




Abstract
Protein scaffolds derived from non-immunoglobulin sources are increasingly being adapted and engineered to provide unique binding molecules with a diverse range of targeting specificities. The ColE7 immunity protein (Im7) from Escherichia coli is potentially one such molecule, as it combines the advantages of (i) small size, (ii) stability conferred by a conserved four anti-parallel α-helical framework and (iii) availability of variable surface loops evolved to inactivate members of the DNase family of bacterial toxins, forming one of the tightest known protein–protein interactions. Here we describe initial cloning and protein expression of Im7 and its cognate partner the 15 kDa DNase domain of the colicin E7. Both proteins were produced efficiently in E.coli, and their in vitro binding interactions were validated using ELISA and biosensor. In order to assess the capacity of the Im7 protein to accommodate extensive loop region modifications, we performed extensive molecular modelling and constructed a series of loop graft variants, based on transfer of the extended CDR3 loop from the IgG1b12 antibody, which targets the gp120 antigen from HIV-1. Loop grafting in various configurations resulted in chimeric proteins exhibiting retention of the underlying framework conformation, as measured using far-UV circular dichroism spectroscopy. Importantly, there was low but measurable transfer of antigen-specific affinity. Finally, to validate Im7 as a selectable scaffold for the generation of molecular libraries, we displayed Im7 as a gene 3 fusion protein on the surface of fd bacteriophages, the most common library display format. The fusion was successfully detected using an anti-Im7 rabbit polyclonal antibody, and the recombinant phage specifically recognized the immobilized DNase. Thus, Im7 scaffold is an ideal protein display scaffold for the future generation and for the selection of libraries of novel binding proteins.
Edited and Reviewed by P. Hudson (co-author), H. Hoogenboom and D. Burton
Introduction
Development and production of highly specific protein-based binding molecules is crucial to the ever-expanding diagnostics, therapeutics and protein array fields. Traditionally, such reagents have been sourced from the immunoglobulins of the vertebrate immune system, exemplified by antibodies where millennia of natural selection has evolved highly effective and adaptable molecules of immune surveillance, capable of targeting a huge range of targets in response to infection and disease. Antibodies are adaptable to many forms, from polyclonal mixes, monoclonal lines to a range of chemically and recombinantly produced binding fragments (Glennie and Johnson, 2000; Holliger and Hudson, 2005). Now, a growing number of alternative protein scaffolds are being investigated, for the advantages they offer over immunoglobulin-based molecules as specific binding molecules incorporating a diverse and powerful range of binding and recognition interfaces (Binz and Plückthun, 2005).
The rise of such protein scaffolds has been at least partly driven by technology, with a combination of advanced protein engineering techniques and in vitro molecular library display and selection technologies allowing a fair (but improving) mimic of the natural immune system, which can then be applied to the protein of choice. The wish list of desirable scaffold characteristics is long, but includes such factors as ease of access to deeply recessed critical epitopes on protein targets; ability to be efficiently produced and purified to high levels; and favourable pharmacokinetics, bio-distribution and activity per unit mass. However, most emphasis is justifiably focused on the intrinsic structure of the protein itself, especially possession of a conserved tertiary structure with a stable hydrophobic core, combined with a solvent accessible surface or ligand binding site capable of tolerating mutations, deletions and insertions (Fernandez-Carneado et al., 2000; Skerra, 2000). Maintenance of such scaffold stability while accommodating extensive variability is particularly evident in the guiding example of antibodies, where six variable complementarity determining region (CDR) loops are displayed on the top of the adjacent heavy and light variable domains. The ability to be displayed in an in vitro library selection system such as bacteriophage- or ribosome-display is also vital for the isolation of variants with specific binding specificities (Irving et al., 2001; Kohl et al., 2003; Hoogenboom, 2005), though this may be superseded in the future by the power of multiplexing through protein microarrays.
To date, scaffold selection has focused on adaptation of naturally occurring protein folds, with an emphasis on modification of natural binding functions and a reduction in the ‘functional size’ of the protein scaffold to more appropriately match the scale and nature of the intended target (Ladner and Ley, 2001). Examples include ankyrin repeat proteins (Sedgwick and Smerdon, 1999; Binz et al., 2004); fibronectin type III (FN3) domains (Koide et al., 1998; Batori et al., 2002); lipocalins and the related anticalins (Skerra, 2000; Schlehuber and Skerra, 2005); knottins (Smith et al., 1998; Souriau et al., 2005) and the Z domain of Staphylococcal protein A (Nord et al., 1997). This list is by no means exhaustive (Binz and Plückthun, 2005), but these scaffolds illustrate many favourable characteristics, including the absence of disulphide bridges allowing production in a reducing environment, ease of protein expression, natural variability in loop length and conformation, and varying combinations of α-helical and β-sheet content. In this study, based on one of the highest known natural affinity interactions, we have identified the soluble, stable Im7 immunity protein from Escherichia coli as a new scaffold candidate.
Im7 in its natural form binds the ColE7 colicin, a 60 kDa bacterial toxin produced and exported by E.coli strains carrying the colE7 plasmid, which effectively attacks non-immune strains, providing a distinct selective advantage to the host. ColE7 is a member of the E7 family of toxins, which consists of enzymatic DNases and Rnases; other related E-group toxins alternatively act through formation of pores or by inhibiting peptidoglycan synthesis (Lakey et al., 1994; James et al., 1996). The ColE7 colicin can be divided into three domains, of which only the DNase domain is translocated into the victim cell. In order to prevent auto-degradation of the host-cell chromosome by the nuclease toxin prior to export and release into the environment, host E.coli also encodes an immunity protein, termed ImmE7 (Im7). The Im7 coding sequence also resides on the colE7 plasmid and is co-regulated and co-expressed with the toxin, the two proteins associate and are exported from the cell as a complex, with dissociation occurring only once the toxin encounters a susceptible cell. The Im7–DNase complex is formed by shape complementarity and electrostatic contacts (Figure 1a) and is one of the tightest protein–protein interactions known, with dissociation constants approaching 10−14 M (Wallis et al., 1995a,b).
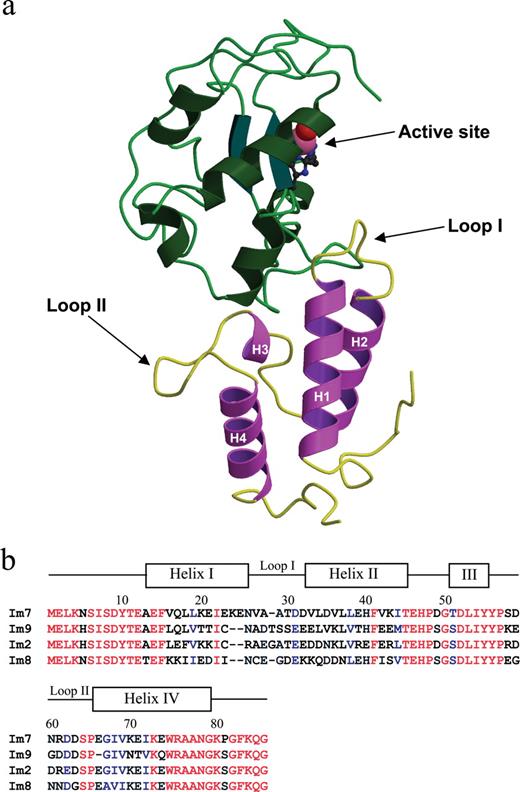
(a) Ribbon diagram representation of the crystal structure of Im7 and its associated DNase domain (PDB ID: 7CEI) (Ko et al., 1999). Helix I (residues 13–26), helix II (residues 33–45), helix III (residues 51–55), helix IV (residues 66–79), and variable loop I (residues 27–32) and variable loop II (residues 56–64) are indicated. For the DNase domain, the bound Zn2+ ion co-ordinated to three histidine residues and a water molecule in the active site are indicated. The figure was generated using Molscript and Raster 3D (Kraulis, 1991; Merritt and Bacon, 1997). (b) Sequence alignment of Im7, Im9, Im2 and Im8. The location of the helices and loops in the structure of Im7 are shown, and identical (red) and conserved (blue) residues are indicated.
Structurally, the Im7 protein and its homologues Im9, Im8 and Im2 are composed of four anti-parallel α-helices wrapped around a central hydrophobic core, which stabilizes folding of the soluble protein (Chak et al., 1996). Sequences of these related immunity proteins show ∼50% identity at the amino acid level (Figure 1b); however, they bind with high affinity only to their cognate DNase domains (Ko et al., 1999), with sequence variability within the solvent-exposed variable loop l (predominantly) and loop 2 (to a lesser extent) accounting for the discrimination between binding partners. In contrast, the underlying framework is well conserved, in a manner reminiscent of the underlying immunoglobulin fold of antibodies. Previously, genetic manipulation of these DNase type immunity proteins (Bernath et al., 2005), in particular Im2 (ColE2) and Im9 (ColE9) immunity proteins, has investigated folding and kinetics and elucidated the structural basis of their specificities (Li et al., 1997; Ferguson et al., 2001). Through genetic manipulation involving direct exchange of domains between these two immunity proteins, i.e. helix II of Im9 was grafted onto Im2 and vice versa, it was demonstrated that these proteins have the potential to be tolerant of loop exchange, and that they can readily form hybrid proteins (a result reflecting their clear divergence from an ancestral protein) (Ferguson et al., 2001).
Encouraged by these results, we hypothesized that the Im7 four-helix bundle would present an ideal compact protein scaffold for the display of molecular repertoires. In the present study, we describe initial cloning and expression of the Im7 protein and validation of its display in the context of fd-bacteriophage display, and demonstrate that it is tolerant to extensive loop substitutions.
Material and methods
General reagents and techniques
General chemicals and reagents were purchased from Sigma® (St Louis, MO, USA), BDH Laboratories (Poole, England) or BDH chemicals (Merck Pty Ltd, Victoria, Australia), unless otherwise stated. All media ingredients were purchased from Oxoid Unipath Pty Ltd (Hampshire, UK). Restriction endonucleases and Vent DNA polymerase (Australian supplier for New England Biolabs, Gene Search, Arundel, Qld, Australia) and T4 DNA ligase (Invitrogen, Mount Waverly, VIC, Australia) were used according to the manufacturer's instructions. DNA fragment recovery and purification was performed using the Machery-Nagel NucleoSpin® Extract kit (GmbH & Co., Düren, Germany). Small-scale preparations of DNA from E.coli were performed using the QIAprep Spin Miniprep Kit, Qiagen Pty. Ltd (Doncaster, VIC, Australia). Monoclonal anti-FLAG antibody was purified from hybridoma cell line KM5-1C7-8-5 (provided by Dr N.Nicola, CRC for Cellular Growth Factors, WEHI, Australia) and coupled to Mini-Leak™ Low resin (Ken-En-Tec, Denmark) to generate anti-FLAG affinity resin. Nickel-charged resin, Ni IDA-Agarose, was purchased from Scientifix Pty. Ltd (Cheltenham, VIC, Australia). Recombinant gp120 HIV-1 strain LAV, full-length oligomeric glycoprotein gp120 (Baculovirus), Cat. No. 2003LAV (HIV-1) was from Protein Sciences Corporation (Meridan, Connecticut, USA). Benchmark™ Prestained Protein Ladder Cat. No. 1078-010 was from Gibco BRL Life Technologies (Gaithersburg, MA, USA).
The following is a list of commercial antibodies used for Enzyme linked immunosorbent assays (ELISAs) and Westerns and Surface Plasmon Resonance: Anti-FLAG monoclonal antibody (WEHI), 0.35 mg/ml in 50% glycerol; ImmunoPure® Goat anti-mouse IgG, Fc specific, horseradish peroxidase conjugated, (Pierce, Rockford, IL, USA); Monoclonal anti-polyhistidine (mouse IgG2a isotype), (Sigma®); Polyclonal anti-fd bacteriophage biotin conjugate, (Sigma®); ImmunoPure® Streptavidin, horseradish peroxidase conjugated (Pierce); Sheep anti-rabbit horseradish peroxidase conjugated, (Silenus, AMRAD Biotech, Boronia, VIC., Australia); Monoclonal anti-Penta-HIS antibody, (Qiagen Pty. Ltd, Doncaster, VIC., Australia).
Standard molecular biological techniques were performed as described (Ausubel et al., 1989). DNA sequencing analysis was performed on both strands using the ABI Prism Big Dye Terminator Sequencing Reaction kit, version 2.0, following the manufacturer's instructions, and was analysed using capillary separation at the Australian Genome Research Facility (AGRF, Melbourne, Parkville).
Escherichia coli strains
Escherichia coli K12 B2B110 containing the ColE7 plasmid was a kind gift from David Gordon, Division of Botany and Zoology, Australian National University. Escherichia coli BL21(DE3): pLysS [F−, ompT, hsdSB(rB−mB−), gal(cI857, Ind1, Sam7, nin5,lacUV5-T7gene1), dcm(DE3), pLysS(CmR)] and E.coli TG1 [K12, Δ(lac-proAB), supE, thi, hsdΔ5/F′ traD36, proA+B+, laclq, lacZDM15] were originally purchased from Stratagene (La Jolla, CA, USA).
Preparation of anti-Im7 polyclonal sera
A rabbit α-Im7 polyclonal antiserum was produced by subcutaneous immunization with hexa-histidine tagged Im7 (250 μg; complete Freund's adjuvant). Subsequent boosts were at 4 week intervals (250 μg; incomplete Freund's adjuvant). Sera were collected pre-immunization and after each boost. Sera were prepared using standard techniques (Harlow and Lane, 1988).
Cloning of Im7 wt and DNase domain genes into pGC and fd-tet-DOG 1
PCR amplification of the Im7 and DNase domain constructs used 5′ and 3′ primer combinations and purified ColE7 plasmid (Genbank accession number X63620) as the template. The primer combinations N6382/N6387 and N6451/N6478 (Table I) were used for the Im7 wt and the DNase domain, respectively. The PCR amplified DNA constructs for the Im7 and DNase domain were separated using agarose electrophoresis, purified, digested with restriction endonucleases SfiI and NotI, and ligated into the similarly digested pGC vector (Coia et al., 1996). Following electroporation into competent BL21 (DE3) E.coli cells, clones were identified using colony PCR and were verified using DNA sequencing.
Oligonucleotide primers
| Oligonucleotide | Number | Sequence (5′–3′) | Features |
|---|---|---|---|
| 5′ for wt Im7 | N6382 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCATGGAACTGAAAAATAGTATTAG | SfiI site |
| 3′ for wt Im7 | N6387 (←) | GCTGTGTCTTGATCTGCGGCCGCGCCCTGTTTAAATCCTGGCTTACCGTTAGCAGCTCGCCATTCTT | NotI site |
| 3′ for wt Im7 | N6496 (←) | GGGCTGAATTCTTAGTGATGGTGATGGTGATGTGCGGCCGCGCCCTGTTTAAATCC TGGCTTACC | EcoRI site/hexahistidine tag |
| 5′ for DNase domain | N6451 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCGAGAGTAAACGGAATAAGCCAGG | SfiI site |
| 3′ for DNase domain | N6478 (←) | TAATCTGCGGCCGCTTTACCTCGGTGAATATCAATCGCCCTTTTAGGTGTTACCACGCTGATGTTATCCATATCATAGACACCACCATTTTGGC | NotI site/H126A mutation |
| 5′ for wt Im7 | N7386 (→) | TTACTCCACAGTGCACAGATGGAACTGAAAAATAGTATTAG | ApaLI site |
| 5′ for Im7 | N8639 (→) | TTACTCGCAAGCTTGTAAAGGAAACAGCTATGGAACTGAAAAATAGTATTAG | HindIII site |
| 5′ model 1 internal (1) | N8360 (→) | GATAGCCCCCAGGATAATTATTATATGGATGTGTGGGATGTGTTAGATGTGTTACTCGAACAC | CDR H3 b12 loop |
| 5′ model 1 internal (2) | N8359 (→) | GGAAATTGAAAAAGAGATTGTTGCTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ model 2 internal | N8352 (→) | GAAATTGAAAAAGAGAATGTTCGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATGGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 3 internal | N8355 (→) | GAAATTGAAAAAGAGAATGTTGCTCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATACTGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 4 internal (1) | N8362 (→) | GCCCCCAGGATAATTATTATATGGATGTGTGGGTGTTAGATGTGTTACTC | CDR H3 b12 loop |
| 5′ model 4 internal (2) | N8361 (→) | GGAAATTGAAAAAGAGATTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ final for all models | N6383 (→) | TAGGAACAGACCACCATGGAACTGAAAAATAGTATTAGTGATTACACAGAGGCTGAGTTTGTTCAACTTCTTAAGGAAATTGAAAAAGAGATTG | N-terminus of Im7 |
| Oligonucleotide | Number | Sequence (5′–3′) | Features |
|---|---|---|---|
| 5′ for wt Im7 | N6382 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCATGGAACTGAAAAATAGTATTAG | SfiI site |
| 3′ for wt Im7 | N6387 (←) | GCTGTGTCTTGATCTGCGGCCGCGCCCTGTTTAAATCCTGGCTTACCGTTAGCAGCTCGCCATTCTT | NotI site |
| 3′ for wt Im7 | N6496 (←) | GGGCTGAATTCTTAGTGATGGTGATGGTGATGTGCGGCCGCGCCCTGTTTAAATCC TGGCTTACC | EcoRI site/hexahistidine tag |
| 5′ for DNase domain | N6451 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCGAGAGTAAACGGAATAAGCCAGG | SfiI site |
| 3′ for DNase domain | N6478 (←) | TAATCTGCGGCCGCTTTACCTCGGTGAATATCAATCGCCCTTTTAGGTGTTACCACGCTGATGTTATCCATATCATAGACACCACCATTTTGGC | NotI site/H126A mutation |
| 5′ for wt Im7 | N7386 (→) | TTACTCCACAGTGCACAGATGGAACTGAAAAATAGTATTAG | ApaLI site |
| 5′ for Im7 | N8639 (→) | TTACTCGCAAGCTTGTAAAGGAAACAGCTATGGAACTGAAAAATAGTATTAG | HindIII site |
| 5′ model 1 internal (1) | N8360 (→) | GATAGCCCCCAGGATAATTATTATATGGATGTGTGGGATGTGTTAGATGTGTTACTCGAACAC | CDR H3 b12 loop |
| 5′ model 1 internal (2) | N8359 (→) | GGAAATTGAAAAAGAGATTGTTGCTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ model 2 internal | N8352 (→) | GAAATTGAAAAAGAGAATGTTCGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATGGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 3 internal | N8355 (→) | GAAATTGAAAAAGAGAATGTTGCTCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATACTGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 4 internal (1) | N8362 (→) | GCCCCCAGGATAATTATTATATGGATGTGTGGGTGTTAGATGTGTTACTC | CDR H3 b12 loop |
| 5′ model 4 internal (2) | N8361 (→) | GGAAATTGAAAAAGAGATTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ final for all models | N6383 (→) | TAGGAACAGACCACCATGGAACTGAAAAATAGTATTAGTGATTACACAGAGGCTGAGTTTGTTCAACTTCTTAAGGAAATTGAAAAAGAGATTG | N-terminus of Im7 |
Oligonucleotide primers
| Oligonucleotide | Number | Sequence (5′–3′) | Features |
|---|---|---|---|
| 5′ for wt Im7 | N6382 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCATGGAACTGAAAAATAGTATTAG | SfiI site |
| 3′ for wt Im7 | N6387 (←) | GCTGTGTCTTGATCTGCGGCCGCGCCCTGTTTAAATCCTGGCTTACCGTTAGCAGCTCGCCATTCTT | NotI site |
| 3′ for wt Im7 | N6496 (←) | GGGCTGAATTCTTAGTGATGGTGATGGTGATGTGCGGCCGCGCCCTGTTTAAATCC TGGCTTACC | EcoRI site/hexahistidine tag |
| 5′ for DNase domain | N6451 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCGAGAGTAAACGGAATAAGCCAGG | SfiI site |
| 3′ for DNase domain | N6478 (←) | TAATCTGCGGCCGCTTTACCTCGGTGAATATCAATCGCCCTTTTAGGTGTTACCACGCTGATGTTATCCATATCATAGACACCACCATTTTGGC | NotI site/H126A mutation |
| 5′ for wt Im7 | N7386 (→) | TTACTCCACAGTGCACAGATGGAACTGAAAAATAGTATTAG | ApaLI site |
| 5′ for Im7 | N8639 (→) | TTACTCGCAAGCTTGTAAAGGAAACAGCTATGGAACTGAAAAATAGTATTAG | HindIII site |
| 5′ model 1 internal (1) | N8360 (→) | GATAGCCCCCAGGATAATTATTATATGGATGTGTGGGATGTGTTAGATGTGTTACTCGAACAC | CDR H3 b12 loop |
| 5′ model 1 internal (2) | N8359 (→) | GGAAATTGAAAAAGAGATTGTTGCTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ model 2 internal | N8352 (→) | GAAATTGAAAAAGAGAATGTTCGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATGGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 3 internal | N8355 (→) | GAAATTGAAAAAGAGAATGTTGCTCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATACTGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 4 internal (1) | N8362 (→) | GCCCCCAGGATAATTATTATATGGATGTGTGGGTGTTAGATGTGTTACTC | CDR H3 b12 loop |
| 5′ model 4 internal (2) | N8361 (→) | GGAAATTGAAAAAGAGATTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ final for all models | N6383 (→) | TAGGAACAGACCACCATGGAACTGAAAAATAGTATTAGTGATTACACAGAGGCTGAGTTTGTTCAACTTCTTAAGGAAATTGAAAAAGAGATTG | N-terminus of Im7 |
| Oligonucleotide | Number | Sequence (5′–3′) | Features |
|---|---|---|---|
| 5′ for wt Im7 | N6382 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCATGGAACTGAAAAATAGTATTAG | SfiI site |
| 3′ for wt Im7 | N6387 (←) | GCTGTGTCTTGATCTGCGGCCGCGCCCTGTTTAAATCCTGGCTTACCGTTAGCAGCTCGCCATTCTT | NotI site |
| 3′ for wt Im7 | N6496 (←) | GGGCTGAATTCTTAGTGATGGTGATGGTGATGTGCGGCCGCGCCCTGTTTAAATCC TGGCTTACC | EcoRI site/hexahistidine tag |
| 5′ for DNase domain | N6451 (→) | TTACTCGCGGCCCAGCCGGCCATGGCCGAGAGTAAACGGAATAAGCCAGG | SfiI site |
| 3′ for DNase domain | N6478 (←) | TAATCTGCGGCCGCTTTACCTCGGTGAATATCAATCGCCCTTTTAGGTGTTACCACGCTGATGTTATCCATATCATAGACACCACCATTTTGGC | NotI site/H126A mutation |
| 5′ for wt Im7 | N7386 (→) | TTACTCCACAGTGCACAGATGGAACTGAAAAATAGTATTAG | ApaLI site |
| 5′ for Im7 | N8639 (→) | TTACTCGCAAGCTTGTAAAGGAAACAGCTATGGAACTGAAAAATAGTATTAG | HindIII site |
| 5′ model 1 internal (1) | N8360 (→) | GATAGCCCCCAGGATAATTATTATATGGATGTGTGGGATGTGTTAGATGTGTTACTCGAACAC | CDR H3 b12 loop |
| 5′ model 1 internal (2) | N8359 (→) | GGAAATTGAAAAAGAGATTGTTGCTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ model 2 internal | N8352 (→) | GAAATTGAAAAAGAGAATGTTCGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATGGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 3 internal | N8355 (→) | GAAATTGAAAAAGAGAATGTTGCTCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATACTGATGATGTGTTAGATGTG | CDR H3 b12 loop |
| 5′ model 4 internal (1) | N8362 (→) | GCCCCCAGGATAATTATTATATGGATGTGTGGGTGTTAGATGTGTTACTC | CDR H3 b12 loop |
| 5′ model 4 internal (2) | N8361 (→) | GGAAATTGAAAAAGAGATTGCACGAGTTGGGCCCTACAGTTGGGATGATAGCCCCCAGGATAATTATTATATG | CDR H3 b12 loop |
| 5′ final for all models | N6383 (→) | TAGGAACAGACCACCATGGAACTGAAAAATAGTATTAGTGATTACACAGAGGCTGAGTTTGTTCAACTTCTTAAGGAAATTGAAAAAGAGATTG | N-terminus of Im7 |
Cloning of the Im7 wt coding sequence into bacteriophage display vector fd-tet-DOG1 was performed using PCR amplification using the primer combination of N7386 (introducing the ApaLI restriction cloning site at the 5′ terminus) and N6387 (containing the 3′ NotI restriction site) (Table I). PCR products were purified, digested with restriction endonucleases ApaLI and NotI, and ligated into the similarly digested fd-tet-DOG1 vector. Ligated DNA was electroporated into E.coli TG1 cells and recombinants were selected on YT plates supplemented with tetracycline (15 μg/mL). Random colonies were screened using colony PCR, and positive clones verified using DNA sequencing.
Construction of the four model Im7-CDR H3 b12 chimeras
For construction of the four Im7-H3 b12 chimeric DNA inserts, oligonucleotide primers were designed to reflect E.coli codon biases (Table I). Coding sequences were produced using multi-stage splice overlap extension PCR using Vent DNA, with gel isolation and purification of the intermediate insert following each amplification step. Final amplification for all models was performed using oligonucleotide primers N8639 and N6496 for introduction of HindIII (5′) and EcoRI (3′) restriction sites for cloning into the pGC cytoplasmic expression plasmid. The four newly constructed chimera DNA fragments were gel purified and digested with HindIII and EcoRI restriction enzymes, respectively. Digested fragments for models 1, 2, 3, 4 and Im7 wt were ligated into the HindIII and EcoRI digested pGC vector and transformed into E.coli TG1, and colonies were screened using colony PCR and positive clones verified using DNA sequencing.
Purification of FLAG-tagged proteins from the E.coli periplasm
Starter cultures were grown overnight in 2YT medium supplemented with ampicillin (100 μg/ml) and glucose (2.0% w/v), diluted 1/100 into fresh 2YT/100 μg/ml ampicillin and then grown at 37°C/200 r.p.m. to OD600 = 0.2–0.4. Cultures were induced with IPTG (1 mM final) and grown for further 16 h at 37°C (Im7) or 30°C (DNase domain), and cells were harvested using centrifugation (Beckman JA-10/5000 g/10 min/ml). Periplasmic fractions were isolated using the method of Minsky (Minsky et al., 1986), and the recombinant protein purified using affinity chromatography using an anti-FLAG antibody-Sepharose column (10 × 1 cm) was equilibrated in Tris buffered saline (pH 7.4). Bound protein was eluted using Immunopure® Gentle Ag/Ab Elution buffer (GEB, Cat. No. 21013, Pierce), dialysed against 2× changes of phosphate-buffered saline (PBS; pH 8.0) and concentrated over a 3 kDa ultrafiltration membrane. If necessary, protein samples were further concentrated using Microsep Omega™ microconcentrators (PALL Gelman Laboratory, Sydney, Australia) of MWCO of 1 or 3 kDa. Proteins were quantitated by measurement of absorbance at 280 nm using the theoretical extinction coefficient calculated from the sequence (Gill and von Hippel, 1989).
Protein purity was assayed using electrophoresis through 15% SDS–PAGE gels and using size exclusion gel chromatography through Superose 12 HR 10/30 or Superdex 75 HR 10/30 columns (Amersham Pharmacia Biotech, Uppsala, Sweden) equilibrated with either Tris- or HEPES-buffered saline at a flow rate of 0.5 ml/min.
Purification of His-tagged Im7 proteins from the E.coli cytoplasm
Cytoplasmic expression of Im7 wt and the four Im7-CDR H3 b12 chimeras was essentially as described above. Cells were harvested from 1 l cultures of E.coli TG1 cells, resuspended in lysis buffer (50 mM NaH2PO4, pH 8.0; 300 mM NaCl; 10 mM imidazole) at 5 ml/g wet weight, and lysozyme added to 1 mg/ml (final). Cells were incubated on ice for 30 min, sonicated and centrifuged at 12 000 g, and the soluble fraction was applied to an Ni-IDA affinity column (Scientifix). The column was washed using 10 volumes of wash buffer (50 mM NaH2PO4, pH 8.0; 2 M NaCl; 50 mM imidazole) and the hexahistidine-tagged proteins eluted using elution buffer (50 mM NaH2PO4, pH 8.0; 300 mM NaCl; 250 mM imidazole). Following dialysis, proteins were analysed as described above, and if contaminants were present the affinity purification procedure was repeated.
ELISA and western blot analysis
Protein antigens (20–40 μg/ml) in 1× PBS were coated onto Maxisorp™ microtiter plates (Nunc-Immuno™, Cat. No. 439454, Nunc-Nalge International, Roskilde, Denmark) and incubated at 4°C overnight. Plates were rinsed, blocked with 1× PBS/5% milk protein (5% MPBS) for 2 h at room temperature and incubated with recombinant protein for 1 h at room temperature. Plates were rinsed with 1× PBS and washed three times with 1× PBS/0.05% Tween-20, and the primary antibody (1/1000 in 5% MPBS) was added. Plates were incubated and washed as described above, before the addition of horseradish peroxidase conjugated secondary antibody (1/1000 in 5% MPBS). Plates were washed as described above and developed by the addition of ABTS [2,2′-azino-di(3-ethylbenzothiazoline sulphonate)] (ABTS, Product No. 0120946, Roche, Indianapoulps, IN, USA), and the absorbance was measured at A405 nm.
Recombinant proteins and bacteriophage were analysed using SDS–PAGE through 15% Tris/glycine gels. Samples for immunoblot analysis were transferred onto 0.2 μM nitrocellulose membrane and blocked overnight in 1× PBS/5% MPBS. Antibodies for immunoblotting were diluted to 1:1000 in 5% MPBS (Im7 polyclonal antiserum) and 1:1000 (conjugated anti-rabbit horseradish peroxidase antibody). Membranes were washed with 1× PBS and 1× PBS/0.05% Tween-20, and visualized using the enhanced chemiluminescence system (Product No. RPN2106 Amersham Biosciences, Buckinghamshire, UK).
Purification of recombinant fd-tet-DOG1 bacteriophage
Single phage-expressing TG1 colonies were used to inoculate 3 ml starter cultures (2YT/15 μg/ml tetracycline). Following overnight incubation, cultures were centrifuged at 3500 g for 10 min., the supernatant collected and phage precipitated by the addition of 300 μl of 20% PEG/15% NaCl solution followed by centrifugation, resuspension in 1× PBS and titration. For small-scale phage ELISAs, bacteriophages were diluted in 1× PBS to 1012 virions/ml and ∼5 × 1010 virions coated onto wells by overnight incubation. Standard ELISA protocols for blocking, washing and detection procedures were as described above. Recombinant phages were detected using a polyclonal anti-fd bacteriophage biotin conjugate (1/1000 in 5% MPBS) followed by a secondary streptavidin–horseradish peroxidase conjugate (1/1000 in 5% MPBS), or using the rabbit anti-Im7 antiserum (1:100 and 1:1000 in 5% MPBS) in combination with a sheep anti-rabbit horseradish peroxidase conjugated antibody (1/1000 in 5% MPBS).
Homology modelling
The four proposed Im7-H3 b12 chimeras were homology modelled using the program MODELLER 6v2 (Sali and Blundell, 1993; Sali et al., 1995) using the coordinates of the un-complexed ColE7 immunity protein (PDB ID: 1AYI) and the human antibody IgG1b12 (PDB ID: 1HZH; K chain) as templates. For each Im7-CDR H3 b12 chimera, 10 initial models were produced, and a further 5 graft point energy minimized models were produced for each of these, with the first and last residues of the scaffold and loop at the graft point targeted for energy minimization. The objective function scores for the final top 10 models for each chimera are given in Supplementary Table I available at PEDS online. Analysis of the final top 10 conformers for each of the four models using PROCHECK showed 98% of residues to be within the most favoured regions of the Ramachandran plot, with the remaining 2% within allowed regions. No residues were within the generously allowed or disallowed regions (Laskowski et al., 1993).
Biosensor analysis
Surface plasmon resonance (BIAcore™ 1000 optical biosensor) was used to measure binding of the four Im7-CDR H3 b12 protein chimeras to gp120. A carboxymethyl dextran CM5 BIAsensor chip was first coated with a murine monoclonal anti-penta-histidine antibody (Qiagen) (20 μg/ml/10 mM sodium acetate, pH 3.0) using standard amino-coupling in HBS buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.0005% surfactant P-20). Approximately 1850 response units were immobilized, resulting in the effective capture of all five hexahistidine tagged proteins (Im7 wt and 4 Im7-CDR H3 b12 chimeras) to similar levels (∼200 RU). Surfaces were washed in HBS/P20 for 1500 s prior to injection of the HIV-1 gp120 analyte (10 μg/ml; 5 μl/min for 300 s), followed by 3000 s dissociation.
Helical content and stability measurements using CD spectroscopy
An AVIV® circular dichroism (CD) spectrophotometer (Lakewood, NJ, USA) (model 62DS) was used to measure the helical content and thermostability of the four Im7-CDR H3 b12 chimeric proteins (including the wild-type Im7 as a comparison). Protein samples were adjusted to 200 μg/ml in 0.1 M phosphate buffer (pH 7.2) and the far-UV CD spectra recorded at 10°C (scan window 250–196 nm, intervals of 0.2 nm). A buffer baseline was subtracted from the protein CD data before conversion to the mean residue ellipticity [𝛉] (degrees squared centimeters per decimole) and calculation of the percentage α-helical content (Hirst and Brooks, 1994; Myers et al., 1997; Morrow et al., 2000; Hatters et al., 2001).
For measurement of thermostabilities of Im7 wild type and Im7-CDR H3 b12 chimeras, data were collected at 222 nm, scanning from 10 to 80°C (1 degree increments) with 1 min equilibration between measurements. Data were recorded over a 10 s averaging period, and the far-UV CD thermal denaturation curves for each protein were fitted using nonlinear regression in SIGMAPLOT (version 4.01) as described by Gursky and Atkinson (1998).
Results
Cloning and expression of the wild-type Im7 protein
Escherichia coli strain ColE7-K12 B2B110 harbours plasmid-borne copies of the cei (Im7) and cea (DNase toxin) genes (Soong et al., 1992, 1994). The cei coding sequence was PCR amplified from total bacterial DNA and cloned directionally into the bacterial expression vector pGC, which directs protein to the periplasmic space (Coia et al., 1996). Following verification by DNA sequencing and small-scale protein expression, recombinant Im7 protein was harvested from the E.coli periplasm and purified using anti-FLAG affinity chromatography. Yields were consistently ∼2 mg/l of culture, and size exclusion gel chromatography showed little evidence of protein aggregation (Figure 2a). To further enhance protein expression, and to allow purification using a second affinity tag system, the Im7 coding sequence was transferred into a pGC variant modified for high-level cytoplasmic expression with a C-terminal hexa-histidine tag sequence. His-tagged recombinant Im7 was readily isolated from the bacterial cytoplasm, and it again showed excellent gel filtration and solubility characteristics (>27 mg/l of culture).
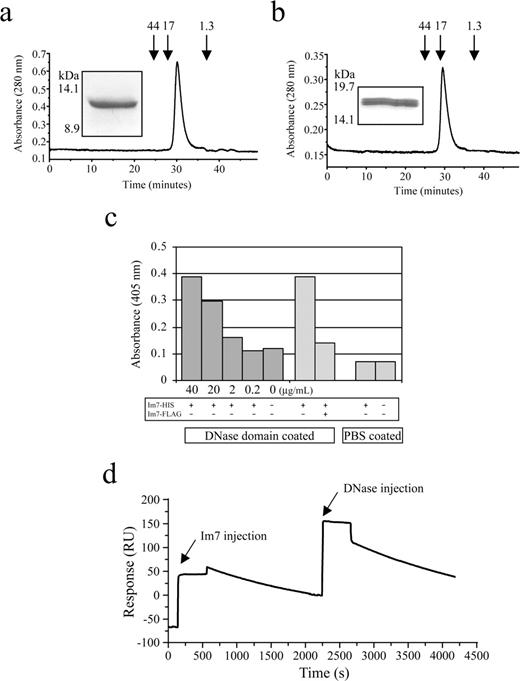
Recombinant Im7 and DNase proteins. (a) Size exclusion chromatograph of the Im7 post-anti-FLAG antibody column fraction, analysed using FPLC through a precalibrated Superose-12 gel-filtration column. The Im7 protein elutes from the column at ∼30 min consistent with the predicted molecular mass of ∼12 kDa including dual FLAG affinity tags. Arrows indicate approximate molecular masses of protein standards, and the A280 is given in arbitrary units. The insets show the same sample analysed using SDS–PAGE through a 15% (w/v) polyacrylamide Tris/glycine gel and stained using Coomassie Brilliant Blue. (b) As for (a), except the analysis of the recombinant DNase domain. The protein elutes from the column at ∼29 min consistent with the predicted molecular mass of ∼17 kDa including dual FLAG affinity tags. (c) ELISA of the interaction between immobilized DNase domain (20 μg/ml) and recombinant hexa-histidine tagged Im7. The interaction was successfully competed by the addition of excess FLAG-tagged Im7. Results represent the average of triplicate wells. (d) BIAcore trace of the interaction between Im7 wt and DNase domain. An anti-penta-histidine tag antibody was first covalently coupled to the sensor surface, and used to capture his-tagged Im7, which displays slow dissociation relative to baseline over 2000 s. The sensogram shows the binding of the DNase domain injected at the ∼2250 s time point, with the dissociation from 2500 to 4000 s due almost entirely to the dissociation between the anti-penta-histidine tag antibody and the his-tagged Im7.
To validate the folding and functionality of the recombinant Im7 protein using binding assays, we next sought to isolate the 17.5 kDa DNase domain component of the ColE7 toxin. This domain is located at the C-terminal end of the colicin E7 protein (residues 444–576) and contains a 32 amino acid HNH motif, the site of DNA binding (Pommer et al., 2001), including a transition metal ion (Zn2+) within the active sites coordinated by three histidine residues (Kleanthous et al., 1999; Ko et al., 1999; Sui et al., 2002). Normally, Im7 and DNase domain proteins are co-expressed to prevent auto-degradation of the host cell chromosome; however, in this case independent isolation was required. Thus, the DNase domain's nuclease activity was abrogated by introduction of the site-directed mutation His-126-Ala to prevent coordination of the zinc ion within the DNase domain active site (Figure 1a). Introduction of this mutation effectively eliminated the ability of the recombinant DNase to non-specifically cleave nucleic acid (results not shown). Protein yields for the DNase domain protein were comparatively poor (<1 mg/l of culture), but size exclusion chromatography of the anti-FLAG affinity purified fraction indicated a single, non-aggregating species (Figure 2b). Upon SDS–PAGE electrophoresis, a second recombinant DNase, apparently a truncated form, was also visible (Figure 2b, inset). While this did not affect the ability of the DNase to interact with Im7, the appearance of this form was consistent with the specific cleavage product reported to occur within the E.coli periplasm, either in the presence or absence of Im7 (Shi et al., 2005).
Next, to validate the folding of our recombinant Im7, we tested its ability to interact with the immobilized DNase domain using ELISA (Figure 2c). Titration of varying amounts of Im7 showed positive binding, an interaction that was successfully competed by addition of excess free FLAG-tagged Im7. These results are consistent with the reported extremely strong binding between the cognate partners. This was confirmed by surface plasmon resonance measurements using Im7 immobilized on a biosensor chip through its C-terminal hexa-histidine tag and using measurement of the interaction with recombinant DNase domain (analyte), which indicated very slow off-rate kinetics (Figure 2d).
Display of Im7 on the surface of fd-bacteriophage
We sought to determine whether Im7 could be efficiently displayed on the surface of fd-bacteriophage as a fusion with the phage gene3 protein (p3), for eventual production and screening of molecular libraries. Compared with other phage-displayed proteins such as antibodies, Im7 is small (∼90 amino acids), evolved to be efficiently expressed in E.coli and requires no disulphide bond formation, factors which offer a decided advantage in the context of this particular E.coli-based library display system. In addition, the crystallographic structure of Im7 suggests that when coupled to the N-terminus of p3, the Im7 loop regions will be orientated outward and away from the phage virion surface (Figure 1a). To test this hypothesis, the wild-type Im7 coding sequence was subcloned into phage vector fd-tet-DOG1 which allows multivalent display (3–5 copies per phage) and thus increased sensitivity in binding assays due to avidity affects (Clackson et al., 1991; Nuttall et al., 1999). In order to test the localization of Im7 on the phage surface, rabbit polyclonal antisera were first raised against the hexa-histidine tagged Im7. These antisera reacted specifically with the Im7 protein, and did not interact with the associated hexa-histidine tag sequence (Figure 3a).
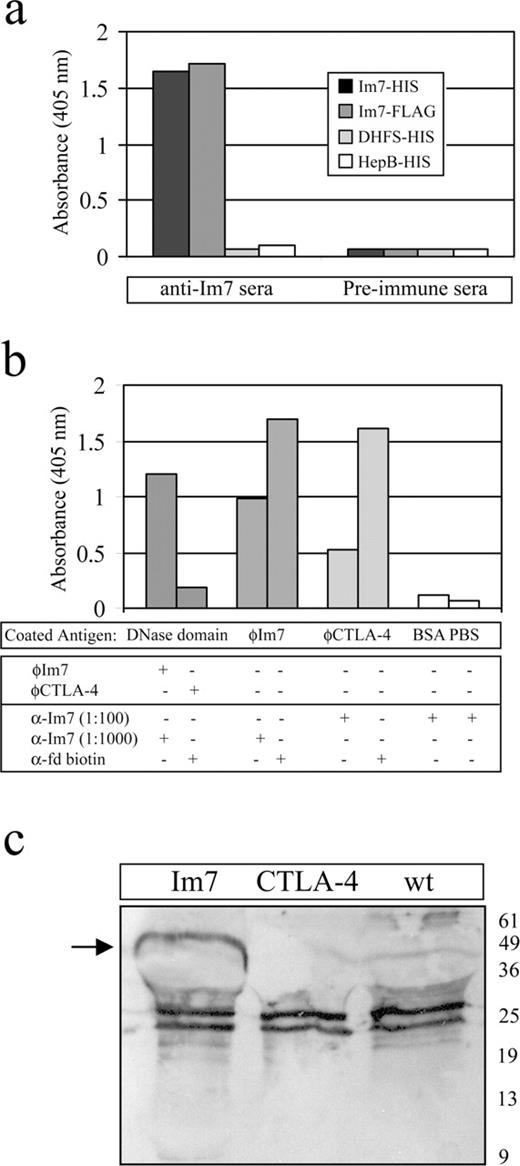
Im7 is displayed on the surface of fd bacteriophages. (a) ELISA validating rabbit polyclonal antisera raised against hexa-histidine tagged Im7. Immune and pre-immune sera at 1:100 dilutions were tested for binding to hexa-histidine and FLAG epitope tagged Im7, and against two hexa-histidine tagged negative control proteins. All antigens were coated at 40 μg/ml, and results represent the average of triplicate wells. (b) ELISA indicating display of Im7 on the surface of fd bacteriophage. Wells were coated with recombinant DNase domain (∼2 μg per well), probed with Im7 or CTLA-4 phage and detected using the anti-Im7 or anti-fd phage secondary reagents (panels 1 and 2). Similarly, ELISA wells were coated with equal amounts of fd-tet-DOG1 bacteriophage displaying Im7 or CTLA-4 gene 3 protein fusions (∼5 × 1010 virions/well) and probed with the anti-Im7 antisera. Equal amounts of the phages were immobilized as shown by detection using an anti-fd phage antibody (panels 3–6). (c) Equal amounts of fd-tet-DOG1 virions displaying wild-type gene 3 protein, or fusions with Im7 or CTLA-4, were separated by SDS–PAGE, transferred to nitrocellulose membrane, and probed with the anti-Im7 polyclonal antisera. The Im7-gene 3 protein fusion (∼50 kDa; arrowed) is specifically immunodecorated, while the minor band visible at ∼10 kDa most probably represents Im7 proteolytically cleaved from the fusion protein. Cross-reacting bands present in all three preparations are most probably due to non-specific interactions from the polyclonal antiserum.
Recombinant Im7 and recombinant CTLA-4 (negative control) bacteriophages were purified and in a series of ELISA-based experiments used to demonstrate that Im7 is displayed on the bacteriophage surface. For example, when recombinant DNase domain was coated onto ELISA wells and probed with these preparations, only the bacteriophage displaying Im7 demonstrated positive binding (Figure 3b). Conversely, when equal numbers of bacteriophages displaying Im7 or CTLA-4 were immobilized and probed with the anti-Im7 antisera, there was a strong positive response at 1:1000 dilution for the Im7 phage, whereas there was only slight cross-reactivity of the CTLA-4-bearing phage at 10-fold greater amounts of antisera (Figure 3b). Finally, bacteriophages displaying wild-type p3, Im7 and CTLA-4 were immobilized onto a nitrocellulose support, and when they were probed with the anti-Im7 antisera only the Im7–p3 fusion was specifically immunodecorated (Figure 3c). Further, an Im7 bacteriophage-displayed library with complete randomization of 7 residues within the loop 1 region (Figure 1a; S.M.J., unpublished data) was spiked at a ratio of 1:1000, with the phage displaying the wild-type Im7 protein. Following biopanning against immobilized DNase domain, >50% of randomly selected clones contained the wt sequence following one round of selection, a figure that increased to >90% following round 3. The remaining clones showed only single residue conservative variations from wt. These results provide preliminary evidence that the Im7 protein can be successfully displayed and selected in an fd bacteriophage-based system.
Can Im7 accommodate structural diversity in its variable loop 1 region?
Next, we sought to determine whether the Im7 protein could tolerate extensive modifications and additions to its framework structure. A relatively simple and powerful technique to demonstrate such functionality is loop grafting of a bioactive epitope from, for example an antibody, onto the scaffold of interest (Jones et al., 1986). Such experiments are especially successful where disruption to the scaffold is minimized and the grafting encompasses all critical binding residues (Nuttall et al., 1999; Zondlo and Schepartz, 1999).
An examination of the naturally occurring E.coli immunity proteins, which target different colicins, illustrates the extensive variability within loop regions 1 and 2, making them ideal locations for such loop grafting (Figure 1b). This hypothesis is strengthened by the analysis of immunity protein sequences from other gram negative bacterial species, which reveal similar conserved framework helices and variability within the analogous variable loop regions (Ko et al., 1999). As loop region 1 displays the greatest variability, and the underlying framework is strongly anchored by helices 1 and 2, we elected to first modify this region. We define variable loop 1 as the six solvent exposed residues Val27–Asp32 (Dennis et al., 1998), a highly acidic region which interacts electrostatically with the most basic areas in the DNase domain.
To test the hypothesis that variable loop 1 would be permissive for a wide range of substitutions without undue perturbation of the underlying framework, we elected to graft an apparently sterically constrained and immunodominant antibody loop region. The donor loop chosen was the heavy chain CDR 3 (H3) loop from the anti-HIV-1 human antibody IgG1b12 (Zwick et al., 2003). This antibody was obtained from a combinatorial phage display library screen of monoclonal Fab fragments from a human patient (Barbas et al., 1991; Burton et al., 1991; Burton and Barbas, 1994), and unusually the crystal structure revealed an exceptionally long H3 loop (19 residues N-V95GPYSWDDSPQDNYYMDVW103-C) which docking models suggest targets the viral gp120 canyon, analogous to the gp120-CD4 interaction (Saphire et al., 2001a, b). The antibody paratope also includes the framework-H3 junction residues Ala93 and Arg94 where the side chains project into the solvent, potentially contributing to the protein interaction surface (Zwick et al., 2003).
The b12 H3 loop was thus modelled onto the Im7 scaffold, and four models with different loops were designed, constructed and evaluated to (i) test various substitution points within the helix–loop junctions of Im7; (ii) demonstrate the inherent stability of the Im7 scaffold; and (iii) transfer varying portions of the b12 H3 loop, in properly constrained configurations for interaction with the target gp120 (Figure 4a). For the various chimeric proteins, the Im7 variable loop 1 residues Val27, Ala28, Ala29, Thr30, Asp31 and Asp32 were replaced in part, or in their entirety, by varying portions of the b12 H3 loop. In this article the constructs are termed models 1, 2, 3 and 4. Model 1 has the longest loop graft, three residues from variable loop 1 and the complete b12 loop. Model 4 has variable loop 1 completely removed and the entire b12 loop grafted in its place. Model 3 has the shortest loop graft, a truncated 12-residue b12 H3, and model 2 has a 16-residue b12 loop grafted with part of the Im7 variable loop 1 removed (Figure 4a). In addition, models were designed with regard to mutational data describing critical loop residues implicated in gp120 binding (Saphire et al., 2001a, 2001b; Zwick et al., 2003). For example, an additional mutation was incorporated within model 4 Asn-26-Ile at a position in helix 1 immediately adjacent to the variable loop 1 region, in order to increase binding affinity via this hydrophobic residue (Porte et al., 1997). The homology modelling modeller objective function scores (MOS) for the 10 lowest energy homology models of each Im7-H3 chimera are shown in Supplementary Table I available at PEDS online, and the lowest energy conformation for each model is shown in Figure 4b.
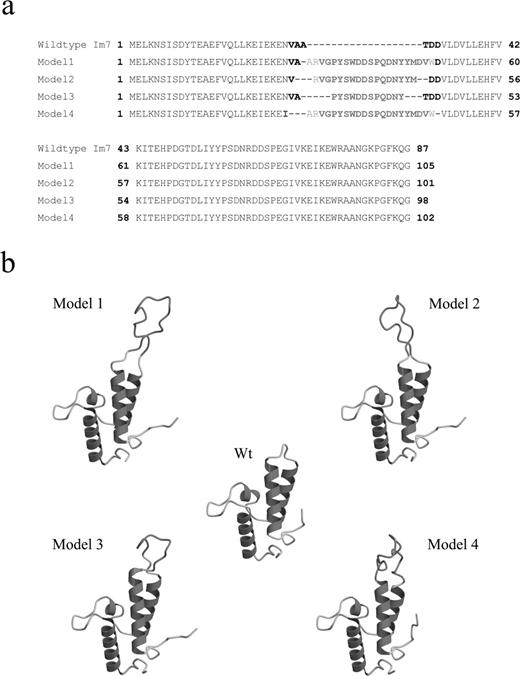
Im7 chimeric proteins. (a) Amino acid sequence alignments of the Im7 wt and the four Im7- H3 chimeras. The variable loop 1 residues conserved from the Im7 scaffold are given in boldface, residues from the IgG1b12 CDR H3 loop are shown in boldface in red, while additional adjacent residues on either end of the inserted loop (originally from the parent b12 antibody) are given in red. For model 4, residue Asn 26 was mutated to the aliphatic residue isoleucine (given in blue). (b) Molscript depictions of the lowest energy conformers of the four Im7-H3 chimeras. The b12 CDR H3 loop component for each conformer is given in orange. A colour version of this figure is available at PEDS online.
The lowest energy chimera was model 2 (MOS = −14.2) < model 1 (MOS = 12.6) < models 3 and 4 (MOS = 21.1 and 21.5, respectively), and comparison of the homology modelling results with the templates suggests that model 2 should best mimic the Im7 loop–helix junction conformation and loop take-off angle from the globular protein scaffold of the b12 antibody (Figure 4b). In contrast, models 3 and 4 showed the greatest deviation in the take-off angle from the globular protein scaffold compared with the b12 antibody when the H3 residues of the template and chimeras were aligned (data not shown). These differences are apparent in Figure 4, where models 1 and 2 have their H3 loops mainly within the plane of the paper but the model 3 and 4 loops project out of the plane of the diagram.
Construction and expression of the four Im7-H3 chimeras
The four Im7-b12 chimeras described in the previous section were constructed using splice overlap PCR (see Materials and Methods for details), cloned into pGC for cytoplasmic expression with C-terminal hexa-histidine tags, and their DNA sequences confirmed. Compared with the wild-type Im7, expression levels for all four chimeric proteins were reduced between 2- and 10-fold (Table II). Notwithstanding, all proteins could be efficiently purified from the E.coli cytoplasm (Figure 5a) and showed essentially identical behaviour using gel filtration chromatography (results not shown). To further validate the folding of the underlying Im7 scaffold, proteins were first immobilized and probed with the anti-Im7 rabbit polyclonal sera (Figure 5b). All models were recognized, though to varying extents, for example models 1 and 4 possessed the lowest parental Im7 structural content (Table II) and accordingly produced the weakest ELISA responses.
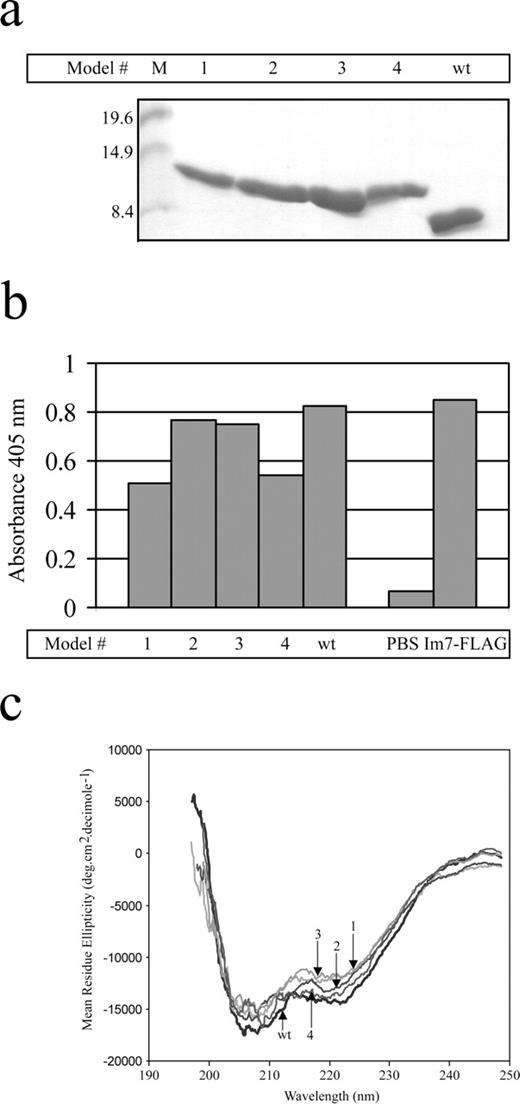
Expression characteristics of Im7 chimeric proteins. (a) Cytoplasmically expressed hexa-histidine tagged Im7 wt and the four Im7-b12 H3 chimeras were purified using Ni-IDA chromatography, electrophoresized using SDS/PAGE through a 15% (w/v) polyacrylamide Tris/glycine gel and stained with Coomassie Brilliant Blue. Approximate molecular weight markers (in kDa) are indicated. (b) ELISA of proteins from (a), immobilized at 1 μg per well and probed with the polyclonal anti-Im7 antiserum. Results represent the average of duplicate wells. (c) Far-UV CD spectral analysis of Im7 wt and the four Im7-H3 chimeras. Spectra were measured at 10°C, in 0.1 M phosphate buffer (pH 7.2) from 250 to 196 nm.
Summary of hexa-histidine tagged Im7 wt and chimeric proteins
| Protein | Molecular weight (Da) | No. of amino acids | % of b12 loop | Expression level (mg/l) |
|---|---|---|---|---|
| Im7 wt | 10 930 | 96 | 0 | 27.5 |
| Model 1 | 13 190 | 114 | 25 | 2.0 |
| Model 2 | 12 762 | 110 | 20 | 5.7 |
| Model 3 | 12 327 | 107 | 15 | 13.1 |
| Model 4 | 12 903 | 111 | 26 | 9.2 |
| Protein | Molecular weight (Da) | No. of amino acids | % of b12 loop | Expression level (mg/l) |
|---|---|---|---|---|
| Im7 wt | 10 930 | 96 | 0 | 27.5 |
| Model 1 | 13 190 | 114 | 25 | 2.0 |
| Model 2 | 12 762 | 110 | 20 | 5.7 |
| Model 3 | 12 327 | 107 | 15 | 13.1 |
| Model 4 | 12 903 | 111 | 26 | 9.2 |
Summary of hexa-histidine tagged Im7 wt and chimeric proteins
| Protein | Molecular weight (Da) | No. of amino acids | % of b12 loop | Expression level (mg/l) |
|---|---|---|---|---|
| Im7 wt | 10 930 | 96 | 0 | 27.5 |
| Model 1 | 13 190 | 114 | 25 | 2.0 |
| Model 2 | 12 762 | 110 | 20 | 5.7 |
| Model 3 | 12 327 | 107 | 15 | 13.1 |
| Model 4 | 12 903 | 111 | 26 | 9.2 |
| Protein | Molecular weight (Da) | No. of amino acids | % of b12 loop | Expression level (mg/l) |
|---|---|---|---|---|
| Im7 wt | 10 930 | 96 | 0 | 27.5 |
| Model 1 | 13 190 | 114 | 25 | 2.0 |
| Model 2 | 12 762 | 110 | 20 | 5.7 |
| Model 3 | 12 327 | 107 | 15 | 13.1 |
| Model 4 | 12 903 | 111 | 26 | 9.2 |
Next, the helical content and stability of the wild-type Im7 and chimeras were measured using far-UV CD spectroscopy, to determine whether the transfer of the H3 loop adversely affected the Im7 secondary structure. CD scans from 250 to 196 nm showed no significant deviation from the wild-type α-helical content, indicating that the underlying α-helical framework was maintained in all four grafted structures (Figure 5c). The spectra showed the signature characteristics for proteins composed primarily of α-helices, with a double minimum at 222 and 208–210 nm (Yang et al., 1986) in the wavelength scan range. The conformational stability of the chimeric proteins was compared by measurement of thermal denaturation in solution (Figure 6a–e). The thermal denaturation of Im7 wt and the four Im7-H3 chimeras was fully reversible. In descending order, the melting temperatures (Tm) were model 4 > Im7 wt∼model 1 > model 2 > model 3 (Table III). While the thermostabilities of models 2 and 3 were slightly reduced compared with the wild-type Im7 protein, this was not unexpected: most mutations in the form of inserted sequences or single amino acid substitutions that diverge from a consensus for the protein family do appear to adversely affect the cohesive folding and packing of a naturally selected protein (Lehmann et al., 2002). Surprisingly, the model 4 chimera appeared more stable than the wild type, which may be attributed to the complete absence of the Im7 variable loop 1 residues Val27, Ala28, Ala 9, Thr30, Asp31 and Asp32 and the burial of the hydrophobic H3 framework proximal residues. The absence of these residues results in an immediate transition from the highly structured Im7 α-helices into the loop region in a manner analogous to the framework-CDR junctions in the Ig framework of the parental IgG1b12 antibody (Zwick et al., 2003).
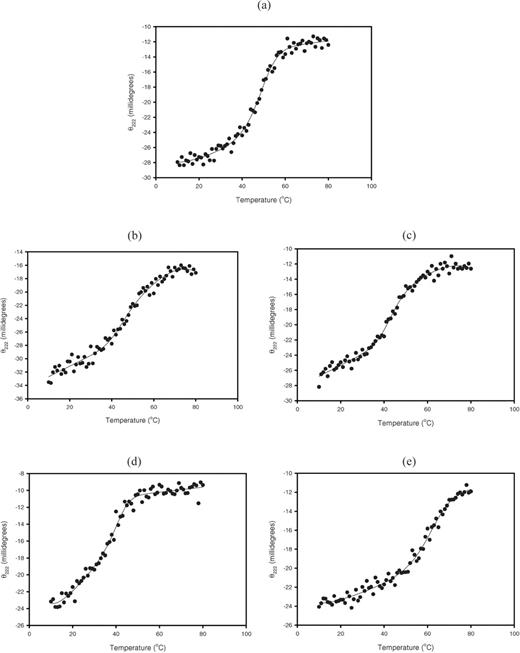
Thermal denaturation characteristics of Im7 wt and chimeras. Recombinant proteins in 10 mM phosphate buffer (pH 7.2) were monitored using CD at 222 nm, from 10 to 50°C. Data points are designated by solid circles, the solid curve illustrates the non-linear regression analysis fit by a sigmoidal function. (a) Im7 wt (b) Model 1, (c) Model 2, (d) Model 3 and (e) Model 4.
Thermal stability of Im7 wt and chimeric proteins
| Protein | Tm (°C) |
|---|---|
| Wildtype Im7 | 48.1 ± 0.8 |
| Model 1 | 47.1 ± 4.9 |
| Model 2 | 45.0 ± 1.8 |
| Model 3 | 40.9 ± 1.6 |
| Model 4 | 61.1 ± 2.8 |
| Protein | Tm (°C) |
|---|---|
| Wildtype Im7 | 48.1 ± 0.8 |
| Model 1 | 47.1 ± 4.9 |
| Model 2 | 45.0 ± 1.8 |
| Model 3 | 40.9 ± 1.6 |
| Model 4 | 61.1 ± 2.8 |
Thermal stability of Im7 wt and chimeric proteins
| Protein | Tm (°C) |
|---|---|
| Wildtype Im7 | 48.1 ± 0.8 |
| Model 1 | 47.1 ± 4.9 |
| Model 2 | 45.0 ± 1.8 |
| Model 3 | 40.9 ± 1.6 |
| Model 4 | 61.1 ± 2.8 |
| Protein | Tm (°C) |
|---|---|
| Wildtype Im7 | 48.1 ± 0.8 |
| Model 1 | 47.1 ± 4.9 |
| Model 2 | 45.0 ± 1.8 |
| Model 3 | 40.9 ± 1.6 |
| Model 4 | 61.1 ± 2.8 |
Im7-H3 chimeras target gp120
Having demonstrated that loop grafting to the Im7 scaffold retained the beneficial expression and folding characteristics of the parental protein, we then asked whether the same loop grafting experiments resulted in transfer of affinity for antigen. The Im7 wild type and H3 chimeras were tested for binding to the original ligand (DNase) and to HIV-1 gp120 using ELISA and biosensor analysis, respectively. As expected, all chimeric proteins retained some affinity for the DNase domain, though significantly less than for the wild type, especially in models 1 and 4 where the b12 loop dominates in the loop 1 region (Figure 7a). Conversely, when the Im7 proteins were immobilized to a biosensor surface and relative affinities for gp120 qualitatively compared using surface plasmon resonance technology, the order of binding was model 1 > model 4 > model 3 > model 2 > Im7 (Figure 7b). This is an almost exact reverse correlation with the results obtained from the DNase binding and Im7 polyclonal antisera.
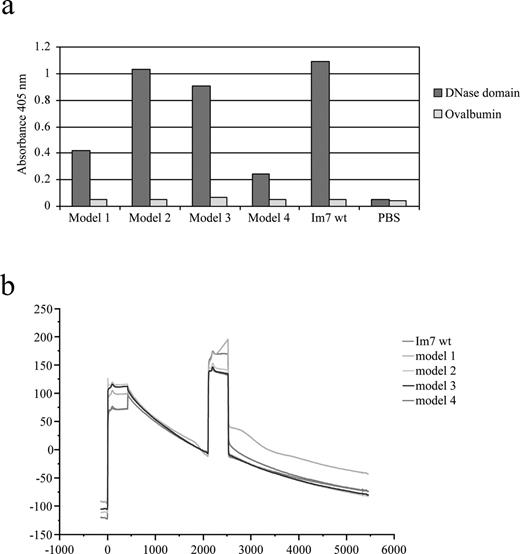
Interaction of Im7 chimeric proteins with DNase and HIV gp120. (a) ELISA of the binding of the Im7 wt and four Im7-H3 chimeras to DNase domain and ovalbumin (negative control antigen). Results represent the average of duplicate wells. (b) BIAcore traces of the interaction between Im7 wt and chimeric model proteins and HIV gp120. An anti-penta-histidine tag antibody was first covalently coupled to the sensor surface and used to capture equal amounts (∼200 RU) of Im7 recombinant proteins, which display slow dissociation relative to baseline over 6000 s. Sensograms show the binding of the HIV-1 gp120 analyte (10 μg/mL) injected at the ∼2000 s time point at a constant flow rate of 5 μl/min with an injection volume of 35 μl. No specific interactions were observed when the full-length oligomeric gp120 was injected over a flow cell with immobilized anti-penta-histidine antibody alone. A colour version of this figure is available at PEDS online.
Discussion
We have presented the Im7 protein from E.coli as a novel compact protein domain scaffold ideal for the display of molecular libraries. There are several features that make Im7 appropriate for such an application: first, its robust helical structure, with no requirement for internal or stabilizing disulphide linkages, offers distinct advantages in both ease of in vitro protein purification and in future adaptation to an in vivo intracellular environment in the manner of ‘intrabody’ immunoglobulins. This aspect may have particular utility in the targeting of, for example, viral antigens normally sequestered from the endogenous immune response, or in addressing molecules to intracellular organelles. Second, the small size of Im7 suggests that it may be able to target antigenic epitopes inaccessible to larger binding molecules. This avoidance of steric hindrance must be balanced against the retention of sufficient underlying scaffold to allow concurrent maintenance of scaffold integrity and establishment of important framework-antigen contacts, which can represent an important component of binding affinity beyond the major specificity-determining loop regions. An examination of the Im7 structure and chimera models suggests that such regions are retained, for example the loopII–helix3 and loopI–helix1 junctions (Figure 1a) are available to contribute both side-chain and main-chain interactions that we assume are responsible for the observed residual binding of the chimeras to colicin E7 DNase. Third, the bacterial origin of Im7 means that the framework has been pre-selected and evolved for expression in E.coli, and thus is ideally suited for use in the powerful bacteriophage-based display and selection systems available for this host. Finally, the absence of disulphide linkages and the relatively small size also make Im7 an attractive candidate for incorporation into the increasingly popular in vitro selection systems, such as ribosome display where individual proteins are directly coupled to the translation ribosome, and the intact complex panned and recovered while retaining the information encoded within the associated mRNA. Indeed, a recent report describes Im7/Im9 as the model system of choice for testing selection for directed evolution of nuclease proteins within an in vitro compartmentalization system (Bernath et al., 2005).
The experiments and results we have presented confirm these hypotheses, particularly in the context of successful display upon the surface of fd bacteriophages. More important, we have demonstrated that the underlying Im7 structure is not significantly perturbed by four extensive substitutions/insertions into the loop 1 region, these additions being of sufficient size and sequence variability to warrant classification as ‘drastic’ modifications, extending well beyond the naturally occurring variability that mediates the toxin-specific binding of the related family of E.coli immunity proteins. Our choice for these loop grafting experiments was the CDR H3 loop from the anti-HIV-1 gp120 IgG1b12 antibody, predicated by the ability of both the complete antibody and a peptide mimic of the H3 loop (coupled to BSA) to apparently penetrate the recessed CD4-binding site of gp120, to prevent viral membrane fusion and to protect against viral challenge in vivo (Saphire et al., 2001a). This epitope is not otherwise generally accessible to human/murine antibodies due to the nature of the recessed cavity on the functional (oligomeric) viral spike (Sattentau and Moore, 1995; Saphire et al., 2001b; Pantophlet and Burton, 2003). Indeed, our modelling of the Im7-H3 chimeras suggests that H3 should be available for binding to the gp120-CD4 without steric hindrance.
In our loop grafting experiments varying portions of the b12 loop were first modelled onto the Im7 scaffold by reference to both the parent antibody and the structure of the target antigen. While the most energetically favourable projections of the models appeared quite divergent from each other, it is likely that this degree of flexibility and variability was reduced through induced fit upon binding into the gp120 target cleft. Following successful recombinant protein production, there was a low (but measurable) transfer of antigen-specific affinity, which probably reflects the difficulties associated with using the intact gp120 trimer as a target analyte, combined with the expected drop in affinity typical of intra-scaffold loop grafting experiments. The Im7 protein being a much smaller protein than the parental antibody, we predict that the underlying scaffold may naturally come into close proximity to gp120, depending on the length and orientation of the H3 loop in the chimeras. Possible future experiments may involve mutation of H3 loop proximal residues for stabilizing the loop presentation or modification of the helix II loop to perhaps achieve a higher electrostatic and shape complementarity for access to adjacent clefts and pockets on the viral surface. Certainly, residues have similarly been identified on the parental b12 antibody scaffold adjacent to the CDR H3 loop that appear important in maintaining its upright position and optimal presentation to the gp120-CD4 binding site cavity (Zwick et al., 2003). Thus, further modification of the underlying scaffold may ameliorate disruption to the b12 antibody H3 loop caused in the loop grafting by removal or misalignment of intra-loop H-bonds or the tightly packed aromatic framework residues at its base.
The next step is the development of molecular libraries based around the loop structures of Im7. Randomization of only a relatively small component, for example 7 residues at the tip of loop 1 and the adjacent framework-loop junctions, will generate a potential diversity of >109 combinations. When combined with simultaneous modification of loop region 2 a binding surface capable of targeting a broad range of antigens with potentially high affinity could be formed. It is notable that the ColE7–Im7 natural interaction achieves an exceedingly robust binding affinity (KD ∼10−14 M) through a relatively compact binding interface of ∼700Å (as calculated for the ColE7-Im7 complex)(Ko et al., 1999) similar to the interface that we envisage will be produced in randomized molecular libraries. The stabilities of the loop graft models presented here suggest not only that the underlying framework maintains its structure with variable loops but also that some conformations may even have stability (and folding) advantages over the wild-type Im7. This suggests a strategy for further library diversification, by incorporation of additional residues, again randomized, into the loop regions, similar to the extensive heterogeneity observed in the heavy chain CDRs found in naturally occurring single domain antibodies (Nuttall et al., 2002, 2003, 2004). Such molecular libraries, we envisage, will form powerful resources for the generation of binding reagents of non-antibody origin.
The authors thank Professor Nick Hoogenraad and Ms Kylie Pearson for rabbit immunizations, blood collection and sera preparation. The authors express their gratitude to Professor Dennis Burton for his kind hospitality while S.M.J. was a visitor in his laboratory and for a critical reading of the manuscript. We thank Ms Ann Hessell, Dr Erica Saphire and Dr Michael Zwick for their advice and discussions.
References
Ausubel,F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (
Barbas,C.F.III, Kang,A.S., Lerner,R.A. and Benkovic,S.J. (
Binz,H.K., Amstutz,P., Kohl,A., Stumpp,M.T., Briand,C., Forrer,P., Grutter,M.G. and Plückthun,A. (
Burton,D.R., Barbas,C.F. III, Persson,M.A., Koenig,S., Chanock,R.M. and Lerner,R.A. (
Chak,K.F., Safo,M.K., Ku,W.Y., Hsieh,S.Y. and Yuan,H.S. (
Dennis,C.A., Videler,H., Pauptit,R.A., Wallis,R., James,R., Moore,G.R. and Kleanthous,C. (
Ferguson,N., Li,W., Capaldi,A.P., Kleanthous,C. and Radford,S.E. (
Fernandez-Carneado,J., Grell,D., Durieux,P., Hauert,J., Kovacsovics,T. and Tuchscherer,G. (
Harlow,E. and Lane,D. (
Hatters,D.M., Wilson,L., Atcliffe,B.W., Mulhern,T.D., Guzzo-Pernell,N. and Howlett,G.J. (
Irving,R.A., Coia,G., Roberts,A., Nuttall,S.D. and Hudson,P.J. (
Kleanthous,C., Kuhlmann,U.C., Pommer,A.J., Ferguson,N., Radford,S.E., Moore,G.R., James,R. and Hemmings,A.M. (
Kohl,A., Binz,H.K., Forrer,P., Stumpp,M.T., Plückthun,A. and Grutter,M.G. (
Laskowski,R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (
Lehmann,M., Loch,C., Middendorf,A., Studer,D., Lassen,S.F., Pasamontes,L., van Loon,A.P. and Wyss,M. (
Morrow,J.A., Segall,M.L., Lund-Katz,S., Phillips,M.C., Knapp,M., Rupp,B. and Weisgraber,K.H. (
Nord,K., Gunneriusson,E., Ringdahl,J., Stahl,S., Uhlen,M. and Nygren,P.A. (
Nuttall,S.D., Rousch,M.J., Irving,R.A., Hufton,S.E., Hoogenboom,H.R. and Hudson,P.J. (
Nuttall,S.D., Krishnan,U.V., Doughty,L., Nathanielsz,A., Ally,N., Pike,R.N., Hudson,P.J., Kortt,A.A. and Irving,R.A. (
Nuttall,S.D., Krishnan,U.V., Doughty,L., Pearson,K., Ryan,M.T., Hoogenraad,N.J., Hattarki,M., Carmichael,J.A., Irving,R.A. and Hudson,P.J. (
Porte,D., Oertel-Buchheit,P., John,M., Granger-Schnarr,M. and Schnarr,M. (
Saphire,E.O., Parren,P.W., Barbas,C.F. III, Burton,D.R. and Wilson,I.A. (
Saphire,E.O., Parren,P.W., Pantophlet,R., Zwick,M.B., Morris,G.M., Rudd,P.M., Dwek,R.A., Stanfield,R.L., Burton,D.R. and Wilson,I.A. (
Smith,G.P., Patel,S.U., Windass,J.D., Thornton,J.M., Winter,G. and Griffiths,A.D. (
Sui,M.J., Tsai,L.C., Hsia,K.C., Doudeva,L.G., Ku,W.Y., Han,G.W. and Yuan,H.S. (
Wallis,R., Leung,K.Y., Pommer,A.J., Videler,H., Moore,G.R., James,R. and Kleanthous,C. (
Author notes
1Cooperative Research Centre for Diagnostics 343 Royal Parade, Parkville, Victoria 3052, 2CSIRO Molecular and Health Technologies, 343 Royal Parade, Parkville, Victoria 3052 and 3Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Victoria 3010, Australia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}