as a ‘selective Ras barrier’. Merlin-deficient Schwann cells require the Ras–Erk pathway activity for proliferation. Accordingly, suppression of the Ras–Erk pathway likely contributes to merlin’s tumor suppressor activity. Taken together, our results, and studies by others, support targeting or co-targeting of this pathway as a therapy for NF2 inactivation-related tumors.
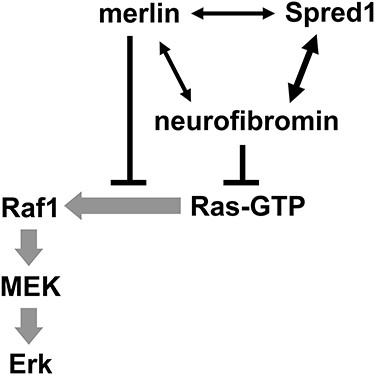
Introduction
The small GTPase Ras is a molecular switch controlling diverse cellular processes including proliferation, differentiation and survival (1). Ras is activated by its guanine nucleotide exchange factors, which promote GTP loading, and inactivated by its GTPase activating proteins (GAPs), which accelerate intrinsic Ras-GTP hydrolysis into Ras-GDP. Ras transduces, for example, growth factor receptor activation signals to effector pathways. The Ras–Raf–MEK–Erk pathway is the most frequently activated pathway in human tumors. Germline mutations in this pathway cause developmental syndromes, collectively termed RASopathies (1).
Neurofibromatosis types 1 and 2 (NF1, NF2) are both genetic disorders characterized by the development of multiple nerve sheath tumors of Schwann cell origin, featuring neurofibromas and schwannomas, respectively (2). The NF1 gene product neurofibromin is a principal Ras-GAP, which is also inactivated in many other types of tumors. The NF2 gene product merlin is a scaffold protein, critically involved in contact inhibition of cell proliferation (CICP) (2). Although merlin appears to coordinate multiple signaling pathways, including Hippo, Ras, Rac, mTORC1 and Wnt (3), many details of how exactly merlin regulates these pathways remain obscure. We have previously reported that merlin can interact with Ras and p120RasGAP (4). Here, we show that it can also interact with the neurofibromin/Spred1 complex and Raf1. Merlin does not increase the Ras-GAP activity of neurofibromin, but it does cooperate with this activity to suppress Raf1 activation. Suppression of the Ras–Erk pathway likely contributes to merlin’s tumor suppressor activity. Our results, in tandem with other studies, support targeting or co-targeting of this pathway as a therapy for NF2 inactivation-related tumors.
Results
Increased Erk1/2 activity in merlin-deficient rat Schwann cells in the absence of general upregulation of receptor tyrosine kinases
Because the cells do not undergo replicative senescence in culture (5), we employed rat Schwann cells (RSCs) to investigate merlin signaling. RSCs were hypersensitive to CICP, which triggered morphologic transition from round to bipolar or tripolar shape (Fig. 1A). Stable knockdown (KD) of merlin by lentivirus consistently resulted in loss of CICP (Fig. 1A) and even focus formation (Supplementary Material, Fig. S1). Merlin has two major isoforms (Iso; Supplementary Material, Fig. S2A and B) (6). Confluent RSCs and SV40 large T antigen partially transformed RSCs (NSLTs) (7) predominantly expressed merlin Iso2 at the mRNA level (Supplementary Material, Fig. S2A). Re-expression of merlin Iso1 or Iso2 in merlin-KD RSCs reversed the loss of CICP phenotype (Supplementary Material, Fig. S2C and D), indicating that loss of CICP by merlin-KD was not an off-target effect.
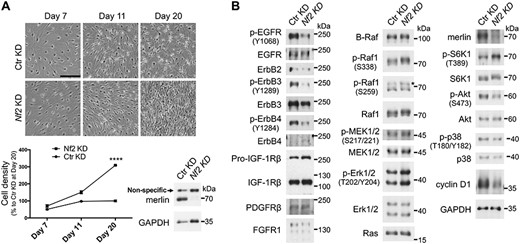
Loss of merlin causes loss of CICP in RSCs. (A) Cells were infected with lentiviruses on day 0 and selected and monitored in situ without passaging. Cell number was counted from at least three representative regions, normalized to that of Ctr KD at day 20 and represented as mean ± SD. ****P ≤ 0.0001 (day 20, t-test, n = 3). Representative results of more than three independent experiments are shown. Scale bar, 250 μm. (B) Immunoblot analysis of signaling profiles of RSC derivatives. The asterisk indicates the bands with correct size. Cells were transduced, amplified, replated, grown to confluence and directly lysed on dish with 2× SDS sample buffer. See Supplementary Material, Fig. S3 for the cell status before lysis. Note that the p-EGFR antibody may cross-react weakly with other tyrosine-phosphorylated proteins and that both the p-EGFR and the p-ErbB4 antibodies have strong cross-reactivity with p-ErbB2 (data not shown). This figure was assembled from blots of replicated loading of the same samples; many of the targets have been probed twice or more for confidence.
The signaling changes due to merlin loss were analyzed by immunoblotting (Fig. 1B). Most of the assessed receptor tyrosine kinases (RTKs) were downregulated in merlin-KD cells, including ErbB3 and ErbB2, the receptor pair for neuregulin 1 (NRG1), the major growth factor for Schwann cells (8). Quantification of replicate blots revealed a ~15 to 20% and ~30% decrease of ErbB2 and ErbB3 in merlin-KD RSCs, respectively (Supplementary Material, Fig. S4). Only IGF-1Rβ was clearly upregulated. No change of B-Raf was observed, whereas Raf1 level was slightly increased in merlin-KD cells. Raf1 also showed a mobility shift, consistent with the hyperphosphorylation at multiple sites mainly mediated by Erk1/2 as a negative feedback (9). Normalization of p-Raf1 (S338) to the total Raf1 levels showed a ~10% increase in merlin-KD RSCs (Supplementary Material, Fig. S5), suggesting that the mobility shift largely resulted from phosphorylation at other sites. Consistent with Raf1 hyperphosphorylation, the active p-Erk1/2 was increased in merlin-KD cells, whereas the change in p-MEK1/2 was very subtle. By contrast, p38 MAPK level and phosphorylation were decreased in merlin-KD cells. S6K1 phosphorylation at T389, a surrogate marker for mTORC1 activity, was increased concomitantly with decreased Akt activity in merlin-KD cells. This is consistent with Akt-independent activation of mTORC1 also observed in other cell systems (10) and reminiscent of mTORC1 activation by TSC1 or TSC2 loss, where suppression of Akt signaling via a negative feedback mechanism has been well documented (11). Unexpectedly, cyclin D1 was dramatically reduced in merlin-KD RSCs. Employing a different antibody, we confirmed a ~61% decrease (Supplementary Material, Fig. S6).
Schwann cells normally do not express ErbB4 (12,13). Indeed, ErbB4 was barely detectable in our RSCs, whereas merlin-KD RSCs appeared to have elevated ErbB4 (Fig. 1B). Using a different batch of the cells, we also detected elevated ErbB4 (by ~34%) in merlin-KD RSCs (Supplementary Material, Fig. S7). However, p-ErbB4 (Y1284) was much lower in merlin-KD RSCs (Fig. 1B). It should be noted that this p-ErbB4 antibody has strong cross reactivity with p-Y1248 (and possibly p-Y1196) in ErbB2 (data not shown). Although the absolute expression levels of ErbBs were unknown, ErbB4 was likely expressed at a much lower level than ErbB2/3, and the p-Y1284 signals might be largely from ErbB2.
Merlin interacts with neurofibromin
Given the increased phosphorylation in Raf1 in the absence of general RTK upregulation or overactivation in merlin-KD RSCs, we hypothesized that Ras activity might be increased. Having recently identified that merlin can directly interact with Ras and p120RasGAP (4), we asked whether it might also interact with neurofibromin. Indeed, endogenous neurofibromin could be co-immunoprecipitated with endogenous merlin from RSCs, NSLTs and 293T cells (Fig. 2A and Supplementary Material, Fig. S8). Omitting the lysate, or using a lysis buffer that could not efficiently extract neurofibromin [n-Octyl-β-D-glucopyranoside (NOG) as the detergent], eliminated the neurofibromin signal from merlin immunoprecipitation (IP; Supplementary Material, Fig. S8A and C), excluding that the signal was from cross-reactivity with the antibody for IP. In contrast, p120RasGAP was more efficiently co-immunoprecipitated using the NOG buffer, likely due to different biochemical properties between neurofibromin and p120RasGAP. Although merlin could not be detected from neurofibromin IP, our further data support this interaction (see below).
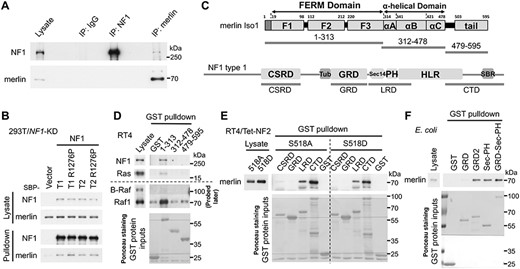
Merlin interacts with neurofibromin. (A) Immunoblot analysis of Co-IP of merlin and neurofibromin using lysate from confluent NSLTs. (B) Immunoblot analysis of streptavidin pulldown of SBP-tagged NF1 proteins. Here, a weak promoter was used to drive ectopic NF1 expression close to the endogenous level (59). Cells were lysed ~48 h after transfection. R1276P: GAP-inactive mutation. (C) Domain organization of merlin iso1 and neurofibromin type 1. Fragments for GST pulldown are indicated. Tub, tubulin-binding region; HLR, HEAT-like repeats; SBR, syndecan-binding region. (D–F) Immunoblot analysis of GST pulldown using indicated fragments and lysates from confluent cells or merlin-expressing E. coli. RT4/Tet-NF2 inducibly overexpress merlin iso1. S518A, non-phosphorylatable mutant; S518D, phosphomimetic mutant. GST-proteins were detected by Ponceau S staining. Representative results of at least two independent experiments are shown for each panel.
Neurofibromin also has two major isoforms (Supplementary Material, Fig. S9) (14). Confluent RSCs and NSLTs almost exclusively expressed neurofibromin type 2 (Supplementary Material, Fig. S9A). Both neurofibromin isoforms interacted with both merlin isoforms to similar extents (Fig. 2B and Supplementary Material, Fig. S10). In all the experiments, we detected only trace amounts of neurofibromin from merlin pull-down assays and vice versa, indicating a low-stoichiometry interaction.
The FERM domain of merlin interacts with the Sec14-PH domains of neurofibromin
The interaction domains were mapped by GST pulldown (Fig. 2C–F). Only merlin 1–313, which contains the FERM domain, could pull down neurofibromin and Ras from the lysate of a rat schwannoma cell line RT4-D6P2T (RT4; Fig. 2C and D). Four GST-tagged neurofibromin fragments that could be purified from Escherichia coli (15) were used to pull down merlin S518A (active) and S518D (inactive) mutants (16) from cell lysates (Fig. 2C and E). The leucine-rich repeat domain (LRD) and the C-terminal domain (CTD) demonstrated a clear pulldown of merlin, with slightly better binding to merlin S518A than to the S518D mutant. The GAP-related domain (GRD) did not demonstrate any binding, whereas the cysteine/serine-rich domain (CSRD) binding was very weak. Due to the poor purity of CTD, the high cysteine content of CSRD and a lack of structural information, we are more confident about the specificity of the binding of LRD, which contains the Sec14-PH domains. The isolated Sec14-PH domains could also bind to merlin expressed in RT4 (Supplementary Material, Fig. S11A) or E. coli (Fig. 2F), suggesting a direct interaction. The GAP-related domain type 2 (GRD2) could also pull down merlin (Fig. 2F). However, merlin 1–313 bound similarly to full-length neurofibromin types 1 and 2 (Supplementary Material, Fig. S11B). Combined with the results in Fig. 2B and Supplementary Material, Fig. S10, these data suggest that GRD2’s binding was likely an artifact. Of note, we cannot exclude that additional merlin binding sites may exist in neurofibromin.
Merlin cannot increase the Ras-GAP activity of neurofibromin
In pursuit of an initial hypothesis that merlin might increase the Ras-GAP activity of neurofibromin by contributing an additional Ras-binding site, we purified streptavidin-binding peptide (SBP)-tagged full-length neurofibromin types 1 and 2 from 293T/NF(1 + 2)-KD cells and merlin 1–313 from E. coli. The GAP activity was tested using a GST-Raf1 RBD pulldown-based approach (Supplementary Material, Fig. S12A–C). Neurofibromin type 1 demonstrated robust GAP activity, whereas the activity of neurofibromin type 2 was undetectable with this level of input. Merlin did not increase the GAP activity of either neurofibromin isoform; instead it showed a mild interference with the activity of neurofibromin type 1 at an early time point (5 min), reminiscent of the results with p120RasGAP (4).
To test whether merlin could increase the GAP activity of GRD2 in the context of an isolated fragment, merlin 1–313 was fused with GRD2-Sec14-PH (Supplementary Material, Fig. S12D) and purified from 293T/NF(1 + 2)-KD cells. Weak Ras-GAP activity of GRD2-Sec14-PH was detectable with increased input; fusion with merlin 1-313 decreased the GAP activity (Supplementary Material, Fig. S12E).
Furthermore, the dynamics of Ras activation and inactivation following NRG1 stimulation were similar in control (Ctr) KD and merlin-KD NSLTs (Supplementary Material, Fig. S13A); reconstituted neurofibromin demonstrated similar GAP activity in 293T/NF1-KD and 293T/NF(1 + 2)-KD cells (Supplementary Material, Fig. S13B and C). Collectively, the data challenge merlin’s positive effect on the Ras-GAP activity of neurofibromin in vitro and in vivo.
Merlin acts downstream of Ras to suppress MEK1/2 and Erk1/2 activation
As described above, the merlin/neurofibromin complex exhibits very low stoichiometry. Moreover, transient transfection of merlin constructs into 293T cells always resulted in very high expression (even with merlin under a weak promoter, due to the SV40 large T antigen in 293T cells and the SV40 origin in the vectors), which severely affected the expression of co-transfected constructs (data not shown). To circumvent these issues and promote the formation of the merlin/neurofibromin complex to better reveal its functional relevance, we fused full-length merlin with neurofibromin; however, the fusion proteins were poorly expressed (data not shown). The merlin 1–313-neurofibromin fusion proteins were then tested (Fig. 3A). These constructs, under the weak EFS promoter (a short version of EF1a promoter), were expressed well when transiently transfected into 293T-derived cells (Supplementary Material, Fig. S14). Fusion with merlin 1–313 did not increase the Ras-GAP activity of neurofibromin under all the tested conditions (Supplementary Material, Fig. S14 and Fig. 3B). We noticed that EGF stimulation did not appreciably increase HRas-GTP level in neurofibromin type 1 (with or without merlin 1–313 fusion) expressing cells, likely due to the strong GAP activity. It still activated the downstream effectors Raf1 and Erk1/2 (Supplementary Material, Fig. S14)—indicating that Ras-GTP level itself may not faithfully reflect its signaling output. With culture time shortened to ~24 h after transfection and no serum starvation implemented, merlin 1–313-fused neurofibromin suppressed MEK1/2 and Erk1/2 activity more efficiently, occurring to both isoforms despite a major difference in their GAP activity (Fig. 3B). Notably, only merlin 1–313-fused neurofibromin, as well as merlin 1–313 itself, was co-precipitated with GST-Raf1 RBD, suggesting an interaction between merlin FERM and Raf1 RBD.
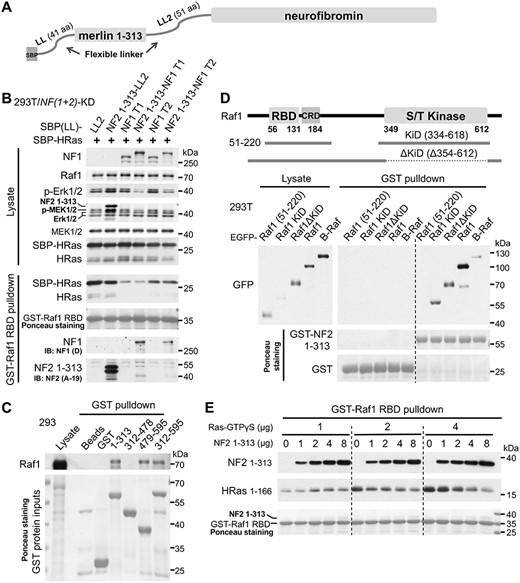
Merlin cooperates with neurofibromin to suppress the Ras–Erk pathway by blocking Ras binding to Raf1. (A) Schematic diagram of fusion proteins. (B) Constructs were co-transfected into 293T/NF(1 + 2)-KD cells; ~24 h after transfection, cells were lysed for pulldown and analyzed by immunoblotting. (C–E) Immunoblot analysis of GST pulldown using indicated fragments and lysates from confluent cells (C, D) or purified proteins (E). GST-proteins were detected by Ponceau S staining. Cells were lysed ~26 h after transfection (D). IB, immunoblotting; GTPγS, a non-hydrolyzable analog of GTP. Representative results of at least two independent experiments are shown for each panel.
The FERM domain and the tail of merlin interact with the RBD and the KiD of Raf1
Reprobing a previous pull-down blot corroborated the interaction between merlin FERM and Raf1, but not B-Raf (Fig. 2D). Unexpectedly, upon repeating the pulldown using 293 lysate, merlin tail also bound to Raf1 (Fig. 3C). This discrepancy might be explained by the different status of Raf1, as RT4 cells presumably have high Ras–Erk activity due to an activating mutation in ErbB2 (17). We compared six RBD/Ras-association (RA) domains from Ras effectors for pulldown of merlin from 293T lysate—only Raf1 RBD demonstrated a clear, albeit weak binding, whereas the binding of B-Raf RBD was marginal (Supplementary Material, Fig. S15A). Using purified merlin 1–313, we confirmed a direct interaction and the preference for Raf1 RBD over B-Raf RBD (Supplementary Material, Fig. S15B). Merlin FERM also robustly bound to the KiD of Raf1 (Fig. 3D). Unexpectedly, Raf1 51–220 containing the RBD-CRD (Cys-rich domain) did not show detectable binding to merlin FERM, indicating that the CRD may block merlin binding in this isolated fragment.
Merlin FERM blocks Ras-GTP binding to Raf1 RBD
Merlin binding to Raf1 RBD raises the possibility that merlin blocks Ras-GTP binding to Raf1 RBD, which could indeed be supported by the competition experiment in Fig. 3E. Surprisingly, when the concentration of Ras-GTPγS was relatively low, merlin instead promoted Ras-GTPγS binding to Raf1-RBD. This probably denotes a ‘trans-enhancing’ artifact caused by the dimerization of the GST domain and existence of a Ras-binding site in merlin, which could override the blocking effect when Ras-GTP concentration is low.
Merlin FERM does not block the isolated Raf1 KiD dimerization or interaction with MEK
Homodimerization or heterodimerization between Raf proteins mediated by their KiDs is essential for their activation (18). Using proteins expressed in E. coli, we confirmed a direct interaction between Raf1 KiD and merlin FERM (Supplementary Material, Fig. S16A). However, the latter failed to demonstrate any effect on Raf1 KiD dimerization (Supplementary Material, Fig. S16A) or the interaction with its substrate MEK1 (Supplementary Material, Fig. S16B), suggesting that merlin FERM binds to a different region in Raf1 KiD.
Merlin does not affect the formation of the neurofibromin/Spred1 complex
Spred proteins recruit neurofibromin to the plasma membrane via a high-affinity interaction between the EVH1 domain of Spred proteins and the GRD of neurofibromin (19–22). All three Spred proteins can interact with neurofibromin, with Spred1 and Spred2 being more efficient (19,21). The importance of Spred1 in neurofibromin functionality is highlighted by the fact that germline mutations in SPRED1 cause Legius syndrome, a NF1-like disease but with a milder phenotype (23). The merlin-binding site in neurofibromin, Sec14-PH, neighbors GRD. We tested whether merlin affects the interaction between neurofibromin and Spred1. Neurofibromin overexpressed in 293 cells could only co-precipitate a trace amount of endogenous Spred1 (Supplementary Material, Fig. S17A) but a prominent amount of overexpressed Spred1 (Supplementary Material, Fig. S17B). Overexpression of merlin did not show an appreciable effect on the neurofibromin/Spred1 complex. Incidentally, no discernible difference was observed between the neurofibromin type 1 and 2 isoforms regarding Spred1 binding (Supplementary Material, Fig. S17A), consistent with the in vitro binding result using purified EVH1 domain and GRD type 1/2 (22).
Merlin also interacts with Spred1 and binds more efficiently to the neurofibromin/Spred1 complex
We repeated the experiment the other way around (Fig. 4A). This time, endogenous neurofibromin was readily detected in the pulldown of overexpressed Spred1. Overexpression of merlin did not convincingly reduce the quantity of neurofibromin co-precipitated with wild-type Spred1, when the precipitated amount of Spred1 and the protein levels of neurofibromin in the lysates were taken into consideration. Of note, Spred1 also co-precipitated overexpressed merlin. The pathogenic T102R mutation in Spred1 severely impairs interaction with neurofibromin (19–22,24), as also seen here, but has no effect on the interaction with merlin, suggesting that Spred1 uses a different interface to interact with merlin. Endogenous merlin was not convincingly detectable in Spred1 pulldown, whereas three previously reported Spred1 interaction partners—Ras, Raf1 and B-Raf (25–27)—were undetectable in Spred1 pulldown in this experiment (data not shown)—indicating that, among these, neurofibromin is the prominent interaction partner of Spred1.
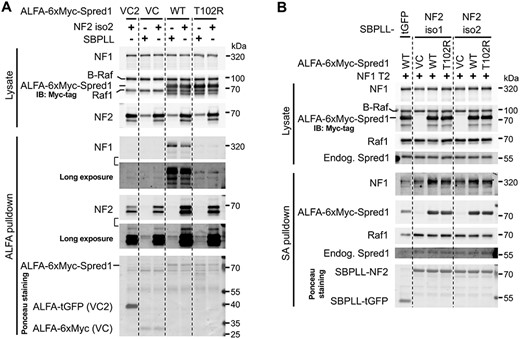
Merlin also interacts with Spred1 and binds more efficiently to the neurofibromin/Spred1 complex. (A, B) Immunoblot analysis of pulldown. Indicated constructs were co-transfected into 293 cells; ~24 h later, half volume of fresh medium was further added; ~48 h after transfection, cells were lysed for ALFA pulldown (A) or streptavidin (SA) pulldown (B). Proteins on the blots were also detected by Ponceau S staining. Note that untagged NF2 and NF1 were used in (A) and (B), respectively; all NF2 constructs used the S518A mutants. tGFP, TurboGFP. Representative results of two independent experiments for (B).
Next, we tested whether Spred1 affects the interaction between merlin and neurofibromin (Fig. 4B). Overexpression of wild-type Spred1 markedly increased the amount of overexpressed neurofibromin co-precipitated with overexpressed merlin, occurring to both merlin isoforms. The Spred1 T102R mutant also promoted the merlin/neurofibromin complex formation, but less efficiently, in line with the fact that it can still bind to neurofibromin weakly. Spred1 was also co-precipitated with merlin; even endogenous Spred1 was detectable in the pulldown, corroborating the interaction with merlin. Raf1 was also detectable in merlin pulldown, at very low abundance, whereas B-Raf was not (data not shown).
Activation of the Ras–Erk pathway by KD of neurofibromin or overexpression of active Ras or Raf is not sufficient to drive loss of CICP in RSCs
Our next step was to test whether activation of the Ras–Erk pathway can drive loss of CICP in RSCs. Neither KD of neurofibromin nor overexpression of active Ras or Raf mutants of different potency increased the cell density, despite clear activation of the Ras–Erk pathway (Supplementary Material, Fig. S18). In contrast, knockout of neurofibromin or merlin, or overexpression of active Raf proteins, drove loss of CICP in a spontaneously immortalized MEF line (Supplementary Material, Fig. S19)—all indicating cell type-specific responses to the activation of the Ras–Erk pathway. Of note, here is that loss of neurofibromin effected greater impact on the Ras–Erk pathway than loss of merlin (Supplementary Material, Figs S18A and S19A).
KD of Raf fails to sustainably suppress the cell density of merlin-deficient RSCs
To assess the contribution of Raf proteins to the loss of CICP in merlin-KD RSCs, we further knocked down Raf1 and B-Raf individually or in combination. While the cells appeared to adapt quickly to the KD, we did not consistently observe a reduction of cell density (data not shown). Also, the Raf-KD cells maintained the basic MEK and Erk activity, unlike overexpression of merlin iso2 (Supplementary Material, Fig. S20).
Merlin-deficient RSCs depend on the Ras–Erk pathway activity for proliferation
Determined to improve upon the results achieved by genetic approaches, we activated Raf by sorafenib (a Raf inhibitor that at low concentration paradoxically activates Raf proteins by inducing Raf dimerization) (28) or attenuated ErbB2/ErbB3 activity by omitting NRG1 from the culture. In both parental and merlin-KD RSCs, sorafenib increased, while NRG1 omission decreased, the cell density (Fig. 5A). We next tested an ErbB inhibitor lapatinib and a highly selective MEK inhibitor PD0325901 (29), both efficiently blocked the proliferation of merlin-KD RSCs (Fig. 5B). Our data suggest that the activity of the Ras–Erk pathway is required for the proliferation of merlin-deficient RSCs and that modestly increased activity of this pathway may contribute to loss of CICP in merlin-deficient RSCs.
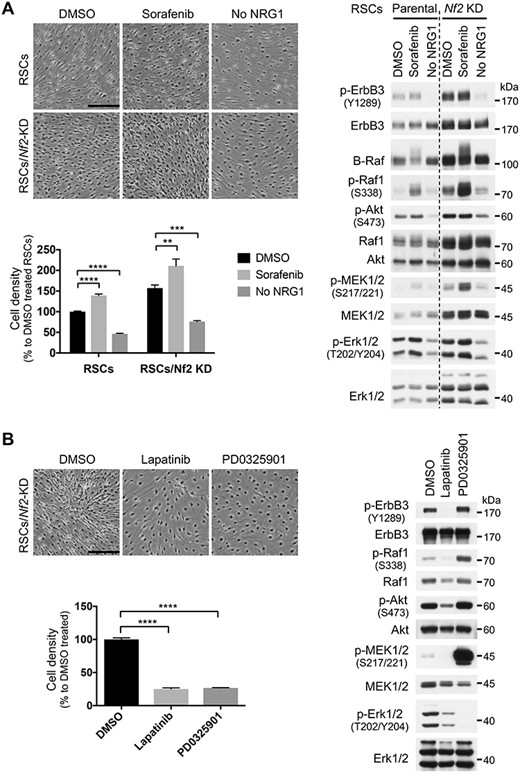
Merlin-deficient RSCs depend on the Ras–Erk pathway activity for proliferation. (A) RSCs and merlin-KD RSCs were grown to near confluence, then treated with 2 μm sorafenib or cultured in the absence of exogenous NRG1 for 12 days and directly lysed on dish with 2× SDS sample buffer for immunoblotting. Photos shown were at 9 days of treatment. Note that the lysates were not fully normalized for loading; both Akt and Erk1/2 can serve as loading control. (B) Merlin-KD RSCs were grown to near confluence and then treated with 0.5 μm lapatinib or 1 μm PD0325901 for 7 days. Media (with the inhibitors) were changed every 3 days, and after photography on the last day, cells were lysed for immunoblotting as described above. Cell number was counted from at least three representative regions, normalized to the respective controls and represented as mean ± SD. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001; ordinary one-way analysis of variance followed by Dunnett’s multiple comparison test. Scale bars, 250 μm. Representative results of at least two independent experiments are shown for each panel.
In line with our in vitro data, activation of Raf by Raf inhibitors in vivo drove hyperplasia of multiple tissues in the rat, including the sciatic nerve (30). However, activation of endogenous Raf1 by a heterozygous L613V mutation in a Noonan syndrome (one form of RASopathy) mouse model (31) did not drive sciatic nerve hyperplasia (Supplementary Material, Fig. S21), although it can drive glial cell increase in the cortex (32), again highlighting cell type-specific responses. These results also indicate that Raf inhibitors and activating mutations of Raf1 may elicit different consequences for Schwann cells.
Discussion
Previous studies in Drosophila, mouse Schwann cells and human schwannomas reported elevated RTK levels in merlin-deficient cells, tissues and tumors (merlin; expanded double mutants in Drosophila) (33,34). Yet, our in vitro data from RSCs do not support the generality of this notion. In fact, a follow-up study using 68 human schwannomas revealed quite a variable pattern of RTK expression between tumors (35). Nevertheless, RTK overexpression is indeed often observed in human schwannomas (36). Notably, an invariable ErbB2 activating mutation (V664E) has been identified in the ethylnitrosourea-induced rat schwannoma model, establishing its role as a schwannoma driver in rats (17). Moreover, cells from ethylnitrosourea-induced schwannomas acquired homozygosity for the mutant ErbB2 (37), highlighting the importance of ErbB2 dosage during the development of ErbB2-driven schwannomas. Thus, elevated RTKs in human schwannomas are more likely a consequence of the selection for proliferative advantage than of merlin loss. The RTK–Ras–Erk pathway is extremely complicated and subject to extensive negative feedback (38). Downregulation of RTKs in merlin-deficient RSCs may emanate from a negative feedback mechanism in response to intensified Ras–Raf signaling. It should be noted that merlin may directly or indirectly associate with some RTKs and affect their signaling and turnover (39–41). It is also worthy of mention that among the investigated RTKs, only IGF-1Rβ was convincingly upregulated in merlin-deficient RSCs—the upregulation of which has also been reported in merlin-deficient mouse Schwann cells and human schwannoma cells (34,42)—thus, IGF-1Rβ upregulation may be a consequence of merlin loss.
Contradicting a previous report suggesting that merlin represses cyclin D1 expression (43), merlin-deficient RSCs showed much decreased cyclin D1, tallying with the fact that at an early stage of merlin loss we did not observe increased proliferation of the cells. An implication for the underlying mechanism comes from a study on Nf2 deletion in the mouse liver, which revealed that Nf2 loss induced not only tumorigenesis but also senescence programs, due to activation of the Rac1–PAK1 pathway, which in turn induced reactive oxygen species and a DNA-damage response (44).
Recent evidence reinforces a prominent role of merlin in suppressing the Hippo pathway effectors YAP/TAZ (45). The importance of dysregulation of the Hippo pathway in Schwann cell tumorigenesis has been highlighted by two recent studies, in which combined inactivation of Lats1/2 in the Schwann cell lineage in mice resulted in the development of peripheral nerve sheath tumors (there were histopathological differences in the tumors between the two studies, due to different Cre lines and maybe different genetic backgrounds of the mice), whereas combined (but not complete) ablation of Yap/Taz in these mice partially rescued tumor development and progression (46,47). Both studies support YAP/TAZ as a driver for Schwann cell tumorigenesis. They also highlight a contributing role of RTK–Ras–Erk in Schwann cell tumorigenesis. Particularly, Chen et al. (47) showed that additional inactivation of Nf1 in the Lats1/2 mutant mice exacerbated the tumor phenotype. Although our work on the Ras–Erk pathway alone could not clearly attribute a significant contribution of this pathway to Schwann cell loss of CICP, their in vivo work complements our mechanistic findings.
Our data suggest that the RTK–Ras–Erk pathway is a potential therapeutic target for NF2 inactivation-related tumors, which is also supported by other studies. Lapatinib has been shown to decrease proliferation of human schwannoma cells in vitro (48). A recent work comprehensively evaluated six MEK1/2 inhibitors, including PD0325901, in a number of merlin-deficient cellular and mouse models and demonstrated variable efficacy between the inhibitors, with significant tumor growth inhibition by the most potent inhibitors (49). As multiple lines of evidence support YAP/TAZ as a driver for merlin-deficient tumors (not limited to the Schwann cell lineage) (44,46,47,50), combination therapy co-targeting YAP/TAZ and the RTK–Ras–Erk pathway will likely offer better efficacy; this has been demonstrated in the Schwann cell tumor models with Lats1/2 inactivation.
Our previous study identified that merlin FERM and the tail interact with the PH-C2 domains of p120RasGAP (4). Interestingly, PH-C2 and Sec14-PH take a similar position relative to the GAP domain or GRD (Supplementary Material, Fig. S22), hinting that merlin may employ a common mode to interact with these two Ras-GAPs. Spred proteins do not interact with p120RasGAP (19,22); however, we speculate that a functionally similar co-factor may exist to facilitate the interaction between p120RasGAP and merlin.
The high affinity between Spred1 and neurofibromin enables a stable complex (20,22). Although merlin’s affinity to Spred1 or neurofibromin is unknown, neither appeared high. Merlin does not affect the complexation between Spred1 and neurofibromin, whereas Spred1 promotes the complexation between merlin and neurofibromin, pointing to the order of the complex assembly—Spred1 and neurofibromin form a complex first, then merlin joins (Supplementary Material, Fig. S23A and B). Our efforts also provide an explanation for the early observation that overexpression of Spred1 blocked the signaling from Ras to Raf (25). Of note, Spred1 may interact with Ras, Raf1 and B-Raf (25–27), allowing these proteins to build an intricate network for Ras regulation. Whether merlin can interact with Spred2/3 requires future study.
Integration of two recent structural studies on full-length B-Raf and the RBD-CRD of Raf1, respectively, revealed that although Raf proteins are autoinhibited by a 14-3-3 dimer [through the interaction with p-S365 and p-S729 (p-S259 and p-S621 in Raf1) that flank the KiD] and the CRD (through the interaction with the 14-3-3 dimer and the KiD), the RBD and most of the CRD are accessible by Ras (Supplementary Material, Fig. S23C) (51,52). Thus, Raf proteins are susceptible to Ras-mediated activation and merlin can execute an interception (Supplementary Material, Fig. S23D). The B-Raf:Raf1 heterodimer is the most efficient combination for activating the downstream (18). It is intriguing that merlin targets Raf1 instead of B-Raf, given that B-Raf RBD has higher affinity to Ras-GTP than Raf1 RBD (53). We failed to demonstrate a blocking effect by merlin FERM on Raf1 KiD dimerization or interaction with MEK; however, the possibility of a blocking effect by full-length merlin cannot be excluded, given that merlin FERM only accounts for ~50% of full-length merlin and the tail of merlin can also bind to Raf1 at an undefined region. Nevertheless, Ras-GTP binding initiates Raf activation (18), making it more plausible to surmise that merlin’s interaction with both RBD and KiD involves stabilizing the merlin/Raf1 complex and the autoinhibitory conformation of Raf1.
The interaction between merlin and Raf1 is complicated and may also serve other purposes, e.g. release of Mst1/2 to activate the Hippo pathway (41). Interestingly, Ji et al. identified that merlin phosphorylation at Y207 is required for the interaction with Raf1. In our study, merlin FERM purified from E. coli also demonstrated a robust binding to Raf1, negating the requirement for p-Y207. However, we found it difficult to demonstrate their in vivo interaction. So, phosphorylation at Y207 may either enhance the affinity or play a role in the regulated assembly of the complex.
Given the preferential binding of Ras-GTP to the effectors (53), our efforts provide an insight into a long-standing puzzle of how Ras-GAPs compete with Ras effectors. It is generally held that Ras-GAPs accomplish this by depleting the Ras-GTP pool; however, the signaling may not be efficiently prevented. Our investigations propose that merlin functions as a ‘selective Ras barrier’ in concert with the neurofibromin/Spred1 complex to suppress the Raf branch. The output of different Ras effector pathways may have a critical role in cell fate determination, e.g. proliferation or differentiation. We anticipate a widespread regulatory mode in small GTPase signaling whereby scaffold proteins cooperate with GAP proteins to fine-tune signaling outputs to achieve specific biological responses. Our data also highlight that merlin loss, but not neurofibromin loss, promotes autonomous Schwann cell proliferation. This is in line with the cell constitution difference between human schwannomas and neurofibromas (2) and the difference between NF1 and NF2 mouse model studies (54,55).
Materials and Methods
Materials
All chemicals were from Carl Roth GmbH & Co. KG (Karlsruhe, Germany), unless otherwise stated. Protease inhibitor cocktail tablets (cOmplete, EDTA-free) were from Roche Diagnostics (Mannheim, Germany). Recombinant human heregulinβ-1 (NRG1; the EGF-like domain part) and EGF was from PeproTech (Hamburg, Germany). Poly-L-lysine (PLL) was from Sigma-Aldrich (Munich, Germany). Forskolin was from Apollo Scientific Ltd (Cheshire, UK) or LC Laboratories (Woburn, Massachusetts, USA). PD0325901, sorafenib (p-toluenesulfonate salt) and lapatinib (di-p-toluenesulfonate salt) were from LC Laboratories. GammaBind G Sepharose, Glutathione Sepharose 4B Fast Flow and Ni-Sepharose 6 Fast Flow were from GE Healthcare Life Sciences (Freiburg, Germany). Pierce High Capacity Streptavidin Agarose and NeutrAvidin were from Thermo Scientific (Schwerte, Germany). ALFA Selector ST was from NanoTag Biotechnologies GmbH (Göttingen, Germany). Polyethylenimine (PEI, linear, mol. wt. ~25 000) was from Polysciences (Hirschberg an der Bergstrasse, Germany). Blasticidin (solution) was from InvivoGen (Toulouse, France). Puromycin was from PAA Laboratories (Pasching, Austria). GDP and GTPγS were from Thermo Scientific’s Active Ras Pull-Down and Detection Kit.
Protease inhibitor cocktail tablets were dissolved in H2O as 100× stock, aliquoted and stored at −20°C. PLL was dissolved in autoclaved H2O at 1 mg/ml (20× stock), sterilely filtrated and stored at 4°C. Small molecular inhibitors were dissolved in DMSO, diluted as 1000× stock and stored at −80°C. Forskolin was dissolved in ethanol at 4 or 10 mm and stored at −20°C. PEI was dissolved in H2O at 1 μg/μl, pH 7.0, sterilely filtrated and stored at −20°C; the stock in use was stored at 4°C.
Antibodies are listed in Supplementary Material, Table S1.
Plasmids
Throughout this manuscript, NF1 and neurofibromin and NF2 and merlin may be used interchangeably.
For stable KD, a series of lentiviral vectors modified from the pGIPZ vector (Open Biosystems) was used. The targeting sequences embedded in an optimized miRNA scaffold were cloned into these vectors. The artificial miRNA(s) was embedded in a big transcript under a CMV promoter. Details of the design will be published elsewhere. The target sequences are listed in Supplementary Material, Table S2. Empty vectors were used for Ctr KD.
For expression of GST-tagged merlin fragments in E. coli, full-length human merlin iso1 was cloned into pGEX-4T-1 between BamHI and NotI sites and the fragments subsequently generated by deletion mutagenesis.
pGEX-NF1 CSRD (aa 543-909), LRD (aa 1545-1950) and CTD (aa 2260-2818) have been described in (15); additional fragments were generated by deletion mutagenesis. For construction of GST-NF1 GRD (aa 1198-1530), the GRD fragment from pGEX-4T-1-NF1 (333) (from Dr Mohammad R. Ahmadian) was subcloned into pGEX-4P-1, a vector mutated from pGEX-4T-1 to have the same ORF and MCS as pGEX-6P-1 (GE Healthcare Life Sciences). All numbering is according to human neurofibromin type 1.
pGEX-Raf1-RBD (aa 51-131) has been described in (56). pET15TEV-HRas 1-166 has been described in (57). pGEX-RalGDS-RA (pGEX-RGF97) (58) was from Dr Ignacio Rubio. pGEX-A-Raf (aa 17-94), pGEX-B-Raf (aa 150-233), pGEX-RGL1 (aa 681-773) and pGEX-RIN1 (aa 622-745) (53) were from Professor Mitsu Ikura and Dr Matthew J. Smith.
The assembly of GRD2, GRD-Sec-PH (aa 1198-1816), GRD2-Sec-PH and full-length NF1 minigenes containing an intron has been described in (59).
Lentiviral vectors modified from pCDH-CMV-MCS-EF1a-Puro (System Biosciences) have been described in (59). In this study, long flexible linker(s) (GS type) were further introduced. The ALFA-tag (60) vector was generated by replacing the SBP-tag2 in the customized lentiviral vector. The respective cDNAs were cloned into these vectors; the mutants were generated by either direct mutagenesis or by subcloning fragments containing the mutation(s). The templates for HRas, NRas, KRas 4A and KRas 4B (WT, G12V, Q22R, D153V) (61) were from Dr Ion C. Cirstea. The templates for Raf1 (WT, S259A) and B-Raf (WT, S365A) (62) were from Professor Philip Stork. pEGFP-Raf1, pEGFP-B-Raf (63) and the templates for 6xMyc-mouse Spred1 (25) were from Dr Ignacio Rubio. An I191M mutation found in both sources of B-Raf was corrected in pEGFP-B-Raf by Dr Ion C. Cirstea. Corrected B-Raf was subcloned to generate lentiviral SBP-B-Raf; the S365A mutant was generated by replacing the EcoRI-XhoI fragment.
The pET-A-SBP vector for expression of SBP-tagged protein in E. coli has been described in (4). Merlin 1–313 was subcloned into pET-A-SBP at the BamHI and NotI sites.
LentiCRISPR (Addgene #49535) (64) or LentiCRISPR v2 (Addgene #52961) (65) vectors were used to knock out Nf1 and Nf2 in MEF1 cells. The oligos for the sgRNAs are listed in Supplementary Material, Table S3.
More details are available upon request.
Cell culture
RSCs and NSLTs (from Professor Alison Lloyd) were cultured in Dulbecco's modified eagle medium (DMEM) (with glutamine, high glucose) supplemented with 3% FBS, 10 ng/ml NRG1 and 1 μm forskolin. RSCs were cultured in PLL-coated plates. Of note, the proliferation of RSCs is affected by the coating material types and intensity. In most cases, PLL of molecular weight 70 000–150 000 was used, unless otherwise stated. The plates were coated with PLL at a concentration of 0.05 mg/ml in H2O (1 ml for each well of a 6-well plate, 5 ml for a 10 cm plate, 10 ml for a 15 cm plate) for 1 h at room temperature. The PLL solution was removed and the plates washed once with an equal amount of H2O. The H2O was removed and the plates dried and stored at room temperature until use. Plates from the same batch of coating were used for each independent experiment.
293, 293T cells and its derivatives; RT4 cells and its derivatives and MEF1 cells (from Dr Zhong-Wei Zhou) (66) were cultured in DMEM (with glutamine, high glucose) supplemented with 10% FBS.
Cell counting
Cells were either counted manually using photographs and the ‘Cell Counter’ plugin of ImageJ or trypsinized and counted using an automated Beckman Coulter Z2 Cell and Particle Counter. For the former, multiple representative regions with a defined area were chosen and counted. For counting using the Z2 Counter, cells were grown in 6-well plates with three or four replicates; three replicates were trypsinized and counted, one replicate was lysed for immunoblotting or the trypsinized cells were pelleted and lysed for immunoblotting if only three replicates were plated; cell size limit was set at 150–8000 fl (volume) for RSCs and its derivatives and 9–25 μm (diameter) for MEF1 cells and its derivatives.
Transient transfection
293T and 293 cells were transfected with PEI at a ratio of DNA (μg) to PEI (μg) of 1:3 or 4; 150 mm NaCl was used as the diluent.
Lentivirus production and transduction
The procedure has been described in (4,59).
Immunoblotting and quantification
Immunoblotting procedure has been described in (4); signals were detected with either films or a ChemiDoc MP imager (Bio-Rad). Tricine–SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) (7% gels) was used for Fig. 4 and Supplementary Material, Figs S7 and S17; all other data were generated with Glycine–SDS-PAGE. Some of the pre-stained protein ladders run differently in these two gel systems; annotated ladder sizes were not calibrated. For sequential probing using horseradish peroxidase (HRP)-based secondary antibodies, if the primary antibodies are from different species, 30% H2O2 (~30 min at 37°C) was used to inactivate the HRP from the first round of probing (67). For fluorescent detection, 5% milk in Tris-buffered saline with 0.1% Tween 20 was used for blocking, and only IRDye 800CW-labelled secondary antibodies were used (68). Stripping was performed only when necessary (25 mm glycine-HCl, pH 1.92, 1% SDS with constant gentle shaking at room temperature); stripping efficiency was monitored by remaining IRDye 800CW signals. Quantification exclusively used data by fluorescent detection; areas were manually selected with the volume tool (Rectangle) of Image Lab v6.01 for Mac (Bio-Rad), and identical rectangles were applied to the other lanes; background subtraction used ‘Local’.
Reverse transcription PCR
Total RNA was isolated with RNeasy Mini Kit (QIAGEN) and reverse transcription was carried out with Omniscript RT Kit (QIAGEN) supplied with RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen). A gene-specific primer ratNf1-RTP (5′-catgtgctcttcctttgtgaac, only for subsequent Nf1 amplification) or oligo dT was used for the first-strand cDNA synthesis. For the subsequent PCR, the following primer pairs were used: ratNf1-isoquantF (5′-cttgaggagaaccagaggaac), ratNf1-isoquantR (5′-cctgcttcatacggtgagac); mmNf2ex15-F (5′-agcgagagctcagacagagg), mmNf2ex17-R (5′-caaacaagccagccctctac). Note that the Nf2 primers were initially designed to amplify mouse Nf2; there are two mismatches (underlined) for rat Nf2.
Co-IP and pulldown
Basic lysis buffer: PBS containing 0.6–0.7% CHAPS or 0.1% dodecyl-β-D-maltoside. 2 mm MgCl2 was included if Ras was to be detected. 2% NOG was only used in the lysis buffer for Supplementary Material, Fig. S8C. For the lysing step (but not for washing), 1× protease inhibitor cocktail and 1–2 mm DTT were included. The detailed procedure has been described in (4,59).
Protein expression and purification
The procedure has been described in (4,59).
Nucleotide loading into purified HRas 1–166 and in vitro Ras-GAP activity assay by GST-Raf1 RBD pulldown
The procedure has been described in (4,59).
Histopathology analysis
All animal studies were approved by the Animal Care Committees of University Health Network and the University of Guelph and performed in accordance with the standards of the Canadian Council on Animal Care. Sciatic nerves of both sides were from 14-week-old Raf1 L613V/+ mice (129Sv × C57BL/B6) (31) and the wild-type littermates. The nerves were fixed in 4% paraformaldehyde for 24 h and then placed in PBS. The fixed nerves were processed for HE staining by standard histology procedures and evaluated by a pathologist blinded to animal genotype.
Statistics
Statistics were performed using GraphPad Prism 6 or 7 for Mac.
Data Availability
All data supporting the findings are available within the article and its Supplementary Material or upon request.
Acknowledgements
We thank A. Lloyd for RSCs and NSLTs; Z.W. Zhou for MEF1 cells; S. Shibahara for rat Nf1 type 1 cDNA; G.E. Bollag and K. Scheffzek for human NF1 type 1 cDNA; M.R. Ahmadian for NF1 GRD cDNA; I.C. Cirstea for Ras cDNAs; P. Stork for Raf1 and B-Raf cDNAs; I. Rubio for EGFP-Raf1, EGFP-B-Raf, pGEX-RalGDS-RA, pGEX-MEK1-His, and mouse Spred1 cDNAs; M. Ikura and M.J. Smith for GST-RBD/RA of A-Raf, B-Raf, RIN1 and RGL1 constructs; B.G. Neel for mouse nerves and comments; C. Hagel for histopathology analysis; S. Groth for assistance with S2 work; B. Pavelka and U. Petz for laboratory support. Special thanks to the anonymous referee for prompting us to investigate Spred1.
Conflict of Interest statement. The authors declare no competing interests.
Funding
Children’s Tumor Foundation (Synodos for NF2) and Deutsche Forschungsgemeinschaft (MO 1421/5-1 to H.M.); National Natural Science Foundation of China (81761138047 to H.J.).
Author contributions
Y.C. and H.M. conceived the study and proposed the model. Y.C., H.M. and H.J. designed the study. Y.C., L.M. and S.S. performed the experiments. Y.-P.H. contributed NF1 constructs. J.C.Y. contributed mouse nerves. Y.C. wrote and H.M. edited the manuscript. All other authors commented on the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}