




Abstract
In this study, we report a novel duplication causing North Carolina macular dystrophy (NCMD) identified applying whole genome sequencing performed on eight affected members of two presumed unrelated families mapping to the MCDR1 locus. In our families, the NCMD phenotype was associated with a 98.4 kb tandem duplication encompassing the entire CCNC and PRDM13 genes and a common DNase 1 hypersensitivity site. To study the impact of PRDM13 or CCNC dysregulation, we used the Drosophila eye development as a model. Knock-down and overexpression of CycC and CG13296, Drosophila orthologues of CCNC and PRDM13, respectively, were induced separately during eye development. In flies, eye development was not affected, while knocking down either CycC or CG13296 mutant models. Overexpression of CycC also had no effect. Strikingly, overexpression of CG13296 in Drosophila leads to a severe loss of the imaginal eye-antennal disc. This study demonstrated for the first time in an animal model that overexpression of PRDM13 alone causes a severe abnormal retinal development. It is noteworthy that mutations associated with this autosomal dominant foveal developmental disorder are frequently duplications always including an entire copy of PRDM13, or variants in one DNase 1 hypersensitivity site at this locus.
Introduction
In 1971, Lefler, Wadsworth and Sidbury described a rare macular dystrophy in a large Irish family settled in North Carolina since 1800 (1). Twenty years later, Small re-examined these patients and stated that North Carolina macular dystrophy (NCMD) is indeed an autosomal dominant non-progressive inherited developmental macular disorder with three steady distinct phenotypes (2–4). In grades 1 and 2, patients are asymptomatic with unspecific drusen within the fovea. In grade 3, bilateral atrophic pigmented and fibrotic macular lesions are very characteristic of NCMD mimicking a ‘macular caldera’ in both eyes. Despite severe macular lesions, visual acuity is relatively preserved in grade 3 through an eccentric novel foveal development. Few families with NCMD phenotype mapping to the MCDR1 locus were identified outside the USA in Europe, Asia, Africa since the initial publication (5,6).
Until recently, this dystrophy was not genetically resolved even if a major locus (MCDR1) was identified on chromosome 6 (6q16) in 1992 (7). In 2015, applying whole genome analysis to 10 families, Small et al. identified three distinct single nucleotide variants in an intergenic region corresponding to a DNase 1 hypersensitivity site (DHS6S1, OMIM 616842, 6q16.2). This DHS6S1 is located upstream of two genes transcribed in opposite directions, PRDM13 (OMIM 616741, PR domain-containing protein 13, 6q16.2, 4 exons) and CCNC (OMIM 123838, Cyclin C, 6q16.2, 13 exons) (8). In one family, a tandem duplication (123 kb) containing only the entire PRDM13 gene and its DHS was identified. Based on transcription function, iPS 3 D cultures, and gene expression, the authors hypothesized that dysregulation of PRDM13 alone causes the disease. Later, in a second publication, a large tandem duplication (69 kb) including the DHS, the whole PRDM13 and a major part of CCNC genes was identified in one family with NCMD phenotype, suggesting once again the presumed causative role of the transcription factor PRDM13 (9). Despite the current major advances made in gene analysis of NCMD, the exact involvement of PRDM13 and CCNC in this disease remains unclear.
In the present study, we report two newly identified NCMD families carrying an even larger tandem duplication than previously described spanning the entire CCNC and PRDM13 genes and their potential DHS. This finding raises the question: whether PRDM13 dysregulation alone explains the observed phenotype. To support this hypothesis, we induced separately either a down-regulation or up-regulation of CCNC and PRDM13 orthologues in Drosophila as a model of eye development.
Results
Identification of two NCMD families, clinical findings
The clinical databases of three French national reference centers specialized in inherited retinal dystrophies were screened for NCMD. Due to its rarity, only two unrelated families could be identified. In the first three-generation family (family A, Fig. 1), four members were examined (I: 2, II: 2, II: 3, III: 2, Fig. 2). Two individuals (I: 2, II: 2) had a visual acuity of 20/20 and displayed grades 1 and 2 NCMD, while II: 2 and III: 2 had a grade 3 NCMD. Patient II: 2 had a visual acuity of 20/40 OD, 20/60 OS, and patient III: 2 had 20/40 OD and 20/25 OS.
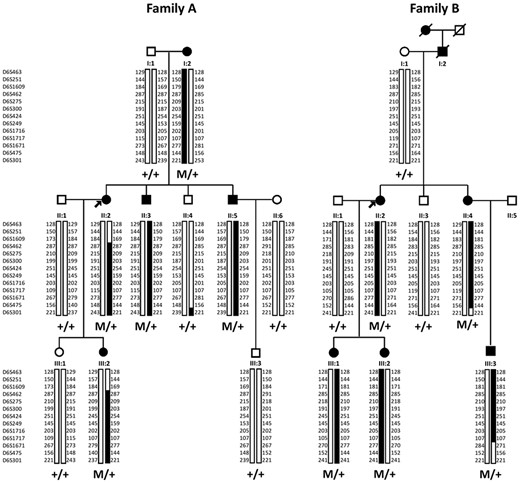
Pedigrees of families affected with autosomal dominant North Carolina macular dystrophy. Filled symbols indicate affected family members; squares: males; circles: females; arrows: index patients. Haplotypes at the MCDR1 locus for 13 microsatellite markers are shown in families (A) and (B). The common haplotype is shown in black. M is for the tandem duplication.
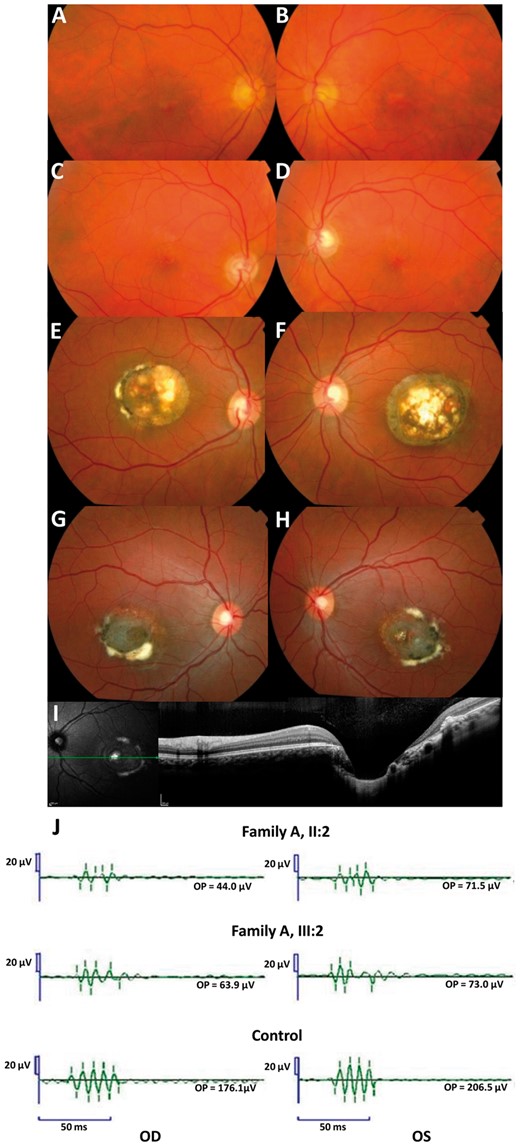
Family A. Fundus photographs of the mother (A-B, I: 2) and the brother (C-D, II: 3) of the index (II: 2). The index (E-F) and her daughter (G-H) have a Grade 3 and a visual acuity of 20/40 in the right eye, 20/60 in the left eye and 20/40 in the right eye and 20/25 in the left eye respectively. For these two last patients, note the severe macular disorganization with atrophy, retinal pigment epithelium tufts, fibrosis and with a profound excavation or ‘caldera’ on OCT frame (I). OPs in two affected subjects and in one control (J): Note the reduction of the late Ops suggesting a dysfunction of rod associated amacrine cells. All latencies are increased.
In the second three-generation family (family B, Fig. 1), five patients were affected with 2 grade 1 (III: 1, III: 2), and 3 grade 3 (II: 2, II: 4, III: 3). Visual acuity ranged from 20/60 to 20/20.
Oscillatory potential analysis from full-field electroretinograms (ERG)
In four affected patients for whom full-field ERGs were available (family A), a decrease in the amplitudes of the potentials OP3, OP4 is noted (Fig. 2J). The latencies of each oscillation are increased. These oscillatory potentials are thought to be generated by neuronal activity of the inner plexiform layer including the amacrine cells (10,11). The other rod- and cone-driven ERG responses were normal.
Genetic linkage analysis
Microsatellite markers originally linked to the MCDR1 locus, were investigated in the available DNA samples in the two families (7). A total of 10 affected and 9 unaffected subjects were genotyped at 13 microsatellite markers on chromosome 6q14-q16.2 (Fig. 1). For each family, we identified a haplotype, which segregated with the disease phenotype (Fig. 1). Several recombination events were found involving proximal and distal markers. For family A, patient II: 2 had a crossover between D6S1609 and D6S462 and the individual II: 4 between D6S475 and D6S301. Two crossovers between D6S475 and D6S301 for II: 4 and between D6S1717 and D6S1671 for III: 3 were also observed in the family B (Fig. 1). Our disease locus is therefore located between D6S462 and D6S475 for family A and between D6S463 and D6S475 for family B, corresponding to approximately 16 Mb and 19 Mb, respectively (Fig. 1). This locus encompassed the 1.8 Mb MCDR1 locus previously reported (7). Only one microsatellite marker, D6S1717, showed a common status (107) between the two morbid haplotypes from the two families suggesting that the two families could be unrelated (Fig. 1).
Whole genome next-generation sequencing (WGS)
WGS did not identify SNP or small Indel in the disease locus among the NCMD patients. In addition, there were no mutations in the DNase 1 hypersensitivity site in these two families. However, a large tandem duplication (98, 389 bp) was identified by WGS. The duplication co-segregated with the disease phenotype in available family members in both families (10 affected patients and 9 healthy relatives).
The exact genomic position of the duplication was confirmed by PCR and Sanger sequencing using primers flanking the duplication junction. The tandem duplication spanned between the positions 99, 984, 309-100, 082, 698 bp on chromosome 6 (Hg19) (Figs 3 and 4, Table 1). No exogenous DNA sequence was inserted in the tandem duplication (Fig. 4). In contrast to previous reports, this new duplication encompassed the DHS and the entire coding sequences of CCNC and PRDM13 genes (Fig. 4, Table 1) (8,9).
Summary of North Carolina macular degeneration known and novel mutations
| Variant number | Type of variant | Chromosomal position (Hg19) | Nucleotide change | Reference |
|---|---|---|---|---|
| V1 | SNP | 6: 100, 040, 906 | G>T | (8) |
| V2 | SNP | 6: 100, 040, 987 | G>C | (8) |
| V3 | SNP | 6: 100, 041, 040 | G>T | (8) |
| V4 | Tandem DUP | 6: 100, 020, 205-100, 143, 306 | 123, 101 bp DUP | (8) |
| V5 | Tandem DUP | 5: 3, 587, 901-4, 486, 027 | 898, 126 bp DUP | (8) |
| V6 | Tandem DUP | 6: 99, 996, 226-100, 065, 137 | 69, 912 bp DUP | (9) |
| V7 | Tandem DUP | 6: 99, 984, 309-100, 082, 698 | 98, 389 bp DUP | Present study |
| Variant number | Type of variant | Chromosomal position (Hg19) | Nucleotide change | Reference |
|---|---|---|---|---|
| V1 | SNP | 6: 100, 040, 906 | G>T | (8) |
| V2 | SNP | 6: 100, 040, 987 | G>C | (8) |
| V3 | SNP | 6: 100, 041, 040 | G>T | (8) |
| V4 | Tandem DUP | 6: 100, 020, 205-100, 143, 306 | 123, 101 bp DUP | (8) |
| V5 | Tandem DUP | 5: 3, 587, 901-4, 486, 027 | 898, 126 bp DUP | (8) |
| V6 | Tandem DUP | 6: 99, 996, 226-100, 065, 137 | 69, 912 bp DUP | (9) |
| V7 | Tandem DUP | 6: 99, 984, 309-100, 082, 698 | 98, 389 bp DUP | Present study |
SNP, single nucleotide polymorphism; DUP, duplication; bp, base pairs.
Summary of North Carolina macular degeneration known and novel mutations
| Variant number | Type of variant | Chromosomal position (Hg19) | Nucleotide change | Reference |
|---|---|---|---|---|
| V1 | SNP | 6: 100, 040, 906 | G>T | (8) |
| V2 | SNP | 6: 100, 040, 987 | G>C | (8) |
| V3 | SNP | 6: 100, 041, 040 | G>T | (8) |
| V4 | Tandem DUP | 6: 100, 020, 205-100, 143, 306 | 123, 101 bp DUP | (8) |
| V5 | Tandem DUP | 5: 3, 587, 901-4, 486, 027 | 898, 126 bp DUP | (8) |
| V6 | Tandem DUP | 6: 99, 996, 226-100, 065, 137 | 69, 912 bp DUP | (9) |
| V7 | Tandem DUP | 6: 99, 984, 309-100, 082, 698 | 98, 389 bp DUP | Present study |
| Variant number | Type of variant | Chromosomal position (Hg19) | Nucleotide change | Reference |
|---|---|---|---|---|
| V1 | SNP | 6: 100, 040, 906 | G>T | (8) |
| V2 | SNP | 6: 100, 040, 987 | G>C | (8) |
| V3 | SNP | 6: 100, 041, 040 | G>T | (8) |
| V4 | Tandem DUP | 6: 100, 020, 205-100, 143, 306 | 123, 101 bp DUP | (8) |
| V5 | Tandem DUP | 5: 3, 587, 901-4, 486, 027 | 898, 126 bp DUP | (8) |
| V6 | Tandem DUP | 6: 99, 996, 226-100, 065, 137 | 69, 912 bp DUP | (9) |
| V7 | Tandem DUP | 6: 99, 984, 309-100, 082, 698 | 98, 389 bp DUP | Present study |
SNP, single nucleotide polymorphism; DUP, duplication; bp, base pairs.
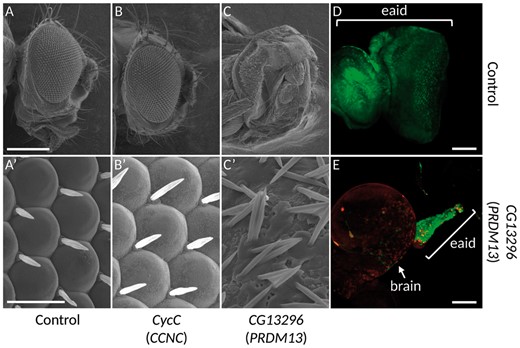
The overexpression of PRDM13 orthologue leads to defects in ocular development. (A-C’) Electron microscopy photographs of adult fly eyes. (A-A’) lgmr-Gal4/+, which drives the expression of the transgene in all photoreceptor cells, shows that the overexpression of the Gal4 by itself, does not lead to any phenotype and constitute the control experiment. Whereas the overexpression of CyclinC does not produce any defect in the eye structure (B-B', lgmr-Gal4/UAS-FLAG-CycC), the overexpression of the orthologue of PRDM13, CG13296, leads to a strong loss of photoreceptors (C-C', lgmr-Gal4/UAS-FLAG-CG13296). (A’-C’): Higher magnifications of Figure A, B and C. (D-E): Third instar larvae eye-antennal imaginal discs overexpressing a control transgene, UAS-Dcr2 (D), or the orthologue of PRDM13, UAS-FLAG-CG13296 (E) at early developmental stages using the FLPout system (14) in conjunction with the ey-FLP (13). Cells expressing the transgenes are visualized through the expression of the GFP (green). In the control experiment (D), all the eye-antennal imaginal disc (eaid) is visible and the structure is unaffected. When the orthologue of PRDM13 is overexpressed, the eaid is almost lost (E), with few remaining cells (compare the size and shape of the discs in D and E). Since the disc is attached to the brain, the brain is usually removed at dissection. Considering the small size of the disc when PRDM13 is overexpressed, please note that the brain in E is shown only to localize the remaining eaid structure. The FLAG-CG13296 is detected with an anti-FLAG antibody (red). The brackets in D and E show the eye-antennal imaginal discs (eaid). Scale bars: (A-C) 231 µm; (A'-C') 16.7 µm; (D-E) 50 µm.
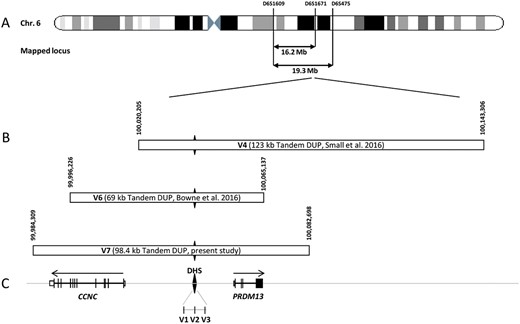
Chromosomal localization of the mapped loci, position of the detected and the referenced mutations on the MCDR1 locus. (A) Schematic representation of the chromosome 6 showing the location of the two mapped loci and, (B) the novel mutation presented in this study (V7), and the known mutations (V1-V6). (C) Schematic representation of the exon-intron structure of CCNC and PRDM13. The black diamond form indicates the DNase I hypersensitivity site (DHS) with the three SNP mutations previously identified in this region (V1-V3). The DHS is present in the three tandem duplications. The horizontal arrows above genes representation indicate the transcriptional direction of each gene on both sides of the DHS.
Defective eye development in Drosophila melanogaster is induced by overexpression of CG13296, the orthologue of PRDM13
To functionally understand the effect of this duplication during development, we used the fruit fly as a model organism and focused our study on the development of its visual system since the NCMD specifically affects the development of the eye in patients. The genome of Drosophila melanogaster contains orthologues for CCNC and PRDM13, namely cyclin-C (CycC) and CG13296 respectively. The phenotype observed in patients bearing the duplication could be explained by an overexpression, or less likely a decrease of the expression of one or both genes. A decreased expression could not be excluded because all the three reported tandem duplications encompassed the DHS, a potential regulator site of both PRDM13 and CCNC genes. To tackle this point, we used the UAS/Gal4 system to overexpress or knock down the expression of these genes specifically in photoreceptors using the lgmr-Gal4 driver (Fig. 3D and 3E) (12). To knock down the expression of these genes, two different UAS-RNAi lines were used for each of them. Neither the knock down of CycC nor of CG13296 led to any eye defect (data available on request). We did the reverse experiment by overexpressing these two genes using UAS-ORF constructs designed for this study. Two different lines for each construct were used. Whereas the overexpression of CycC did not lead to any phenotype compared to the control (Fig. 3A and B’), the overexpression of CG13296 led to a strong phenotypic characteristic of photoreceptor degeneration (Fig. 3C and C’) that can be related to NCMD grade 3. Moreover, the overexpression of CG13296 in the third instar larva eye-antennal imaginal disc with the ey-FLPout system leads to an almost complete loss of the eye-antennal imaginal disc, compared to the control experiment (Fig. 3D and E) (13,14).
All together, these data suggest that the phenotype observed in patients bearing the duplication encompassing CCNC and PRDM13 genes is likely due to an overexpression of PRDM13 transcription factor, which leads, in Drosophila, to the malformation of photoreceptors.
Discussion
Here, we identified a third tandem duplication (V7) in MCDR1 that encompasses for the first time the entire sequence of CCNC, PRDM13 genes and an intergenic DHS. These results contribute to the increasing evidence that this rare macular developmental disease appears to be frequently linked to large duplications, accounting for 4 out of 14 families published with genetic results and for 50% of mutation types (V1, V2, V3 as SNPs, V4 V6 V7 as duplications, Table 1).
According to Small, NCMD is linked to a dysregulation of PRDM13, because of the following observations: (i) PRDM13 is a transcription factor specifically expressed in the retina whereas CCNC displays ubiquitous expression, (ii) large duplication (V4) include a complete copy of PRDM13 excluding CCNC (V4), (iii) during 3 D iPSC culture with neural retinal differentiation, CCNC expression is constant whereas PRDM13 expression is correlated with time or neural differentiation (8).
However, all three described duplications including the present findings involve the DHS being a potential regulator of both PRDM13 and CCNC genes, thus an associated role of CCNC could not be excluded. Our fly model did not support a role of CCNC since in the CycC knock down flies no eye anomalies could be detected. Similarly, CycC overexpression had no effect on ocular structures. Moreover, the fly model clearly showed that down regulation of CG13296, PRMD13 orthologue, had no effect on eye development whereas its overexpression induced a severe eye malformation (Fig. 3). These data are supported by the fact that all duplications included a complete sequence of PRDM13 and the DHS (Fig. 4). In addition, the DHS SNPs might also upregulate the level of PRDM13 transcription.
PRDM13 is a transcription factor involved in neuronal differentiation of the retina particularly in amacrine cell fate. A sustained expression followed by a reduction of PRDM13 levels after or during retinal cell differentiation is probably necessary. In the mouse, Prdm13 expression begins at E12.5 in the neuroblastic layer, and is gradually reduced from P9 onwards with a lower level maintained in adulthood (15). In human wild type iPSCs, PRDM13 mRNA levels also decreased during differentiation towards retinal cells (8). The above observations, together with the phenotype induced by overexpression of the Drosophila PRDM13 orthologue on fly retinal development, suggest that NCMD results from increased levels of PRDM13 transcription factor with possibly an insufficient decrease during retinal differentiation. The NCMD phenotype could also be explained by spatial and time difference sequences of retinal development. Indeed, in monkey retina, the sequence of retinal cell differentiation and synaptic development is dissimilar between the cone-dominated fovea and the rod-dominated peripheral retina. In the fovea, the amacrine synapses appeared at fetal day 88, following those of cones (fetal day 60) and bipolar cells (fetal day 55) (16). In peripheral retina, the sequence is the opposite with amacrine synapses formed at fetal day 78, bipolar at 99, and photoreceptors at 105 (16). Interestingly, the second NCMD locus, i.e. MCDR3, the underlying mutation (V5) is also a duplication including IRX1, another transcription factor (8). Dysregulation of this transcription factor in Drosophila led to an abnormal eye development with a mirror pattern (17).
The anomalies of oscillatory potentials (OP) noted in our family A suggested an abnormal amacrine cell subtype specification. Abnormal amacrine number and subtype specification of GABAergic and glycinergic amacrine cells were reported in Prdm13-/- mice without any effect on other retinal cell types (15). Forced expression of Prdm13 in chimeric mice demonstrated that Prdm13 induced these two subtypes and not the cholinergic subtypes. Nevertheless, the Prdm13-/- mice had normal OPs waveforms and amplitudes. As OP3, OP4 are possibly generated by amacrine cells linked to rods, the decrease of OP3, OP4 with normal OP1-OP2 amplitudes noted in our patients could reflect a rod associated amacrine cell dysfunction or subtype differentiation (10,11).
To conclude, this study provides major evidence reinforcing the hypothesis that the NCMD phenotype results exclusively from the upregulation of PRDM13, both in patients with variants affecting a DNase 1 hypersensitivity site located between CCNC and PRDM13 or with large duplications spanning an entire copy of PRDM13. An upregulation and/or an insufficient decrease of PRDM13 during or after retinal cell fate specification are potentially the cause of the severe abnormal macular development. As this singular developmental dystrophy is frequently caused by large duplications accounting for 50% of mutation types, specific genetic analyses are required in NCMD patients.
Materials and Methods
Informed consent was obtained for clinical examination and genetic analysis from all patients. All methods were carried out in accordance with approved protocols of Montpellier and Lille University Hospitals, and in agreement with the Declaration of Helsinki. The Ministry of Public Health accorded approval for biomedical research under the authorization number 11018 S.
For each patient, age at examination, refraction, initial and final best-corrected visual acuity were noted. The best-corrected visual acuity was obtained with Snellen charts. Color fundus photographs were performed with Topcon Imagenet (Ophthalmic Imaging Systems, Japan) or Nidek non-mydriatic automated fundus camera AFC 330 (Nidek Inc, Japan). Autofluorescence imaging and spectral domain optical coherent tomography were performed with Combined Heidelberg Retina Angiograph + OCT Spectralis device (Heidelberg Engineering, Dossenheim, Germany). The retinal lesions were classified according to the three grades: grade 1 with small drusen within the fovea, grade 2 with confluent macular drusen, grade 3 with characteristic large macular calderas (18).
Analysis of OPs
Full-field electroretinography (ERG) was performed according to the guidelines of the International Society for Clinical Electrophysiology of Vision using a Ganzfeld apparatus (Ophthalmologic Monitor, Métrovision, Pérenchies, France) (19). Scotopic OPs were extracted by applying a Fast Fourier Transform filter to remove all Fourier components below 80 Hz then calculating the amplitudes and the sum of the four peaks of oscillations.
Genotyping of microsatellite markers and linkage analysis
PCR was carried out in a 25 μl final volume containing 50 ng genomic DNA, 5 pmol of each primer, 0.2 mM dNTPs (MP Biochemicals, Asse-Relegen, Belgium), 2 mM MgCl2, PCR buffer and 1 unit of DNA polymerase (AmpliTaq Gold; Applied Biosystems, Foster City, CA). Initial denaturation at 95 °C for 10 min was followed by 35 cycles of denaturation at 94 °C for 30 s, specific annealing temperature for 30 s, and extension at 72 °C for 1 min. A final extension step was performed at 72 °C for 10 min. The PCR products were diluted and mixed with Genescan 400HD ROX size standard, and subsequently analyzed on an Applied Biosystems 3130xL genetic analyzer (Applied Biosystems, Foster City, CA). Results were analyzed with GeneMapper software (version 4.0, Applied Biosystems, Foster City, CA). Two-point LOD scores were calculated with Superlink-online (http://bioinfo.cs.technion.ac.il/superlink-online/; date last accessed June 28, 2017). The phenotype was analyzed as an autosomal dominant and fully penetrant trait with an affected allele frequency of 0.001.
Whole genome sequencing
Whole genome sequencing was performed on six members of family A and five members of family B by the Centre National de Recherche en Génomique Humaine (Institut de Biologie François Jacob, CEA). After a complete quality control, genomic DNA (1 µg) was used to prepare a library for whole genome sequencing, using the Illumina TruSeq DNA PCR-Free Library Preparation Kit, according to the manufacturer's instructions. After normalization and quality control, qualified libraries were sequenced on a HiSeq2000 platform from Illumina (Illumina Inc., CA, USA), as paired-end 100 bp reads. At least 3 lanes of HiSeq2000 flow cell were produced for each sample with an average sequencing depth of 30x. Sequence quality parameters were assessed throughout the sequencing run and standard bioinformatics analysis of sequencing data was based on the Illumina pipeline to generate FASTQ file for each sample.
The following treatments were performed on the fastQfiles alignment on the human genome (GRCh37) [and decoy (Heng Li's hs37d5 genome for 1000 genomes project, ftp://ftp.ncbi.nlm.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz)] was performed using bwa software (mem + default option, https://github.com/lh3/bwa; date last accessed June 28, 2017, version 0.7.12).
Duplicate sequences were then referenced and eliminated from the bam files using Sambamba tools (http://lomereiter.github.io/sambamba/docs/sambamba-view.html; date last accessed June 28, 2017). An additional step of realignment was performed on the bam file using GATK programs (RealignerTargetCreator/IndelRealigner). Coverage analyses were generated using an in house pipeline based on metrics generated by Bedtools programs (http://code.google.com/p/bedtools/; date last accessed June 28, 2017).
Identification of variants was then performed [using HC only if they do not use multicall+. g.vcf if used] using 4 programs: UnifiedGenotyper and HaplotypeCaller from GATK, Platypus (http://www.well.ox.ac.uk/platypus; date last accessed June 28, 2017) and Samtools. Results generated by these 4 programs were then grouped in a vcf file. Annotation of the vcf file is carried out using and annotated using snpEff and snpSift (http://snpeff.sourceforge.net and http://snpeff.sourceforge.net/SnpSift.html; date last accessed June 28, 2017) based on data available in the Ensembl database (http://www.ensembl.org/index.html; date last accessed June 28, 2017) and dbNSFP database (https://sites.google.com/site/jpopgen/dbNSFP; date last accessed June 28, 2017).
Detection of genetic variants
Cleaned sequence data were aligned and mapped to the reference genome (hg19) by Burrows-Wheeler aligner (BWA, http://bio-bwa.sourceforge.net/; date last accessed June 28, 2017) using mem option (20). FreeBayes variant caller version 0.9.10-3-g47a713e was used on the ready-to-use alignments to call both SNVs and INDELs (21). ERDS (Estimation by Read Depth with SNVs) software was used with default parameters to call CNVs from WGS data on each individual (22). It uses WGS data along with previously generated VCF files using the read depth, paired end mapping, soft-clip signature, and number of contiguous heterozygous and homozygous SNVs to call CNVs.
Duplication confirmation
Genomic DNA was isolated from 10 ml peripheral blood leucocytes using standard salting out procedure (23). PCR primers were designed to amplify the duplication junction (primer pairs and PCR conditions are available on request) which was subsequently sequenced with an Applied Biosystems 3130xL genetic analyser (Applied Biosystems, Foster City, CA), using a BigDye Terminator cycle sequencing ready reaction kit V3.1 (Applied Biosystems, Foster City, CA) following manufacturer’s instructions. Sequence analysis was performed using Collection and Sequence Analysis software package (Applied Biosystems, Foster City, CA).
Eye development in D. melanogaster
The genome of D. melanogaster contains orthologues for CCNC and PRDM13 genes i.e. cyclin-C (CycC) and CG13296 respectively. We used the UAS/Gal4 system to overexpress or knock down the expression level of these genes specifically in photoreceptors using the lgmr-Gal4 driver (12). To knock down the expression of these genes, two different UAS-RNAi lines where used for each of them.
Flies were raised at 25 °C. For targeted misexpression we used the UAS/GAL4 system (24). UAS strains used were: UAS-CycC-RNAi (VDRC, 27937 and 48834), UAS-CG13296-RNAi (VDRC, 106770 and 17132), UAS-FLAG-CycC and UAS-FLAG-CG13296 (this study). The FLPout system (14), in conjunction with the ey-FLP line (13) was used for FLAG-CG13296 overexpression in the third instar eye-antennal imaginal discs. The lgmr-Gal4 was used to overexpress the different constructs in photoreceptor cells. The UAS-FLAG-CycC was built as follows: the cDNA of CycC was PCR amplified from the LD35705 clone (DGRC) with primers containing NotI and XhoI restriction sites (GCGGCCGCGGGCAATTTTTGGC, CTCGAGCTAACGCTGAGGCGGTGGT) and clone into the pUAST. After sequencing, the FLAG tag was inserted as a linker with EcoRI and NotI restriction sites (AATTCATGGACTACAAGGACGACGATGACAAGGC, GGCCGCCTTGTCATCGTCCTGTAGTCCATG). The UAS-FLAG-CG13296 was built as follows: the cDNA of CG13296 was PCR amplified from the RT01047 clone (DGRC) with primers containing NotI and XhoI restriction sites (GCGGCCGCACACACGACAGCTCAACTG, CTCGAGCTAGCTGGATTTCGAAGA) and clone into the pUAST. The FLAG tag was inserted as a linker with EcoRI and NotI restriction sites and after sequencing, both constructs were then processed for standard P-element transgenesis (BestGene, Chino Hills, CA).
The samples were sputter coated with an approximative 10 nm thick gold film and then examined under a scanning electron microscope (Hitachi S4000, at COMET, MRI facilities at INM, Montpellier, France) using a lens detector with an acceleration voltage of 10 kV at calibrated magnifications.
Acknowledgements
We thank all the family members who participated in this study. We acknowledge the support from the INSERM. We address special thanks to SOS Rétinite Pigmentaire foundation, which supports fellowship for GM. WJ received financial support from Fondation de France (programme Berthe Fouassier). We would like to thank the French Institute of Bioinformatics (IFB, ANR-11-INBS-0013) for providing storage and computing resources on its national life science Cloud. IFB acknowledges funding by the call ‘Infrastructures in Biology and Health’ in the framework of the French ‘Investments for the Future’ (ANR-11-INBS-0013) initiative, and EU H2020 projects CYCLONE (644925), EXCELERATE (676559) and EGI-Engage (654142). IFB is the French ELIXIR node. Patrick Carroll, a native English speaker made English corrections of the manuscript.
Conflict of Interest statement. None declared.
Funding
Laboratory of Excellence GENMED (Medical Genomics) grant no. ANR-10-LABX-0013 managed by the National Research Agency (ANR) part of the Investment for the Future program.

{kind=link}
{kind=link}
{kind=link}
{kind=link}