




Abstract
Pten controls a signaling axis that is implicated to regulate cell proliferation, growth, survival, migration, and metabolism. The molecular mechanisms underlying the specificity of Pten responses to such diverse cellular functions are currently poorly understood. Here we report the control of Pten activity and signaling specificity during the cell cycle by Ndfip1 regulation of Pten spatial distribution. Genetic deletion of Ndfip1 resulted in a loss of Pten nuclear compartmentalization and increased cell proliferation, despite cytoplasmic Pten remaining active in regulating PI3K/Akt signaling. Cells lacking nuclear Pten were found to have dysregulated levels of Plk1 and cyclin D1 that could drive cell proliferation. In vivo, transgene expression of Ndfip1 in the developing brain increased nuclear Pten and lengthened the cell cycle of neuronal progenitors, resulting in microencephaly. Our results show that local partitioning of Pten from the cytoplasm to the nucleus represents a key mechanism contributing to the specificity of Pten signaling during cell proliferation.
Introduction
Phosphatase and tensin homolog deleted on chromosome 10 (Pten) is a master regulator of cell function and one of the most commonly mutated or lost tumor suppressor proteins in cancer (Song et al., 2012). Besides cancer, Pten dysfunction has also been shown to be important in autism spectrum disorders (Butler et al., 2005; O'Roak et al., 2012), epilepsy (Pun et al., 2012), and diabetes (Butler et al., 2002; Pal et al., 2012), indicating the pleiotropic effects of Pten in disease. Interestingly, Pten mutations in disease states are not functionally equivalent (Rodriguez-Escudero et al., 2011), suggesting context-dependent effects of Pten activity. The main catalytic role for Pten resides in the cytoplasm of the cell where Pten negatively regulates signaling by phosphatidylinositol 3-kinase (PI3K) through dephosphorylation of the plasma membrane lipid phosphoinositide-3,4,5-trisphosphate (PIP3) (Maehama and Dixon, 1998; Stambolic et al., 1998). Pten is also localized to the nucleus of cells; however, nuclear pools of PIP3 appear insensitive to catalysis by Pten, suggesting that the enzyme function is dispensable in the nucleus (Lindsay et al., 2006). Supporting the non-enzymatic function of Pten are phosphatase inactive mutants that have been shown to maintain tumor-suppressive function (Maier et al., 1999; Gildea et al., 2004), as well as discrepancies between mouse models of Pten loss compared with transgenic mice overexpressing Akt (Blanco-Aparicio et al., 2007). Together, this evidence indicates that Pten can function independent of the PI3K signaling pathway. If Pten functions via multiple signaling networks, it currently remains unknown how Pten can specifically regulate selected signaling components whilst excluding others.
A central concept for the regulation of signaling networks is the compartmentalization of the effector machinery. Consistent with this, it is now well established that Pten can be localized to a number of organelles in the cell. Pten is found at the plasma membrane (Vazquez et al., 2006), nucleus (Gimm et al., 2000), ER (Bononi et al., 2013), mitochondria (Zhu et al., 2006), and even secreted outside of the cell (Putz et al., 2012; Hopkins et al., 2013). Therefore, the subcellular distribution of Pten appears important for determining the signaling components regulated. The recent identification of Pten trafficking on endosomes (Li et al., 2014) has added to our current understanding of the spatial regulation of Pten in the cell and also provides a platform for differential Pten signaling. Rab5 GTPase in combination with the adaptor protein Nedd4-family interacting protein 1 (Ndfip1) were shown to be required for both the ubiquitination and trafficking of Pten on endosomes from the cytoplasm to the nucleus of the cell. However, how the compartmentalization of Pten functions under physiological settings to confer the regulation of separate networks besides PIP3 signaling is currently not fully understood. Ndfip1 is an adaptor and activator of the Nedd4-family of E3 ubiquitin ligases. Together, they form a complex that has been shown to target Pten for ubiquitination and trafficking in the cell (Mund and Pelham, 2009; Howitt et al., 2012). Here we took advantage of the Ndfip1 trafficking system as a model to understand the function of nuclear Pten in a context that maintained Pten abundance and cytoplasmic activity. We find that during the cell cycle, Ndfip1 is required for efficient Pten nuclear compartmentalization and that disruption of this mechanism leads to abnormalities resulting in increased cell proliferation, without affecting PI3K/Akt signaling. Our results show that Pten nuclear compartmentalization alters the signaling specificity of Pten, permitting the targeting of the oncogenic proteins Plk1 and cyclin D1. In vivo, this pathway was shown to have a significant effect on neurogenesis and the regulation of brain size. Together, our data support the hypothesis that spatial localization of Pten enables the regulation of separate signaling networks.
Results
Ndfip1 abundance alters Pten subcellular compartmentalization and cell proliferation
To investigate the role of Ndfip1 in regulating Pten localization in proliferating cells we employed a human neuroblastoma cell line SH-SY5Y that was stably transfected with an inducible expression system for Ndfip1-Flag (referred to as SH-SY5Yi) (Howitt et al., 2009). An interaction between endogenous Pten and inducible-expressed Ndfip1 was observed in cells treated with 4-hydroxytamoxifen (4HT) (Figure 1A). We next examined the relationship between increased Ndfip1 and Pten localization in SH-SY5Yi cells by studying the subcellular distribution of Pten following the induction of Ndfip1-Flag. We observed that nuclear Pten was increased following the induction of Ndfip1 expression compared with non-induced control cells (Figure 1B–D). In contrast, in Ndfip1−/− mouse embryonic fibroblasts (MEFs), we observed a loss of nuclear Pten compared with Ndfip1+/+ cells (Figure 1E–G). Similar results were also observed using immunocytochemical staining for Pten in either SH-SY5Yi cells (Figure 1H) or MEF cells (Figure 1I). To determine whether the abundance of Ndfip1 and its differential regulation of Pten localization had an effect on cell proliferation, we compared the proliferation rates of Ndfip1+/+ and Ndfip1−/− MEFs, SH-SY5Y cells infected with Ndfip1-RNAi or control-RNAi (Howitt et al., 2009), and SH-SY5Yi cells. Genetic loss or RNAi knockdown of Ndfip1 resulted in a significant increase in cell proliferation (Figure 1J and K). Conversely, induced expression of Ndfip1 (SH-SY5Yi + 4HT) was associated with a significant decrease in cell proliferation rates (Figure 1L), which was not linked to cell death (Supplementary Figure S1A). The effect of Ndfip1 abundance in modulating cell proliferation was also observed in the pheochromocytoma cell line PC12 (Supplementary Figure S1B and C), and parallel results were observed in colony forming assays (Supplementary Figure S1D and E).
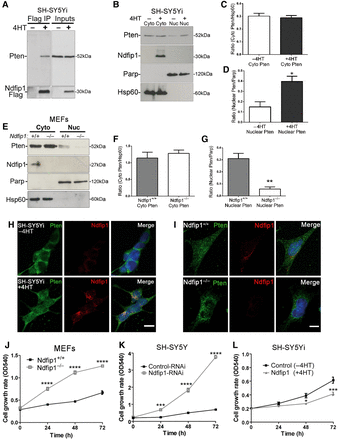
Ndfip1 regulates cell proliferation by controlling Pten nuclear compartmentalization. (A) Pten coprecipitates with Ndfip1 after induction of Ndfip1-Flag expression in SH-SY5Yi cells. (B) Nuclear and cytoplasmic fractionation of SH-SY5Yi cells induced to express Ndfip1 (+4HT) increased the abundance of nuclear Pten compared with control cells (−4HT). PARP and HSP60 are nuclear and cytoplasmic markers, respectively (Cyto, cytoplasmic; Nuc, nuclear). (C and D) Densitometric quantification of both cytoplasmic and nuclear Pten from B. (E) Nuclear and cytoplasmic fractionation of Ndfip1+/+ and Ndfip1−/− MEFs showed that deletion of Ndfip1 resulted in a loss of Pten nuclear compartmentalization. (F and G) Densitometric quantification of both cytoplasmic and nuclear Pten from E. (H) Immunocytochemical staining for Pten and Ndfip1 in SH-SY5Yi cells with and without Ndfip1 induction. (I) Immunocytochemical staining for Pten and Ndfip1 in both Ndfip1+/+ and Ndfip1−/− MEFs. (J) Ndfip1−/− primary MEFs proliferate quicker than Ndfip1+/+ MEFs in MTT assays. (K) Knockdown of Ndfip1 (Ndfip1-RNAi) in SH-SY5Y cells resulted in a significant increase in the proliferation of cells compared with control-RNAi cells in MTT assays. (L) SH-SY5Yi cells expressing Ndfip1 (+4HT) proliferate significantly slower than control cells (−4HT) in MTT assays. Scale bar, 10 µm. Data points are the average ±SEM of three independent experiments.
Ndfip1 regulation of the cell proliferation is Pten-dependent
To test whether Ndfip1 regulation of cell proliferation was dependent on Pten, we performed Ndfip1-RNAi (Figure 2A) and Pten rescue experiments in the Pten-null cell line PC3. Knockdown of Ndfip1 in PC3 cells did not result in increased cell proliferation, suggesting a role for Pten in Ndfip1-mediated cell proliferation (Figure 2B). Rescue experiments re-expressing both Pten and Ndfip1 resulted in a significant decrease in PC3 cell proliferation (Figure 2B and C). Expression of a nuclear-excluded Pten mutant (Pten K289E) (Trotman et al., 2007) with Ndfip1 was unable to change the proliferation rates of PC3 cells (Figure 2B and C), indicating that nuclear Pten function was important for controlling cell proliferation. In SH-SY5Yi cells, partial knockdown of Pten was able to significantly reduce Ndfip1-mediated inhibition of cell proliferation, but had no significant effect on Ndfip1-RNAi SH-SY5Y cells (Figure 2D–F). Taken together, these results strongly indicate that Ndfip1-mediated cell proliferation is mechanistically linked to nuclear Pten function. As genetic loss of Ndfip1 in MEFs resulted in increased cell proliferation, we also performed rescue experiments by either re-introduction of Ndfip1 or introduction of a nuclear localized Pten mutant. In both cases, a significant decrease in cell proliferation was observed (Figure 2G and H, Supplementary Figure S1F and G). In contrast, expression of a cytoplasmic localized Pten mutant did not rescue Ndfip1−/− cell proliferation rates (Figure 2H). Expression of either Pten mutant did not affect the proliferation of Ndfip1+/+ MEFs (Figure 2I). Together, these results show that Ndfip1 controls Pten subcellular distribution and loss of this mechanism results in increased cell proliferation.
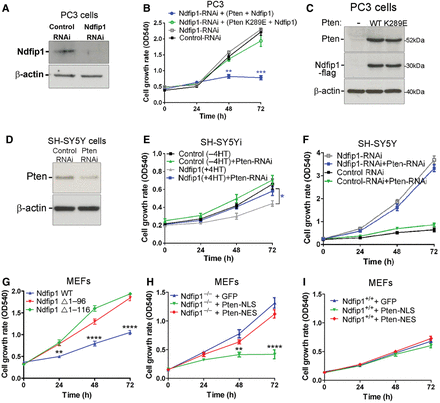
Ndfip1 regulation of cell proliferation is Pten-dependent. (A) RNAi knockdown of Ndfip1 in PC3 cells. (B and C) Knockdown of Ndfip1 in PC3 cells that lack Pten showed no significant change in cell proliferation. Rescue experiments re-expressing Ndfip1 and Pten in PC3 cells inhibited proliferation, but expression of a Pten nuclear-deficient mutant (K289E) with Ndfip1 did not alter proliferation rates. (D) Partial knockdown of Pten (Pten-RNAi) in SH-SY5Y cells. (E) Knockdown of Pten in Ndfip1-expressing SH-SY5Yi cells rescued the inhibition of cell proliferation. (F) Knockdown of Pten did not affect the proliferation of SH-SY5Y Ndfip1-RNAi cells. (G) Ndfip1−/− MEFs hyperproliferation was significantly reduced by expression of WT Ndfip1 but not by mutants lacking the N-terminal region of Ndfip1 (Δ1–96 and Δ1–116) that inhibit binding to Nedd4 ubiquitin ligases. (H) Ndfip1−/− MEFs hyperproliferation was significantly reduced by expression of a nuclear localized Pten mutant (Pten-NLS), but not by a cytoplasmic localized mutant (Pten-NES). (I) Ndfip1+/+ MEFs proliferation rates were not altered after expression of either a nuclear or cytoplasmic localized Pten mutant. Data points are the average ± SEM of three independent experiments.
Ndfip1 control of Pten compartmentalization does not alter the activation of Akt
Previously, we have observed in post-mitotic neurons that upregulation of Ndfip1 after injury resulted in Pten nuclear translocation, which was associated with increased Akt activation and cell survival (Howitt et al., 2012). To determine whether there was a link between Akt signaling and Ndfip1 regulation of nuclear Pten in dividing cells, we assayed pAkt, pS6K, and pErk abundance in Ndfip1+/+ and Ndfip1−/− MEFs following insulin stimulation. In unstimulated cells, pAkt, pS6K, and pErk were similar between Ndfip1+/+ and Ndfip1−/− MEFs (Figure 3A, control lanes). Following insulin stimulation, both cell types showed similar pAkt, pS6K, and pErk increases over the time course of treatment (Figure 3A). These results indicated that Ndfip1−/− MEFs retain similar capacity (compared with wild type cells) to regulate the PI3K/Akt signaling network, and the hyperproliferation phenotype observed in Ndfip1−/− cells is most likely unrelated to Akt activation. In addition, our results suggest that nuclear Pten function can be cell type-dependent, with different functional outcomes observed in mitotic cells compared with our previous findings in post-mitotic neurons (Howitt et al., 2012).
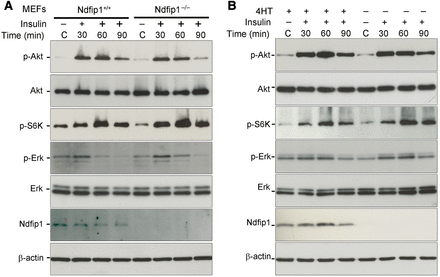
Ndfip1 abundance and Pten nuclear compartmentalization do not affect Akt signaling. (A) Ndfip1+/+ and Ndfip1−/− MEFs were cultured in serum-free media for 3 h before insulin stimulation (2 nM). Both Ndfip1+/+ and Ndfip1−/− MEFs showed similar activation of pAkt (S473), p-S6K, and p-Erk. (B) SH-SY5Yi cells with or without 4HT were cultured for 3 h in serum-free media before insulin stimulation (2 nM). SH-SY5Yi cells with or without Ndfip1 expression (±4HT) showed similar activation of pAkt (S473), p-S6K, and p-Erk.
Further testing for Ndfip1 and nuclear Pten effects on Akt regulation were conducted in SH-SY5Yi cells to artificially induce Ndfip1 and assay the cell response to insulin. As in the MEFs, basal pAkt, pS6K, and pErk were low in the unstimulated cells and elevated over the time course of insulin treatment for both un-induced and induced cells (Figure 3B). Therefore, overexpression of Ndfip1 did not curtail Akt activation in response to insulin. Given that we showed the hyperproliferation phenotype following Ndfip1 loss was Pten-dependent, and that Ndfip1 genotype had no consequence for pAkt abundance or downstream signaling, we conclude that the proliferation changes observed were unrelated to the PI3K/Akt signaling pathway. Instead, we suggest that the loss of Pten nuclear compartmentalization in Ndfip1-deficient cells was responsible for increased cell proliferation. Together, these results indicate that nuclear Pten targets separate signaling networks from cytoplasmic Pten and this pathway has an anti-proliferative function.
Ndfip1 deletion and loss of Pten nuclear compartmentalization alter the cell cycle
Pten has previously been shown to be involved in both G1 and G2/M phases of the cell cycle (Ramaswamy et al., 1999; Jacob et al., 2009). To study changes in the cell cycle following modulation of Ndfip1 abundance, flow cytometric analysis was performed to quantify the percentage distribution of asynchronous Ndfip1+/+ and Ndfip1−/− MEFs in various cell cycle phases. Genetic loss of Ndfip1 resulted in a significant decrease in the G2/M population and the subsequent increase in the G1 population compared with wild type cells (Figure 4A). We observed similar results in SH-SY5Y cells after Ndfip1 knockdown (Supplementary Figure S2A). Conversely, the opposite results were obtained following induced expression of Ndfip1 in SH-SY5Yi cells with an increased G2/M phase of the cell cycle, indicating that cells were stalling at this point of cell division (Supplementary Figure S2B). In contrast, no difference was observed when an Ndfip1 mutant unable to bind Nedd4 ubiquitin ligases was expressed in SH-SY5Y cells (Supplementary Figure S2C). Together, these data indicate that nuclear compartmentalization of Pten under the control of Ndfip1 is important for normal cell cycle regulation, and loss of this signaling network results in the misregulation of the G2/M phase of cell division.
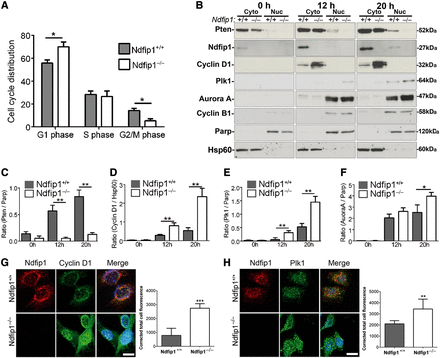
Genetic deletion of Ndfip1 and loss of nuclear Pten alter the cell cycle and result in the dysregulation of oncogenic proteins. (A) Flow cytometry investigating the percentage of cells in each phase of the cell cycle showed that genetic deletion of Ndfip1 in MEFs resulted in a significant increase in the G1 population and significant decrease in the G2/M population when compared with Ndfip1+/+ MEFs. (B) Ndfip1+/+ and Ndfip1−/− MEFs were synchronized using serum withdrawal before cytoplasmic and nuclear fractionation and western blotting performed at various time points. (C–F) Densitometric quantification of Pten, cyclin D1, Plk1, and Aurora A from B. (G) Immunocytochemical analysis of endogenous Ndfip1 and cyclin D1 in Ndfip1+/+ and Ndfip1−/− MEFs showed significantly increased staining for cyclin D1 in Ndfip1−/− MEFs. (H) Immunocytochemical analysis of endogenous Ndfip1 and Plk1 in Ndfip1+/+ and Ndfip1−/− MEFs showed significantly increased staining for Plk1 in Ndfip1−/− MEFs. Scale bar, 10 µm. Data points are the average ± SEM of three independent experiments.
Loss of nuclear Pten is associated with dysregulation of Plk1 and cyclin D1
Our data showed that deletion of Ndfip1 resulted in decreased nuclear Pten and an increase in cell proliferation. To examine for putative cell cycle effectors lying downstream of this pathway, we conducted experiments to study changes in nuclear Pten and mitotic proteins in Ndfip1+/+ and Ndfip1−/− MEFs. Following synchronization and release of MEFs, cells were harvested at various time points for western blotting for Pten, as well as proteins linked to regulation by Pten, including cyclin D1 (Weng et al., 2001), polo-like kinase 1 (Plk1), and Aurora A (Song et al., 2011). We also investigated JunB that has been associated with increased proliferation of immune cells deficient in Ndfip1 (Oliver et al., 2006), as well as another cell cycle regulator cyclin B1. Nuclear compartmentalization of Pten was initially low after release of cells and increased in Ndfip1+/+ MEFs at 12 and 20 h (Figure 4B and C). In contrast, only a small accumulation of nuclear Pten was observed in Ndfip1−/− MEFs at 12 and 20 h, which was significantly reduced compared with Ndfip1+/+ MEFs (Figure 4B and C). Concomitant with these changes, cytoplasmic and nuclear cyclin D1 were significantly increased in Ndfip1−/− MEFs compared with Ndfip1+/+ MEFs (Figure 4B, D and Supplementary Figure S2D). In comparison, cyclin B1 abundance was equivalent between the two genotypes (Figure 4B). In addition to changes in cyclin D1, we also observed an increase in the abundance of Plk1 and Aurora A, proteins involved in the G2/M transition of the cell cycle. Plk1 abundance in the nuclei of Ndfip1−/− MEFs was significantly increased at both 12 and 20 h when compared with Ndfip1+/+ MEFs (Figure 4B and E). Aurora A abundance was significantly increased in Ndfip1−/− MEFs at 20 h compared with Ndfip1+/+ MEFs (Figure 4B and F). The abundance of JunB was increased in Ndfip1−/− MEFs compared with Ndfip1+/+ MEFs, but this difference was not significant between the genotypes (Supplementary Figure S2D).
To further investigate the regulation of cyclin D1 and Plk1, we performed immunocytochemistry on synchronized Ndfip1+/+ and Ndfip1−/− MEFs that were fixed at 20 h after release from cell cycle block. Consistent with the fractionation results, Ndfip1−/− MEFs had significantly increased cyclin D1 and Plk1 immunostaining when compared with Ndfip1+/+ MEFs (Figure 4G and H). Interestingly, immunocytochemistry for cyclin D1 showed increased nuclear staining whereas the cell fractionation experiments indicated increased cytoplasmic cyclin D1. This differential location pattern has been described previously (Alao, 2007) and is thought to be due to technical reasons in fractionation experiments. Importantly, in both experiments, our results showed increased cyclin D1 in Ndfip1−/− MEFs. Together, these data indicate that Pten nuclear compartmentalization during the cell cycle alters Pten signaling specificity, resulting in the regulation of the oncogenic proteins Plk1, cyclin D1, and Aurora A. Loss of this mechanism resulted in the dysregulation of these proteins and subsequent uncontrolled cell proliferation.
Ndfip1-deficient cells are refractory to pharmacological inhibition by PI3K or mTOR inhibitor
Our results revealed that deletion of Ndfip1 inhibited Pten nuclear compartmentalization and this increased cell proliferation without parallel changes in Akt regulation. As such we reasoned that growth factor withdrawal or pharmacological inhibition of the PI3K/Akt/mTOR pathway would have limited effect on the proliferation of Ndfip1-deficient cells. To test this hypothesis, we investigated the effect of growth factor withdrawal as well as PI3K and mTOR inhibitors on the proliferation of control and Ndfip1-RNAi SH-SY5Y cells. SH-SY5Y cells with control-RNAi grown in 1% FBS media exhibited total loss of proliferation after 24 h, whereas Ndfip1-RNAi SH-SY5Y cells did not show any decreased proliferation (Figure 5A). These results indicated that cells lacking Ndfip1 could proliferate independent from growth factor signaling normally required for cell division. Total loss of growth factors (0% FBS media), however, could inhibit the proliferation of Ndfip1-RNAi cells, suggesting that some extrinsic stimulation is required for cell division to occur (Supplementary Figure S3A).
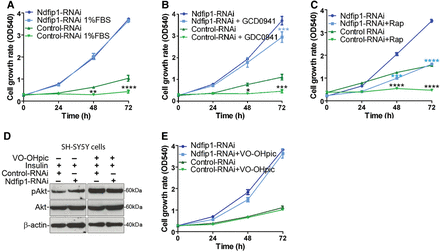
Knockdown of Ndfip1 and loss of nuclear Pten result in cells refractory to PI3K and mTOR inhibitors. (A) The proliferation of control-RNAi SH-SY5Y cells was significantly reduced when grown in 1% FBS. In contrast, the proliferation of Ndfip1-RNAi SH-SY5Y cells was maintained. (B) The proliferation of control-RNAi SH-SY5Y cells was significantly reduced when grown in the presence of the PI3K inhibitor GDC-0941 (20 nM). In contrast, the proliferation of Ndfip1-RNAi SH-SY5Y cells was maintained. (C) The proliferation of control-RNAi SH-SY5Y cells was totally inhibited when grown in the presence of the mTOR inhibitor rapamycin (20 nM). Ndfip1-RNAi SH-SY5Y cell proliferation was significantly inhibited in the presence of rapamycin; however, the cells still maintained a proliferative capacity. (D) Western blot of control-RNAi and Ndfip1-RNAi cells treated with the Pten phosphatase inhibitor VO-OHpic (200 nM). Both cell types after treatment with VO-OHpic increased the pAkt signal by stimulation with insulin (5 nM). (E) The proliferation rate of control-RNAi or Ndfip1-RNAi SH-SY5Y cells was not changed when treated with the Pten phosphatase inhibitor VO-OHpic (200 nM).
To further explore the function of growth factor signaling and the PI3K pathway, we used the drug GDC-0941 that inhibits PI3K and is known to suppress Akt activation and cell proliferation (Folkes et al., 2008; Junttila et al., 2009). After treatment with GDC-0941, the proliferation of control-RNAi cells was significantly inhibited (Figure 5B). In contrast, GDC-0941 treatment had minimal effect on Ndfip1-RNAi cells (Figure 5B, see also Supplementary Figure S3B for similar results with another PI3K inhibitor LY294002). To further test this signaling axis, we targeted the downstream regulator mTOR that integrates several signal transduction pathways, including PI3K signaling, using the drug rapamycin (Sabatini et al., 1994). Control cells treated with rapamycin stopped proliferating within 24 h (Figure 5C). In contrast, Ndfip1-RNAi cells treated with rapamycin continued to proliferate but to a lesser extent than untreated cells (Figure 5C), suggesting partial involvement of mTORC1 signaling in cell proliferation following Ndfip1 loss. Therefore, pharmacological inhibition of cell proliferation through PI3K or mTOR signaling was suppressed in Ndfip1-RNAi cells. These results indicate that the cell proliferation phenotype resulting from Ndfip1 knockdown is unlikely to result from aberrant activation of PI3K/mTOR pathways, but rather a different signaling network is driving cell proliferation.
Next we assayed for a possible relationship between Ndfip1 and the lipid phosphatase function of Pten by treating cells with the specific inhibitor VO-OHpic. We found that VO-OHpic treatment could increase the activation of Akt, but did not alter proliferation rates of either control-RNAi or Ndfip1-RNAi cells (Figure 5D and E). These results suggest that the cell proliferation phenotype observed with Ndfip1 loss is independent of Pten phosphatase activity, and changes in Akt activation are not directly linked to the increased proliferation observed.
Deletion of Ndfip1 and loss of Pten nuclear compartmentalization cause hypersensitivity to Plk1 inhibition
We found that cells lacking Ndfip1 were refractory to traditional inhibitors of the PI3K/mTOR pathway, indicating that the hyperproliferation observed in these cells was driven by other signaling networks. Our results identified that loss of nuclear Pten in these cells was linked to an increase in both Plk1 and cyclin D1 abundance. Plk1 activity can be inhibited by the specific drug BI2536 (Steegmaier et al., 2007), whereas currently there are no known drugs for specifically targeting cyclin D1 activity. We therefore tested whether Ndfip1-deficient cells that maintain normal cytoplasmic Pten function were hypersensitive to Plk1 inhibition. Control and Ndfip1-RNAi SH-SY5Y cells were treated with BI2536. We observed no change in the proliferation of control cells; in contrast, Ndfip1-RNAi cells showed a significant decrease in proliferation (Figure 6A). The BI2536-mediated decrease in Ndfip1-RNAi cell proliferation was not caused by increased cell death during the proliferation assay; however, higher concentrations of BI2536 were observed to preferentially increase the death of Ndfip1-RNAi cells compared with control cells (Figure 6B). After BI2536 treatment, Ndfip1-RNAi cells were observed to have a significantly increased accumulation of cells in the G2/M population of the cell cycle, indicating the stalling of cell division at the G2/M checkpoint (Figure 6C and D). Together, these results suggest that the increased proliferation of cells lacking Ndfip1 and nuclear Pten is driven in part by Plk1 dysregulation, with these cells observed to be hypersensitive to pharmacological inhibition of Plk1 function.
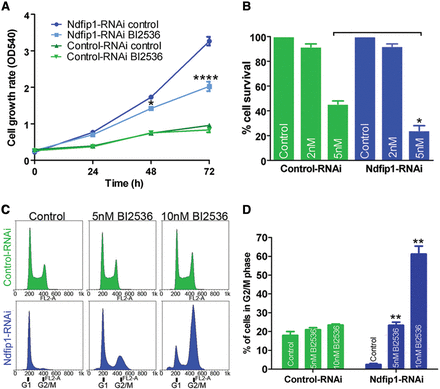
Knockdown of Ndfip1 and loss of nuclear Pten result in hypersensitivity to Plk1 inhibition. (A) The proliferation of control-RNAi SH-SY5Y cells was maintained when treated with the Plk1 inhibitor BI2536 (2 nM). In contrast, the proliferation rate of Ndfip1-RNAi SH-SY5Y cells was significantly reduced when treated with BI2536. (B) Ndfip1-RNAi cells were preferentially sensitive to cell death after treatment with 5 nM BI2536 for 72 h compared with control-RNAi cells. (C) Flow cytometric cell cycle analysis of control-RNAi SH-SY5Y cells treated with BI2536 showed no increase in the G2/M phase of the cell cycle. In contrast, Ndfip1-RNAi SH-SY5Y cells showed an increase in the number of cells in G2/M phase after BI2536 treatment. (D) Quantification of cell cycle analysis in C showed no significant difference in the G2/M phase of control-RNAi SH-SY5Y cells after BI2536 treatment. In contrast, a significant increase in the G2/M population of Ndfip1-RNAi SH-SY5Y cells after BI2536 treatment was observed, indicating a stalling of cells in this phase of the cell cycle. Data points are the average ± SEM of three independent experiments.
Pharmacological targeting of endogenous Ndfip1 inhibits the proliferation of cells in a Pten-dependent manner
Increased Ndfip1 expression resulted in Pten nuclear compartmentalization that could suppress the proliferation of cells. It was therefore relevant to ask whether this pathway could be pharmacologically targeted to suppress cell proliferation. Previously, we have designed and synthesized a small molecule termed C3 that increases Ndfip1 abundance both in vitro and in vivo (Schieber et al., 2011). We therefore treated SH-SY5Y cells with C3 and observed a significant reduction in cell proliferation compared with vehicle treated cells (Figure 7A). The reduction in cell proliferation rates was not related to increased cell death after C3 treatment (Supplementary Figure S3C). Importantly, the function of C3 in increasing Ndfip1 expression was accompanied by increased nuclear compartmentalization of Pten (Figure 7B), and was specific for the presence of Ndfip1 (Figure 7C). Pten was required for inhibition of cell proliferation by C3, as treatment of PC3 cells did not result in the suppression of cell proliferation, but this could be rescued by expression of Pten with C3 treatment (Figure 7D). Therefore, we find that manipulation of endogenous Ndfip1 abundance using a small molecule was able to suppress cell proliferation in a Pten-dependent manner (Figure 7E).
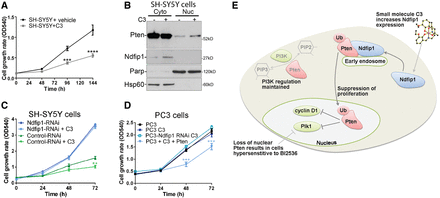
A small molecule that increases endogenous levels of Ndfip1 inhibits cell proliferation in a Pten-dependent manner. (A) Increase in endogenous Ndfip1 by a small molecule, C3 (10 µM), significantly inhibited cell proliferation compared with vehicle treated cells. (B) Western blot of cytoplasmic and nuclear fractionation of cells treated with vehicle or C3 (10 µM). C3-treated cells have an increased abundance of Ndfip1 and an increase in Pten nuclear compartmentalization. (C) The proliferation rate of Ndfip1-RNAi SH-SY5Y cells was not changed when treated with C3 (10 µM). (D) The proliferation rate of PC3 cells that lack Pten was not changed when treated with C3 (10 µM), but expression of Pten with C3 was able to significantly reduce the proliferation of PC3 cells. Data points are the average ± SEM of three independent experiments. (E) A model for the role of Ndfip1 in suppressing cell proliferation through the control of Pten nuclear compartmentalization. Ndfip1 interacts with Pten on early endosomes, resulting in the ubiquitination and trafficking of Pten to the nucleus during the cell cycle. In the nucleus, Pten regulates Plk1 and cyclin D1. The small molecule C3 upregulates Ndfip1 and promotes the nuclear trafficking of Pten. Loss of nuclear Pten results in cells that are hypersensitive to Plk1 inhibition.
Transgene expression of Ndfip1 during brain development increases nuclear Pten and results in microencephaly
To study the function of Ndfip1 abundance in vivo, we created an Ndfip1-transgenic mouse crossed with Cre recombinase under the control of the Nestin promoter (Supplementary Figure S4A). The Nestin Cre transgene is expressed from embryonic day 9.5 and resulted in increased expression of Ndfip1 in the brain (Supplementary Figure S4B). Strikingly, Ndfip1-transgenic mice displayed microencephaly compared with wild type littermates (Figure 8A–C). As transgenic expression of proteins during development can result in cell death, we analyzed embryonic brains for apoptosis using TUNEL labeling. We observed no significant increase in cell death over different embryonic ages indicating that the decreased brain size in Ndfip1-transgenic animals was not related to cell death during development (Supplementary Figure S4C and D). Subcellular fractionation of the developing neocortex showed increased nuclear Pten in Ndfip1-transgenic mice compared with wild type littermates (Figure 8D–F). To examine the differential effects of Ndfip1 abundance on the cell cycle of neuronal progenitors, we performed BrdU pulse experiments combined with phosphohistone-H3 (pHH3) labeling to determine the length of G2/M phase. Consistent with our in vitro findings we observed less double labeling of BrdU and pHH3 in Ndfip1-transgenic embryos compared with wild type littermates, indicating an increased length of the G2/M phase of neuronal progenitor proliferation (Figure 8G and H). Together, these results show that increased Ndfip1 and nuclear Pten suppress neurogenesis in the developing brain, resulting in microencephaly.
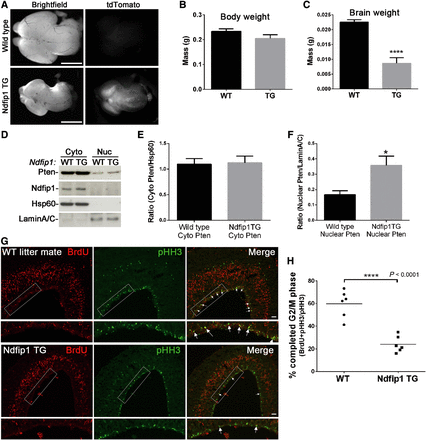
During brain development, Ndfip1 transgene expression increases Pten nuclear compartmentalization and results in microencephaly. (A) Ndfip1-transgenic mice (Ndfip1 TG) crossed with Nestin Cre displayed microencephaly and tdTomato reporter expression compared with a wild type littermate (E14.5). (B and C) Ndfip1 TG embryos at E14.5 were similar in weight to wild type littermates but had significantly smaller brain weights. (D) Cytoplasmic and nuclear fractionation of the E14.5 neocortex showed increased Pten nuclear compartmentalization in Ndfip1 TG mice compared with a wild type littermate. (E and F) Densitometric quantification of both cytoplasmic and nuclear Pten from D. (G) Pregnant Ndfip1 TG × Nestin Cre mice were pulsed for 2 h with BrdU at E14.5 before embryonic brains were collected for immunohistochemistry. Co-labeling of BrdU with pHH3 determines a completed G2/M phase of neuronal progenitors. Highlighted rectangle is magnified in the panel below each image (arrows indicate double labeling). (H) Quantification of the number of neuronal progenitors at the ventricular zone that co-labeled with BrdU and pHH3. A significantly smaller number of double-labeled cells were observed in Ndfip1 TG embryos, indicating a lengthened G2/M phase in comparison with wild type littermates. Scale bar, 1 mm (A); 50 µm (G). Data in B, C, and F were analyzed using an unpaired t-test and represent the mean from three separate litters.
Discussion
The classical role of Pten is to oppose the activity of phosphoinositides at the cell membrane, leading to inhibition of the PI3K/Akt signaling pathway. Loss of Pten in many tissue types has been shown to promote cancer due to hyperactivation of Akt and increased cell survival (Stambolic et al., 1998). However, total loss of Pten abrogates all signaling networks that Pten can potentially regulate, and existing evidence suggests that subcellular location is important for the multifaceted functions of Pten. Here, by modulating an endogenous translocation system, we have identified a role for nuclear Pten in controlling a signaling network involved in cell cycle regulation that is independent from cytoplasmic PIP3 signaling.
Our study points to Pten nuclear compartmentalization by Ndfip1 as a pivotal event for the control of cell proliferation. Supporting this observation, Ndfip1 has previously been identified in a genome-wide siRNA screen for proteins that regulate the cell cycle (Kittler et al., 2007). We and others have also described hyperproliferative phenotypes in immune cells lacking Ndfip1 (Oliver et al., 2006; Altin et al., 2014). This immune phenotype has been attributed to the misregulation of the JunB transcription factor (Oliver et al., 2006); however, here we did not observe significant changes to JunB regulation during the cell cycle. Ndfip1 is an adaptor/activator of Nedd4 E3 ligases for the ubiquitination of substrates and has previously been reported to target Pten for degradation (Mund and Pelham, 2010). This reported role for Ndfip1 in degrading Pten is somewhat paradoxical however, as it was also shown that loss of Ndfip1, which should inhibit the degradation of the tumor suppressor Pten, resulted in increased cell proliferation. In contrast, our in vitro and in vivo studies have indicated that Ndfip1-mediated ubiquitination of Pten (Howitt et al., 2012) is required for subcellular trafficking of the protein rather than degradation. Thus, we find that in mitotic cells, Ndfip1-mediated ubiquitination is required for the control of Pten nuclear signaling networks that regulate cell proliferation.
To identify the signaling networks responsible for the increased proliferation of cells lacking Ndfip1 and nuclear Pten, we investigated a number of candidate mitotic proteins. This search led to two oncogenic proteins, Plk1 and cyclin D1. A direct link between Plk1 abundance and Pten has recently been reported (Song et al., 2011). Pten was found to enhance the E3 ligase activity of APC/C by promoting its association with Cdh1 in the nucleus, and this complex is known to target Plk1 for degradation. In agreement, we found that Ndfip1 knockout cells lacking nuclear Pten accumulated Plk1 during the G2/M phase of the cell cycle, a time when Plk1 is known to function in promoting cell division. In addition to changes in Plk1, we observed that cells lacking nuclear Pten had increased abundance of cyclin D1 in both the nucleus and cytoplasm. An association between Pten loss and cyclin D1 regulation has been reported in a number of cancers, and this regulation is independent of Pten phosphatase activity (Weng et al., 2001). It therefore appears that loss of Pten nuclear compartmentalization has consequences for cell cycle control through dysregulation of the oncogenic proteins Plk1 and cyclin D1. Thus, our findings provide a mechanism that explains earlier evidence showing that loss of nuclear Pten in a number of cancers was correlated with disease progression (Whiteman et al., 2002; Zhou et al., 2002).
In mouse models, Pten deletion during brain development has been shown to increase the proliferation of neuronal progenitors (Groszer et al., 2001). In contrast, Pten deletion in post-mitotic neurons has been shown to increase cell size with no effect on proliferation (Kwon et al., 2006). Thus, loss of Pten activity can have differential effects within the same cell type depending on the mitotic state of the cell. Consistent with this, we have observed differential effects for Pten nuclear compartmentalization in mitotic verses post-mitotic cells. Previously, we observed in adult post-mitotic neurons that Pten is inherently cytoplasmic, and nuclear trafficking of Pten is an injury response, providing a temporary increase in Akt activation for neuron survival (Howitt et al., 2012). In contrast, here we show that Pten nuclear compartmentalization in mitotic cells is a physiological function driven by Ndfip1 during the cell cycle. Significantly, the effectors of nuclear Pten are also divergent between the two differentiation states, whilst the Pten nuclear targets Plk1 and cyclin D1 are found in mitotic cells, these targets are not expressed in post-mitotic neurons of the adult cortex (Clay et al., 1993; Glickstein et al., 2007).
Loss of Pten function results in a variety of clinical manifestations including autism spectrum disorders. In particular, monogenic Pten mutations have been associated with autism with macroencephaly (Butler et al., 2005), and many of these mutations do not impact on the phosphatase function of the enzyme (Rodriguez-Escudero et al., 2011). In correlation with the human disorder, mouse models of Pten deletion in the brain display macroencephaly as well as exhibit altered social behavior (Kwon et al., 2006). Here by promoting the function of nuclear Pten in vivo using Ndfip1 TG mice, we observed microencephaly, the opposite phenotype to that reported for Pten loss. Thus, we find that Ndfip1 functions synergistically with Pten during the cell cycle for the regulation of cell proliferation. Importantly, the decrease in brain size was not related to cell death during development, but rather a reduction in the neuronal progenitor pool due to an increased G2/M phase of the cell cycle. Our findings suggest that loss of nuclear Pten or dysfunctional Pten subcellular compartmentalization can impact on brain development and are possibly related to neurological disorders such as autism with macroencephaly.
In conclusion, we demonstrate that altering Ndfip1 availability in the cell directly affects the subcellular distribution of Pten with consequences for cell proliferation. This complements our recent finding that Ndfip1 is also capable of directing Pten export in microvesicles known as exosomes, which following uptake in recipient cells can inhibit proliferation (Putz et al., 2012). It therefore appears that Ndfip1 can mediate both cell autonomous and non-cell autonomous functions of Pten for the regulation of cell proliferation. Given the importance of Pten in cancer and neurological disorders, our results have broad implications for the pharmacological targeting of Pten in disease settings. We suggest that this new capability to manipulate Pten signaling network specificity can be harnessed for controlling cell proliferation, which is independent from its classical role in opposing the PI3K/Akt pathway.
Materials and methods
Cell culture, plasmid constructs, and transfection
Human SH-SY5Y neuroblastoma (RPMI and 15% FBS), mouse embryonic fibroblasts, PC12, and PC3 cells were cultured in DMEM (Invitrogen), 10% FBS, 2 mM L-glutamine and 50 μg/ml PenStrep. Lentiviral infection was used to make stable SH-SY5Y cell lines of inducible Ndfip1-Flag (SH-SY5Yi) and mutant Ndfip1-Flag (PPxY mutant), as well as stable cell lines of control-RNAi and Ndfip1-RNAi as previously described (Howitt et al., 2009). A clonal cell line for SH-SY5Yi cells that does not express high levels of Ndfip1 was selected for use in all assays. Sustained high expression of Ndfip1 can result in cell death. Cells were transiently transfected with appropriate constructs using the Effectene Transfection Reagent Kit according to the manufacturer's instructions (QIAGEN). Constructs used were Strep-Flag Ndfip1 WT, Strep-Flag Ndfip1 Δ1–96 and Strep-Flag Ndfip1 Δ1–116 in pcDNA3 (both mutants delete the N-terminal region of Ndfip1 that interacts with Nedd4 ubiquitin ligases); GFP Pten-NLS containing the SV40 NLS sequence and GFP Pten-NES containing the NES sequence of the PKI protein were cloned into pEYFP-Nuc (Trotman et al., 2007). Pten knockdown cells were created by lentiviral infection of SH-SY5Y control, Ndfip1-RNAi cells, or SH-SY5Yi cells using PTEN-1047 siRNA or Scrambled siRNA GFP (ABM).
For colony forming assays, cells were plated at 5 × 104 in 100-mm dishes and allowed to grow for 14 days before fixing with glutaraldehyde (6.0% v/v) and stained with crystal violet (0.5% w/v).
Western blotting, insulin stimulation, and cell synchronization experiments
Western blotting was performed as described previously (Howitt et al., 2012). For insulin stimulation assays, cells were grown in FBS-free media for 3 h before being pulsed with 2 nM insulin for 3 min. Cells were collected at various time points following stimulation and analyzed by western blotting.
For cell synchronization experiments, cells were incubated in media without FBS for 24 h before being counted and plated into fresh media containing FBS (0 h time point). Cells were collected at 0, 12, and 20 h after release, representing the G1, S, and G2/M phase of the cell cycle (Elizondo et al., 2000), before nuclear and cytoplasmic fractionation was performed.
Primary antibodies used were: rat monoclonal anti-Ndfip1 clone 1G5 (1:4000); mouse monoclonal anti-Pten 6H2.1 (1:4000, Cascade Bioscience); rabbit polyclonal cyclin D1 SP4 (1:2000, Abcam); mouse monoclonal Plk1 clone 35–206 (1:2000, Millipore); rabbit polyclonal Aurora A (ARK-1 H-130) (1:2000, Santa Cruz); mouse monoclonal cyclin B1V152 (1:1000, Abcam); rabbit polyclonal PARP (1:2000, Abcam); mouse monoclonal HSP60 LK1 (1:4000, Santa Cruz); rabbit polyclonal pS6K (1:1000 Cell Signaling); rabbit polyclonal pERK (1:1000, Cell Signaling). HRP-conjugated secondary antibodies were: goat polyclonal anti-rat (1:5000 or 1:10000, Millipore); goat polyclonal anti-rabbit (1:10000, Millipore); and goat polyclonal anti-mouse (1:15000, Millipore).
Cell proliferation assays
SH-SY5Y cells were seeded in 12-well plates using different cell densities in order to observe significant cell growth of control-RNAi SH-SY5Y cells over the assay period. Control-RNAi SH-SY5Y cells were seeded at 20000 cells per well, whereas Ndfip1-RNAi cells were plated at 5000 cells per well. Other cell types, including PC12, primary Ndfip1+/+ and Ndfip1−/− MEFs, and PC3 cells, were seeded at 5000 cells per well. Cells were synchronized by serum deprivation before plating into 12-well plates in normal growth media to begin the assay. Treatment of cells with GDC-0941, LY294002, rapamycin, VO-OHpic, and BI2536 was performed at the 0 h time point. Cell proliferation was measured each day for 4 days using Thiazolyl Blue Tetrazolium Blue (MTT) assay.
Cell cycle analysis and cell survival assays
To determine cell cycle distribution, 5 × 105 cells were plated in 60-mm dishes. SH-SY5Yi cells were induced with 100 nM 4-hydroxy tamoxifen (4HT) at 48 h before cell cycle analysis to promote the expression of Ndfip1-Flag. Cells were collected by trypsinization, washed in PBS, resuspended in 1 ml of PBS containing 1 mg/ml RNase (Sigma), 0.1% Tween-20, and 50 µg/ml propidium iodide (PI), incubated in the dark for 15 min at room temperature, and analyzed using FACScalibur flow cytometer (Beckman-Coulter). For cell survival assays, all cells (adherent and in suspension) were collected and labeled with 10 µg/ml PI for 15 min at room temperature. At least 40000 cells from each sample were analyzed by FACScalibur flow cytometer. Analysis of cytometry data was performed using Weasel software (WEHI).
Immunoprecipitation and cell fractionation assays
Immunoprecipitations were performed as described previously (Howitt et al., 2012). For immunoprecipitation experiments, Protein G beads (Thermo Fisher Scientific) were used for the precipitation of antibodies and Strep Tactin (Qiagen) for Strep-Flag tagged Ndfip1. Fractionation of cells was performed using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific) according to the manufacturer's instructions with the addition of an ice-cold PBS wash step in between the cytoplasmic and nuclear fraction purification.
Immunocytochemistry
Cells were synchronized by serum depravation for 24 h before being released into G1 phase upon re-addition of serum. Cells were fixed in ice-cold ethanol (10 min) at 20 h after release and blocked and permeabilised in 10% normal goat serum with 0.3% Triton X-100. Cells were stained with primary antibodies overnight before 1 h incubation with the appropriate secondary antibody. Fluorescent images were obtained using a FluoView FV1000 confocal microscope (Olympus) using FV100-ASW software (Olympus). Total cell fluorescence was calculated using ImageJ. Primary antibodies were rat monoclonal anti-Ndfip1, clone 1G5 (1:1000), rabbit polyclonal cyclin D1 SP4, and mouse monoclonal Plk1 clone 35-206. Secondary antibodies were Alexa Fluor 594-conjugated goat anti-rat IgG (1:500, Molecular Probes), Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:500, Molecular Probes), and Alex 488-conjugated goat anti-mouse IgG (1:500, Invitrogen).
Ndfip1-transgenic mice and BrdU immunohistochemistry
Ndfip1-transgenic mice were generated by pronuclear injection of an Ndfip1-IRES-tdTomato construct into fertilized embryos (B6/CBA F1 background). Ndfip1 TG mice were crossed with a second transgenic mouse, in which a transgene-encoding Cre recombinase was driven by the Nestin promoter. Three separate lines of Ndfip1 TG mice were analyzed, all displaying similar phenotypes. Mice were genotyped by PCR of tail biopsies with the Ndfip1 TG allele primers (forward 5′-TCTAGAGCCTCTGCTAACCATGTTCATGCC-3′ and reverse 5′- TATGCTGCACTCTCTGCAGTGATGCTGCTG-3′) and Cre transgene primers (forward 5′-GCATTACCGGTCGATGCAACGAGTGATGAG-3′ and reverse 5′-GAGTGAACGAA CCTGGTCGAAATCAGTGCG- 3′).
Pregnant female mice at 14.5 days post-coitum were injected with BrdU at 2 h before embryos were harvested, and brains were dropped fixed in 4% PFA in 0.1 M phosphate buffer. Brains were then cryoprotected for 24 h in 20% sucrose in 0.1 M phosphate buffer at 4°C before coronal sectioning. Tissue was then washed in PBST (0.1 M PBS and 0.1% Triton X-100) and blocked with 10% normal horse serum in PBST. Sections were incubated with primary antibodies overnight followed by appropriate secondary antibodies for 1 h at room temperature. Cell death was detected using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick-end labeling (TUNEL) as per the manufacturer's instructions (Roche). Sections were counterstained with DAPI (1:10000; Dako) before mounting under glass coverslips with antifade mounting reagent. Primary antibodies used were mouse monoclonal anti-BrdU (1:100 BD Pharmingen) and rabbit polyclonal anti-pHH3 (1:200, Millipore).
Statistical analysis
Proliferation assays were analyzed using two-way ANOVA with Bonferroni post-tests. Cell cycle data and quantification of western blots were analyzed using a student's t-test. All tests were assessed at the 0.05 level of significance. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001.
Supplementary material
Supplementary material is available at Journal of Molecular Cell Biology online.
Funding
This work was supported by the Australia National Health and Medical Research Council through Program and Project Grants (grant numbers 569575 and 1066895), and the Victorian Government through the Operational Infrastructure Scheme.
Conflict of interest: none declared.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}