




Abstract
CHEK2 is a key cell cycle control gene encoding a pluripotent kinase that can cause arrest or apoptosis in response to unrepaired DNA damage. We report a large case-control study of a non-functional variant that had previously been expected to increase cancer rates. Four thousand and fifteen cancer patients (2250 lung, 811 squamous upper aero-digestive and 954 kidney) and 3052 controls in central Europe were genotyped for the mis-sense variant rs17879961 (replacement of T by C), which changes an amino acid (I157T) in an active site of the gene product. The heterozygous (T/C) genotype was associated with a highly significantly lower incidence of lung cancer than the common T/T genotype [relative risk (RR), T/C versus T/T, 0.44, with 95% confidence interval (CI) 0.31–0.63, P < 0.00001] and with a significantly lower incidence of upper aero-digestive cancer (RR 0.44, CI 0.26–0.73, P = 0.001; P = 0.000001 for lung or upper aero-digestive cancer). Protection was significantly greater for squamous than adenomatous lung cancer (P = 0.001). There was an increase of borderline significance in kidney cancer (RR 1.44, CI 0.99–2.00, P = 0.06). This unexpected halving of tobacco-related cancer (since replicated independently) implies much greater absolute risk reduction in smokers than in non-smokers. The mechanism is unknown: perhaps squamous stem cell apoptosis following smoke exposure causes net harm (e.g. by forcing nearby stem cells to divide before they have repaired their own DNA damage from tobacco smoke). If so, reducing the rate of apoptosis by reducing CHEK2 activity could be protective—although not smoking would be far more so.
INTRODUCTION
Checkpoint kinase 2 (CHEK2, also known as Chk2 or Cds1) is a key cell cycle control gene located on chromosome 22, the product of which is activated upon DNA damage (1–3). Activated CHEK2 is a kinase that phosphorylates and hence stabilizes several other proteins that can in turn cause either cell cycle arrest or apoptosis (3). These include the E2F-1 protein (which forms an active complex with the retinoblastoma protein) and the promyelocytic leukaemia (PML) protein (2,3), but perhaps not the p53 protein (4–6). Mutations in CHEK2 have been reported in families with a variant of the Li-Fraumeni syndrome, and it was initially suggested that such mutations could cause the syndrome (7). In two of these families, the defect was a frameshift mutation that would truncate the gene product (deletion of a single nucleotide, C, at position 1100 of the gene: 1100delC). In a further family, it was the mis-sense mutation rs17879961 where T is replaced by C at position 470 of the gene (T470C), causing Isoleucine to be replaced by Threonine at position 157 of the protein (I157T). This change lies in a functionally important domain, and leads to severely reduced or abolished binding to some or all of the main CHEK2 substrates (8). These frameshift and mis-sense mutations in CHEK2 are rare in the general US population (with gene frequencies of 1% or less), and it was proposed that inheritance of a single copy of any of them would be sufficient to cause a high risk of various types of cancer (7).
Subsequent studies showed, however, that the principal frameshift mutation involves at most only a moderate increase in cancer risk (9–12). Further, initial results from a case-control study in Poland (with 13 different types of cancer) found that the mis-sense mutation is not rare in that population, being present in ∼5% of the controls, and that the relative risks (RRs) (T/C versus the common T/T genotype) associated with it are not extreme, ranging in that study from 0.5 (P = 0.1) for lung cancer to 2.1 (P = 0.0006) for kidney cancer (12). We have investigated these associations further in a completely separate study in central Europe of 4000 cases of lung, upper aero-digestive and kidney cancer and 3000 controls.
RESULTS
The overall prevalence of T/C heterozygosity was 5.0% among the 2934 successfully genotyped controls (Table 1), almost identical to the prevalence reported among the 4000 controls in the earlier Polish study (4.8%) (12). However, the prevalence among our controls varied substantially between the six countries, ranging from 7.6% in Russia to 1.0% in Romania (P for heterogeneity = 0.0000001), perhaps indicating a ‘founder’ effect on the prevalence of this polymorphism in different parts of Europe. Within countries, however, the prevalence of heterozygosity among controls did not differ significantly between centres (although the number of heterozygotes at each centre was small) and, after standardizing for country, it was not significantly related to centre, urbanization, age, sex, prevalence of smoking, amount smoked per smoker, alcohol consumption or, among hospital controls, category of disease (data not shown). No controls or cases were C/C homozygotes, although the expected number under Hardy–Weinberg equilibrium, given the country-specific prevalences of heterozygosity among controls, and, separately, among cases, would be only 3.6 (or somewhat more, if there is local clustering). As there are no C/C homozygotes, the odds ratios calculated in a case-control comparison can be used to estimate directly the corresponding RR, i.e. the ratio of the cancer incidence rate among individuals with the T/C heterozygote genotype to that among otherwise similar individuals with the common T/T genotype.
Prevalence of CHEK2 heterozygous (T/C) genotype among controls and among cases of cancer, overall and within each country
| Controls | Lung | Upper aero-digestive | Kidney | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | |
| Country | ||||||||||||
| Russia | 786 | 60 | (7.6) | 399 | 11 | (2.8) | 340 | 12 | (3.5) | 281 | 28 | (10.0) |
| Hungary | 291 | 17 | (5.8) | 339 | 9 | (2.7) | – | – | ||||
| Poland | 790 | 44 | (5.6) | 694 | 20 | (2.9) | 201 | 6 | (3.0) | 79 | 5 | (6.3) |
| Slovakia | 229 | 7 | (3.1) | 300 | 3 | (1.0) | 39 | 0 | (0.0) | – | ||
| Czech Republic | 636 | 16 | (2.5) | 277 | 6 | (2.2) | 55 | 1 | (1.8) | 451 | 17 | (3.8) |
| Romania | 202 | 2 | (1.0) | 156 | 2 | (1.3) | 135 | 1 | (0.7) | 87 | 2 | (2.3) |
| Smoking status | ||||||||||||
| Never smoker | 1037 | 41 | (4.0) | 162 | 4 | (2.5) | 61 | 2 | (3.3) | 424 | 22 | (5.2) |
| Former smoker | 783 | 40 | (5.1) | 409 | 14 | (3.4) | 96 | 2 | (2.1) | 198 | 15 | (7.6) |
| Continuing smoker | 1102 | 64 | (5.8) | 1590 | 33 | (2.1) | 613 | 16 | (2.6) | 273 | 15 | (5.5) |
| Gender | ||||||||||||
| Male | 2140 | 111 | (5.2) | 1689 | 38 | (2.2) | 677 | 18 | (2.7) | 531 | 33 | (6.2) |
| Female | 794 | 35 | (4.4) | 476 | 13 | (2.7) | 93 | 2 | (2.2) | 367 | 19 | (5.2) |
| Age | ||||||||||||
| <55 | 936 | 54 | (5.8) | 616 | 11 | (1.8) | 276 | 7 | (2.5) | 302 | 19 | (6.3) |
| 55–64 | 972 | 40 | (4.1) | 793 | 15 | (1.9) | 279 | 6 | (2.9) | 271 | 15 | (5.5) |
| 65+ | 1026 | 52 | (5.1) | 756 | 25 | (3.3) | 215 | 7 | (3.3) | 325 | 18 | (5.5) |
| Total | 2934 | 146 | (5.0) | 2165 | 51 | (2.4) | 770 | 20 | (2.6) | 898 | 52 | (5.8) |
| RRa (95% CI) | 0.44 (0.31–0.63) | 0.44 (0.26–0.73) | 1.41 (0.99–2.00) | |||||||||
| Two-sided P-value | 0.000007 | 0.001 | 0.06 | |||||||||
| Controls | Lung | Upper aero-digestive | Kidney | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | |
| Country | ||||||||||||
| Russia | 786 | 60 | (7.6) | 399 | 11 | (2.8) | 340 | 12 | (3.5) | 281 | 28 | (10.0) |
| Hungary | 291 | 17 | (5.8) | 339 | 9 | (2.7) | – | – | ||||
| Poland | 790 | 44 | (5.6) | 694 | 20 | (2.9) | 201 | 6 | (3.0) | 79 | 5 | (6.3) |
| Slovakia | 229 | 7 | (3.1) | 300 | 3 | (1.0) | 39 | 0 | (0.0) | – | ||
| Czech Republic | 636 | 16 | (2.5) | 277 | 6 | (2.2) | 55 | 1 | (1.8) | 451 | 17 | (3.8) |
| Romania | 202 | 2 | (1.0) | 156 | 2 | (1.3) | 135 | 1 | (0.7) | 87 | 2 | (2.3) |
| Smoking status | ||||||||||||
| Never smoker | 1037 | 41 | (4.0) | 162 | 4 | (2.5) | 61 | 2 | (3.3) | 424 | 22 | (5.2) |
| Former smoker | 783 | 40 | (5.1) | 409 | 14 | (3.4) | 96 | 2 | (2.1) | 198 | 15 | (7.6) |
| Continuing smoker | 1102 | 64 | (5.8) | 1590 | 33 | (2.1) | 613 | 16 | (2.6) | 273 | 15 | (5.5) |
| Gender | ||||||||||||
| Male | 2140 | 111 | (5.2) | 1689 | 38 | (2.2) | 677 | 18 | (2.7) | 531 | 33 | (6.2) |
| Female | 794 | 35 | (4.4) | 476 | 13 | (2.7) | 93 | 2 | (2.2) | 367 | 19 | (5.2) |
| Age | ||||||||||||
| <55 | 936 | 54 | (5.8) | 616 | 11 | (1.8) | 276 | 7 | (2.5) | 302 | 19 | (6.3) |
| 55–64 | 972 | 40 | (4.1) | 793 | 15 | (1.9) | 279 | 6 | (2.9) | 271 | 15 | (5.5) |
| 65+ | 1026 | 52 | (5.1) | 756 | 25 | (3.3) | 215 | 7 | (3.3) | 325 | 18 | (5.5) |
| Total | 2934 | 146 | (5.0) | 2165 | 51 | (2.4) | 770 | 20 | (2.6) | 898 | 52 | (5.8) |
| RRa (95% CI) | 0.44 (0.31–0.63) | 0.44 (0.26–0.73) | 1.41 (0.99–2.00) | |||||||||
| Two-sided P-value | 0.000007 | 0.001 | 0.06 | |||||||||
T/C denotes the heterozygote (T/C) genotype for the variant rs17879961 in the CHEK2 gene: all other individuals had the common (T/T) genotype.
aAdjusted for age, sex, country and tobacco pack-years: omission of adjustment for tobacco would change these RRs into 0.47, 0.47 and 1.38, respectively.
Prevalence of CHEK2 heterozygous (T/C) genotype among controls and among cases of cancer, overall and within each country
| Controls | Lung | Upper aero-digestive | Kidney | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | |
| Country | ||||||||||||
| Russia | 786 | 60 | (7.6) | 399 | 11 | (2.8) | 340 | 12 | (3.5) | 281 | 28 | (10.0) |
| Hungary | 291 | 17 | (5.8) | 339 | 9 | (2.7) | – | – | ||||
| Poland | 790 | 44 | (5.6) | 694 | 20 | (2.9) | 201 | 6 | (3.0) | 79 | 5 | (6.3) |
| Slovakia | 229 | 7 | (3.1) | 300 | 3 | (1.0) | 39 | 0 | (0.0) | – | ||
| Czech Republic | 636 | 16 | (2.5) | 277 | 6 | (2.2) | 55 | 1 | (1.8) | 451 | 17 | (3.8) |
| Romania | 202 | 2 | (1.0) | 156 | 2 | (1.3) | 135 | 1 | (0.7) | 87 | 2 | (2.3) |
| Smoking status | ||||||||||||
| Never smoker | 1037 | 41 | (4.0) | 162 | 4 | (2.5) | 61 | 2 | (3.3) | 424 | 22 | (5.2) |
| Former smoker | 783 | 40 | (5.1) | 409 | 14 | (3.4) | 96 | 2 | (2.1) | 198 | 15 | (7.6) |
| Continuing smoker | 1102 | 64 | (5.8) | 1590 | 33 | (2.1) | 613 | 16 | (2.6) | 273 | 15 | (5.5) |
| Gender | ||||||||||||
| Male | 2140 | 111 | (5.2) | 1689 | 38 | (2.2) | 677 | 18 | (2.7) | 531 | 33 | (6.2) |
| Female | 794 | 35 | (4.4) | 476 | 13 | (2.7) | 93 | 2 | (2.2) | 367 | 19 | (5.2) |
| Age | ||||||||||||
| <55 | 936 | 54 | (5.8) | 616 | 11 | (1.8) | 276 | 7 | (2.5) | 302 | 19 | (6.3) |
| 55–64 | 972 | 40 | (4.1) | 793 | 15 | (1.9) | 279 | 6 | (2.9) | 271 | 15 | (5.5) |
| 65+ | 1026 | 52 | (5.1) | 756 | 25 | (3.3) | 215 | 7 | (3.3) | 325 | 18 | (5.5) |
| Total | 2934 | 146 | (5.0) | 2165 | 51 | (2.4) | 770 | 20 | (2.6) | 898 | 52 | (5.8) |
| RRa (95% CI) | 0.44 (0.31–0.63) | 0.44 (0.26–0.73) | 1.41 (0.99–2.00) | |||||||||
| Two-sided P-value | 0.000007 | 0.001 | 0.06 | |||||||||
| Controls | Lung | Upper aero-digestive | Kidney | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | n | T/C | (%) | |
| Country | ||||||||||||
| Russia | 786 | 60 | (7.6) | 399 | 11 | (2.8) | 340 | 12 | (3.5) | 281 | 28 | (10.0) |
| Hungary | 291 | 17 | (5.8) | 339 | 9 | (2.7) | – | – | ||||
| Poland | 790 | 44 | (5.6) | 694 | 20 | (2.9) | 201 | 6 | (3.0) | 79 | 5 | (6.3) |
| Slovakia | 229 | 7 | (3.1) | 300 | 3 | (1.0) | 39 | 0 | (0.0) | – | ||
| Czech Republic | 636 | 16 | (2.5) | 277 | 6 | (2.2) | 55 | 1 | (1.8) | 451 | 17 | (3.8) |
| Romania | 202 | 2 | (1.0) | 156 | 2 | (1.3) | 135 | 1 | (0.7) | 87 | 2 | (2.3) |
| Smoking status | ||||||||||||
| Never smoker | 1037 | 41 | (4.0) | 162 | 4 | (2.5) | 61 | 2 | (3.3) | 424 | 22 | (5.2) |
| Former smoker | 783 | 40 | (5.1) | 409 | 14 | (3.4) | 96 | 2 | (2.1) | 198 | 15 | (7.6) |
| Continuing smoker | 1102 | 64 | (5.8) | 1590 | 33 | (2.1) | 613 | 16 | (2.6) | 273 | 15 | (5.5) |
| Gender | ||||||||||||
| Male | 2140 | 111 | (5.2) | 1689 | 38 | (2.2) | 677 | 18 | (2.7) | 531 | 33 | (6.2) |
| Female | 794 | 35 | (4.4) | 476 | 13 | (2.7) | 93 | 2 | (2.2) | 367 | 19 | (5.2) |
| Age | ||||||||||||
| <55 | 936 | 54 | (5.8) | 616 | 11 | (1.8) | 276 | 7 | (2.5) | 302 | 19 | (6.3) |
| 55–64 | 972 | 40 | (4.1) | 793 | 15 | (1.9) | 279 | 6 | (2.9) | 271 | 15 | (5.5) |
| 65+ | 1026 | 52 | (5.1) | 756 | 25 | (3.3) | 215 | 7 | (3.3) | 325 | 18 | (5.5) |
| Total | 2934 | 146 | (5.0) | 2165 | 51 | (2.4) | 770 | 20 | (2.6) | 898 | 52 | (5.8) |
| RRa (95% CI) | 0.44 (0.31–0.63) | 0.44 (0.26–0.73) | 1.41 (0.99–2.00) | |||||||||
| Two-sided P-value | 0.000007 | 0.001 | 0.06 | |||||||||
T/C denotes the heterozygote (T/C) genotype for the variant rs17879961 in the CHEK2 gene: all other individuals had the common (T/T) genotype.
aAdjusted for age, sex, country and tobacco pack-years: omission of adjustment for tobacco would change these RRs into 0.47, 0.47 and 1.38, respectively.
Individuals with the T/C heterozygote genotype had a highly significantly lower risk of primary lung cancer than those with the T/T genotype [RR, T/C versus T/T, 0.44, CI 0.31–0.63, P = 0.000007 for lung cancer of any histological type] (Table 1) and a significantly lower RR of upper aero-digestive cancer (RR 0.44, CI 0.26–0.73, P = 0.001; all were squamous cell carcinomas), but a marginally significantly higher RR of kidney cancer (RR 1.41, CI 0.99–2.00, P = 0.06; all were cancers of the renal parenchyma). For each of these three RRs, no significant heterogeneity was seen with respect to country, age at onset or family history, although the power to identify any such heterogeneity is very limited.
If this genetic characteristic is unrelated to any form of lung cancer, then no significant heterogeneity would be expected between the RRs (T/C versus T/T) for different histological types of lung cancer. There was, however, significant heterogeneity (χ22 = 13.5, P = 0.001) between the RRs for squamous cell (RR 0.20, CI 0.10–0.38), small cell (RR 0.43, CI 0.19–0.98) and adeno-carcinoma of the lung (RR 0.87, CI 0.52–1.45), providing statistically independent additional evidence for some real effect on risk. Only for squamous cell cancers was the protective effect of the T/C genotype highly significant (P = 0.000001) in the present study (Fig. 1). All of the upper aero-digestive cancers were squamous cell carcinomas, although they originated from four different anatomic sites (mouth, larynx, pharynx and oesophagus). The number of cases for each specific upper aero-digestive site were too small for statistical stability, so although the protective effect appears more extreme for oral than oesophageal cancer this is based on only two versus six T/C heterozygotes, and is therefore not informative (overall test of heterogeneity: χ23 = 2.4, P = 0.49). If lung and upper aerodigestive cancer are combined, yielding a total of 2935 genotyped cases, the overall RR (T/C versus T/T, as in Fig. 1) is 0.46 (CI 0.33–0.63).
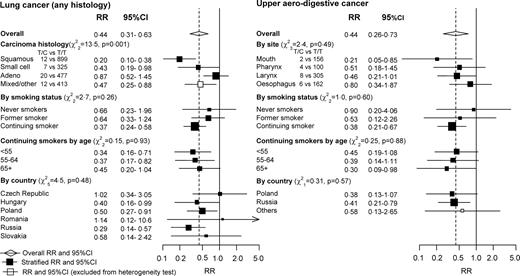
RR of lung cancer lung (any histology) and of upper aero-digestive cancer (all were squamous cell carcinomas) by CHEK2 genotype. Heterozygous (T/C) genotype versus common (T/T) genotype for variant rs17879961 in the CHEK2 gene. RRs are standardized by sex, age and, when relevant, smoking and country. χ2 tests are for heterogeneity. The overall RR is shown by the broken vertical line, and its 95% CI by a diamond. Other RRs are shown by squares (with areas inversely proportional to the variance of log RR), and their 95% CI by horizontal lines. White squares denote mixed categories (which are excluded from heterogeneity test calculations).
Most cases of lung or upper aero-digestive cancer in this study arose among current smokers, and were caused by smoking (13). Comparing current smokers (defined as those who had smoked in the 2 years prior to interview) versus never-smokers, the RRs with respect to smoking were 13.0 (95% CI 10.6–16.0) for lung cancer and 9.3 (6.9–12.6) for upper aero-digestive cancer. Hence, any substantial absolute protective effect against these lung or upper aero-digestive cancers associated with the T/C genotype must chiefly involve a reduction in the hazards of smoking. For both diseases, Figure 1 also illustrates the RRs with respect to genotype (T/C versus T/T) separately among never-smokers, former smokers and current smokers. The apparent protective effect associated with the T/C genotype is substantial and highly significant among current smokers (RR 0.37, CI 0.24–0.58, P = 0.00001 for lung cancer among smokers; RR 0.38, CI 0.21–0.67, P = 0.001 for upper aero-digestive cancer among smokers). The number of cases among former and never smokers were too small to be separately informative about the relevance of genotype, however, and although the results are compatible with there being little or no relevance of this genotype to risk among non-smokers, they are also compatible with the RRs associated with this genotype (T/C versus T/T) being as extreme among non-smokers as among current smokers (Fig. 1). Lung cancer and upper aero-digestive cancer are, however, far more common among smokers than among non-smokers in these populations (14). Hence, turning from RRs to absolute risks, the absolute protective effect associated with the T/C genotype is definitely much greater among smokers than among non-smokers. In terms of absolute risk, therefore, there is a very strong gene-environment interaction.
The deletion mutation (1100delC) was too rare to study properly in this population, being detected in only 10 cases (0.2%) and 6 controls (0.2%). The cases included five with lung cancer, one with oesophageal cancer and four with kidney cancer. While we were not able to calculate meaningful RRs from these small numbers, they do not suggest any substantial benefit or hazard. Because the deletion is rare, the RRs associated with having either functional defect (mis-sense or deletion) versus having neither were similar to those just for the mis-sense mutation.
DISCUSSION
Reliability of evidence of reduced lung cancer risk with the T/C polymorphism
The first report of the T/C polymorphism suggested that it involved a rare mutation, that the heterozygous (T/C) genotype would be a sufficient cause of the Li-Fraumeni syndrome (or a variant of it) and that it would involve a substantially increased RR of various neoplastic diseases, including lung cancer (7). Subsequent studies found, however, that the heterozygous (T/C) genotype is moderately common in parts of central Europe and that for several types of cancer the RRs associated with it are not extreme (9–12). The initial results from the one study that did include some cases of lung cancer actually found a non-significant inverse association (RR, T/C versus T/T, 0.5, P = 0.1), but did not consider the number of cases (272) large enough for this to be good evidence of real protection (12). The present study, with over 2000 cases of lung cancer and 3000 controls (and no overlap with any previous study), finds a similar inverse association that is highly significant (RR 0.44, P = 0.000007). The extreme significance of this P-value may be somewhat distorted by the play of chance, as we have by now analysed 97 genetic polymorphisms in this population, none of which yielded striking findings, and we are emphasizing this one just because it is the most exceptional (Fig. 2). But, even if we were to allow for this being the most extreme of our results by multiplying this P-value for lung cancer by 97, the finding would still remain highly significant (P = 0.0007 instead of P = 0.000007). Moreover, the significant (P = 0.001) heterogeneity between the RRs for the three main histological types of lung cancer provides statistically independent evidence against the hypothesis that there is no real effect on any type of lung cancer of this genetic defect, or of some other genetic factor that is associated (i.e. in linkage disequilibrium) with it on chromosome 22.
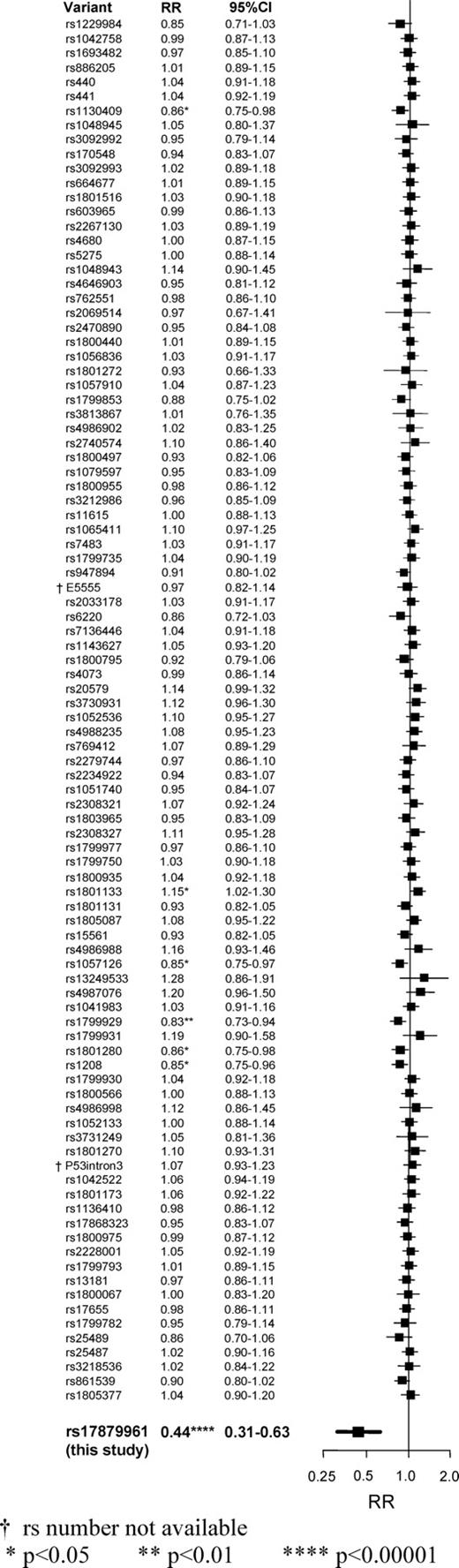
Comparison between the RRs for rs17879961 (bold, this study), and for all of the 96 other genetic polymorphisms analysed in these 2250 cases of lung cancer and 3052 controls. For each separate polymorphism, the RR for lung cancer compares individuals with at least one copy of the less common allele versus those with none. For rs17879961, RR = 0.44 (with 95% CI 0.31–0.63); χ21 = 20 and P = 0.000007. For the 96 other polymorphisms 1 has P < 0.01, 5 others have P < 0.05, none has a CI overlapping that for rs17879961, and the sum of the χ2 test statistics is only 99.1 (as against 96.0 expected if all were irrelevant to risk). †rs number not available; *P < 0.05; **P < 0.01; ****P < 0.00001.
Although our cases and controls are predominantly hospital-based, there is no good reason to expect that this would have materially influenced either the prevalence of the genetic variant in controls, or the apparent protective effect. In our two Polish centres, the study involved hospital controls in Lodz and population controls in Warsaw but the prevalences of the T/C genotype among them (6.0% in Lodz and 5.0% in Warsaw), based on approximately 400 controls in each centre, were similar to each other and to the prevalence of 4.8% among 4000 controls from the only other large Polish study (12). Furthermore, the prevalence of the T/C genotype did not vary significantly between broad diagnostic groups of the hospital controls.
Simulation studies show that population stratification can, under certain assumptions, moderately distort RR calculations, and that this distortion may be statistically significant in large studies (15,16). However, plausible amounts of population mixing in central Europe cannot easily explain the magnitude of the effect we have observed for lung cancer. Furthermore, although we did not genotype for a panel of markers that was chosen deliberately to help control for any potential population stratification (17), the absence of any clear differences between cases and controls in the other 96 genetic polymorphisms that we have already studied (Fig. 2) provides some indirect evidence against population stratification being entirely responsible for the present findings for lung cancer (as does the highly significant heterogeneity between the RRs for different histological types of lung cancer: Fig. 1).
Tobacco related cancers and the T/C polymorphism
Our results for upper aero-digestive squamous cell cancer and the T/C genotype are similar to our overall results for lung cancer, and again indicate an important protective effect. All these squamous cell cancers of the mouth, pharynx, larynx or oesophagus are strongly related to smoking in these populations (13). As, however, the same controls were used for lung and for upper aero-digestive cancer, the two RRs are not independent of each other, so it is difficult to judge from Figure 1 how much they support each other. If, to circumvent this, the 3000 lung and upper aero-digestive cancers are combined and compared to the 3000 controls, the overall RR (T/C versus T/T) is 0.46 (CI 0.33–0.63), and the nominal P-value is 0.000001, which is somewhat more extreme than that for lung cancer alone. This P-value of one in a million (together with the statistically independent P-value of one in a thousand for the heterogeneity between the RRs for different types of lung cancer) provides strong evidence for a real inverse association in this population between the T/C genotype and at least some of the main types of squamous cell cancer that are strongly related to tobacco. If the protective effect is caused by the mis-sense mutation in CHEK2 that we actually studied, then presumably the deletion mutation should have similarly protective effects, but because it was so rare in these populations we were unable to test this.
Kidney cancer and the T/C polymorphism
For kidney cancer, the moderately increased risk (RR 2.1, P = 0.0006) with the T/C genotype that was suggested by the initial Polish study of 13 types of cancer was the most extreme of the 13 RRs in that study, and needed replication (12). The present results also suggest a moderately increased risk (RR 1.5, P = 0.06) of kidney cancer with the T/C genotype. By design, all the genotyped kidney cancers in our study were tumours of the renal parenchyma (predominantly clear cell carcinomas), which are only weakly related to smoking (13). Although our findings are of only borderline significance they, together with evidence from other studies (9–12), suggest that among T/C heterozygotes there are increases in tumours of the renal parenchyma, breast and perhaps other tissues that counterbalance the decrease in the incidence of some of the types of cancer that are strongly related to tobacco smoking.
Paradox of a protective effect
It would not have been particularly remarkable for a functional defect in a key (evolutionarily conserved) cell cycle control gene such as CHEK2 to have increased the incidence of various types of cancer—indeed, this was what was generally expected (1–12; see Introduction). What is really remarkable about the present results is that (at least in smokers) people with one defective copy of this gene have a substantially reduced risk of developing squamous cell cancer of lung and upper aero-digestive tract. If this association is causal (i.e. if a functional defect in one copy of CHEK2 actually causes smokers to have a lower risk), then a novel mechanism must underlie it, and we can only speculate as to what this might be.
Speculation about possible protective mechanism in smokers: interference with squamous stem cell apoptosis?
The CHEK2 gene encodes a multi-functional kinase that is part of a system that can have two opposite effects on damaged stem cells. It can hold back stem cell division until any DNA damage has been repaired, or it can activate cell death by apoptosis if there appears to be damage that cannot be repaired (1–3). Stem cells that have only one good and one defective copy of the relevant CHEK2 gene might have only about half of the normal level of activity of the gene product. The present results strongly suggest that, at least in the very abnormal circumstances of continued DNA damage to squamous epithelia by tobacco smoke, normal stem cell defences (in T/T homozygotes) that involve CHEK2 can on balance have adverse consequences that are ameliorated by a reduction in CHEK2 activity in T/C heterozygotes.
More speculatively, perhaps normal levels of stem cell apoptosis in response to a given level of DNA damage to squamous epithelia are not appropriate if the rate of accumulation of damage (and hence the apoptosis rate) is high and if, in addition, any replacement stem cell has to be produced by a stem cell division in the highly mutagenic environment of the smoker's lung or upper aerodigestive tract (18). If smoking causes widespread stem cell apoptosis then, especially in a mutagenic environment, the resulting proliferation of replacement stem cells might actually increase the rate of fixation of mutations in the stem cell pool from which squamous cell cancers arise and hence eventually increase the cancer incidence rate. T/C heterozygotes might therefore be at somewhat lower risk of the normal mechanisms of tobacco-induced carcinogenesis because they have somewhat lower levels of activity of CHEK2 and hence of stem cell apoptosis.
This particular hypothesis may be wrong, but we hope that our statement of it will stimulate the kind of experimental evidence (for or against it) that is now needed. For, if the association is causal then a very peculiar mechanism is required to explain how a defect in such a key gene could provide substantial protection against tobacco carcinogenesis in a population where tobacco accounts for about half of all male cancer deaths (19).
Gene-environment interactions in the absolute risks
Even if the inverse association that we have observed for the T/C genotype and lung cancer is real, the risk of lung cancer among smokers who are T/C carriers is still substantial. Among men who become habitual cigarette smokers in early adult life and continue to smoke cigarettes, the cumulative risk of death from lung cancer by age 75 (in the absence of other causes of death) is ∼16% in the UK (20), and we have recently demonstrated similar cumulative risks among the continuing smokers in each of our six participating countries (14). [These calculations of cumulative risk merely involve combining national lung cancer mortality rates with the RRs associated with various smoking patterns, and with the prevalences of those patterns among controls (20).] Taking Poland as a representative example, we can calculate by such methods the cumulative risk of lung cancer death by age 75 to be ∼1% among men who never smoke, as opposed to ∼16% among continuing smokers (14). If among these smokers, there is a RR of about 0.44 between those with the heterozygous T/C genotype and those with the common T/T genotype, then the cumulative risks would be about 8 and 17%, respectively (Fig. 3; among smokers, there is no significant dependence on age of the RR, T/C versus T/T). The absolute difference between these shows that if the protective effects associated with the T/C genotype in the present study do represent a real genetic effect then, at least for lung cancer in smokers with this genotype, the absolute effect is substantial, and the absolute reduction in tobacco related cancer mortality would be even greater if there is also some real protection against upper aero-digestive squamous cell cancer in smokers. This does not, however, mean that people with the T/C genotype can smoke safely: for, if they do smoke then, in comparison with non-smokers (or long-term ex-smokers) their cancer hazard is still substantial—and, in addition, smoking causes more deaths from other diseases than from cancer (19).
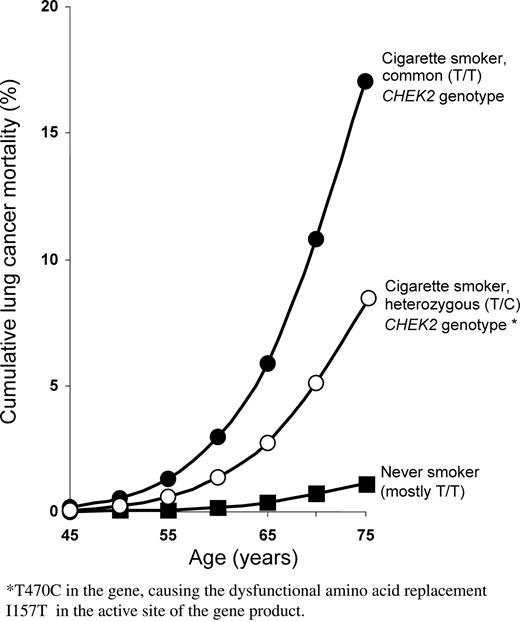
Relevance of smoking and of CHEK2 genotype (T/T or T/C for variant rs17879961) to lung cancer mortality in men aged 45–75 years. Cumulative risk (in the absence of other causes of death) based on national lung cancer death rates for men in Poland in the year 2000, assuming that the prevalences of smoking, ex-smoking and never smoking are as in the controls in this study and that the RRs for lung cancer incidence and mortality are as in this study.
Independent replication
In response to these findings, the Polish study (12) of 272 lung cancer cases that previously showed a non-significant protective effect of the mis-sense mutation has been updated to include a total of 716 cases (272 + 444), and now also shows a highly significant protective effect (RR 0.34, 95% CI 0.19–0.61, P = 0.0002) (C. Cybulski, personal communication).
MATERIALS AND METHODS
The study was conducted in 15 centres in 6 countries of central and eastern Europe, including the Czech Republic (Prague, Olomouc, Brno), Hungary (Borsod, Heves, Szabolcs, Szolnok, Budapest), Poland (Warsaw, Lodz), Romania (Bucharest), Russia (Moscow) and Slovakia (Banska Bystrica, Bratislava, Nitra). Each centre followed an identical protocol and sought to recruit a consecutive group of newly diagnosed cases of primary lung cancer, and, in some centres, upper aero-digestive cancer and/or kidney cancer, as well as a comparable group of population or hospital controls. All participants were recruited between 1998 and 2003.
Lung cancer cases were recruited in all centres, although upper aero-digestive cancer cases were not recruited in Hungary, and kidney cancer cases were not recruited in Slovakia or Hungary. The total number of potential cases for this study, before excluding the small numbers where genotyping failed, was 2250 lung, 811 aero-digestive (168 mouth, 113 pharynx, 326 larynx and 176 oesophagus, with 28 having overlapping sites), and 954 kidney cancers. All cases had been histologically confirmed locally, using standard WHO criteria. Among the lung cancers, 943 were squamous cell, 347 were small cell and 517 were adeno-carcinomas, with the remaining 443 being of mixed, indeterminate or rare histological types. All the aero-digestive cancers were squamous cell carcinomas (as other histological types were excluded from the genotyping); conversely, all of the genotyped kidney cancers were cancers of the renal parenchyma.
Controls in all centres except Warsaw were in-patients or out-patients with non-tobacco-related conditions at the same hospital as the cases, and were frequency matched with the cases by sex, age (within 3 years), centre and area of residence. No diagnostic category made up more than 20% of the overall control group in each centre. In Warsaw, population controls were recruited instead of hospital controls (by random sampling from the Polish electronic list of residents, frequency matched with the cases by sex, age and area of residence). In total, there were 3052 participating controls. The same interviewer-administered questionnaire was used for both cases and controls, except that cases were asked about what their smoking and other habits had been before disease onset, while controls were asked about their current habits. Written consent was obtained from each participant and ethics committee approval was obtained at each study centre as well as at the IARC and the NCI. The overall participation rates were high, being over 75% at each centre for both cases and controls. The ratio of cases to controls differed in each country, however, with a slight deficit of controls when compared with the number of lung cancer cases in Slovakia and Hungary, where collection of blood samples from controls proved more difficult than elsewhere. Further details of recruitment procedures and the questionnaire are given elsewhere (21).
After DNA extraction from lymphocyte samples, genotyping was performed by Taqman. For the deletion mutation, the primers and probes were as published (22). For the mis-sense mutation, the primers and probes used the DNA sequences in the SNP500 database (23). The forward and reverse primers were GGAGAGCTGGTAATTTGGTCA and AAAGGTTCCATTGCCACTGT, respectively, and the probes that complement C and T were TGCAGTGTAA GAGTTTT and ATGCAATGTAAGAGTTT, respectively.
DNA samples from cases and controls were arranged on 384 assay plates, with their positions on the plates chosen randomly to minimize any potential bias between cases and controls and with all recognizable identification of case or control status removed. Duplicate genotyping for quality control was performed for 859 randomly selected individuals (12% of the total series), but no discrepancies were found. As a further check, the genotypes were confirmed by direct DNA sequencing for 72 individuals (7 carriers of the common mis-sense mutation, all 16 carriers of the deletion mutation and 49 non-carriers of either), but again no discrepancies were found. The proportions successfully genotyped (i.e. the call rates) were similar for cases and controls, and were 96% overall for the T/C variant and 97% for the deletion.
The statistical analyses involved SAS software. Calculations of the RR (which in such case-control studies of incident disease can be used interchangeably with odds ratio) and its 95% CI were adjusted by multiple logistic regression for country of residence, age, sex and pack-years of smoking. P-values for all RRs are two-sided. Adjustment of the RRs by centre instead of country made no material difference to the results (data not shown). As RRs for different types of cancers are not mutually independent (because the controls overlap), χ2 tests of heterogeneity between them are based on similarly adjusted logistic regressions that ignore the controls and determine whether the type of cancer is significantly predictive of the genotype.
ACKNOWLEDGEMENTS
The chief acknowledgement is to the thousands of individuals who participated in this study, and to the staff in the collaborating centres. Recruitment was supported by a grant from the European Commission's INCO-COPERNICUS Programme (contract no. IC15-CT96-0313). Genotyping and analysis was funded by a National Cancer Institute R01 grant (contract no. CA 092039-01A2) and an Association for International Cancer Research grant (contract 03–281). J.M. was a CJ Martin research Fellow (NHMRC Australia). CTSU is supported by the Cancer Research UK and the Medical Research Council. No funding agency had any involvement in the study design, in the collection, analysis and interpretation of the data, or in writing the report or deciding to submit the paper. P.B., N.S.-D., J.L., D.Z., A.M., P.R., E.F., D.M., V.B., L.F., V.J., W.-H.C., N.R. and P.B. jointly designed the study and organized the recruitment of participants. J.M., L.M., A.C., F.O., M.S., M.H., R.H. and J.H. organized biological sample storage, DNA extraction and genotyping. J.M., V.G., R.H., R.P. and P.B. conducted the statistical analysis. J.P. suggested the possible explanatory role of apoptosis. P.B. and R.P. drafted the manuscript, and all co-authors contributed to the final draft.
Conflict of Interest statement. The first author declares that all authors have no conflict of interest.

{kind=link}
{kind=link}
{kind=link}