





Abstract
In the global context of arboviral emergence, deep sequencing unlocks the discovery of new mosquito-borne viruses. Mosquitoes of the species Culex pipiens, C. torrentium, and C. hortensis were sampled from 22 locations worldwide for transcriptomic analyses. A virus discovery pipeline was used to analyze the dataset of 0.7 billion reads comprising 22 individual transcriptomes. Two closely related 6.8 kb viral genomes were identified in C. pipiens and named as Culex pipiens associated tunisia virus (CpATV) strains Ayed and Jedaida. The CpATV genome contained four ORFs. ORF1 possessed helicase and RNA-dependent RNA polymerase (RdRp) domains related to new viral sequences recently found mainly in dipterans. ORF2 and 4 contained a capsid protein domain showing strong homology with Virgaviridae plant viruses. ORF3 displayed similarities with eukaryotic Rhoptry domain and a merozoite surface protein (MSP7) domain only found in mosquito-transmitted Plasmodium, suggesting possible interactions between CpATV and vertebrate cells. Estimation of a strong purifying selection exerted on each ORFs and the presence of a polymorphism maintained in the coding region of ORF3 suggested that both CpATV sequences are genuine functional viruses. CpATV is part of an entirely new and highly diversified group of viruses recently found in insects, and that bears the genomic hallmarks of a new viral family.
1. Introduction
Viral biodiversity remains largely unexplored. Viruses are found in all types of organisms (archaea, bacteria, eukaryota and some large dsDNA viruses) and are the most abundant micro-organisms on Earth (Paul and Sullivan 2005). During the past decade, the development of high-throughput next-generation sequencing technologies (NGS) and the use of bioinformatics, metagenomics and phylogenetic analyses have allowed the discovery of many new viruses, particularly of phages in aquatic and mammal gut environments (Phan et al. 2011; Ng et al. 2012). Although NGS have become a sensitive and reliable method for virus discovery, recent virus discoveries in arthropods in general and insects in particular are scarcer compared with other hosts or ecosystems (Cook et al. 2013; Chandler, Liu, and Bennett 2015; Bishop-Lilly et al. 2016) and often target viral families within ssRNA(+) viruses such as Flaviviridae (genus Flavivirus), Togaviridae (genus Alphavirus), Nidovirales (family Mesoniviridae), or ssRNA(−) viruses as Bunyaviridae (Junglen and Drosten 2013). Indeed, arthropods were reported to be a great source of viral diversity (Liu, Chen, and Bonning 2015), especially of RNA viruses (Bichaud et al. 2014; Shi et al. 2016). Still, several recent studies discovered new insect-associated viruses in other recognized viral families (Shi et al. 2016), such as Rhabdoviridae (Quan et al. 2010; Kuwata et al. 2011; Vasilakis et al. 2014), Reoviridae (Attoui et al. 2005; Hermanns et al. 2014; Auguste et al. 2015), Tymoviridae (Wang et al. 2012), Nodaviridae (Schuster et al. 2014) or in nonrecognized families as in the case of the negeviruses (Sandewavirus/Nelorpivirus) (Vasilakis et al. 2013; Auguste et al. 2014; Kallies et al. 2014; Nabeshima et al. 2014; Fujita et al. 2016; Kawakami et al. 2016). The growth of insect-associated virus discoveries provides a fertile ground for the understanding of the complexity and dynamics of arboviral communities in an ecological and evolutionary perspective (Bolling et al. 2015; Hall et al. 2016).
Despite the health interest surrounding the study of mosquitoes, few studies have used NGS for virus-discovery in mosquitoes. In 2011, the first metagenomic approach by 454 pyrosequencing on wild mosquitoes revealed 6 new DNA viruses (Ng et al. 2011). Deep sequencing of small RNA (18–30 bp) also allowed the discovery of new RNA and DNA viruses, in several mosquito species (Ma et al. 2011; Cook et al. 2013; Aguiar et al. 2015). Two novel rhabdoviruses and three novel bunyaviruses were also discovered in several Australian mosquitoes using deep sequencing of pooled insect viromes passaged in cell culture (Coffey et al. 2014). Finally, the used of transcriptome sequencing of a large range of potential hosts has improved and increased the discovery of many new RNA viruses infecting invertebrates (Shi et al. 2016) including the discovery of the new Gamboa mosquito virus from a mix of Culicidae mosquitoes (Shi et al. 2015). Yet, Culex mosquitoes may still host largely unexplored viral diversity and thus be potential reservoir of unknown viruses that might become relevant for human health (Fonseca et al. 2004; Mackenzie and Jeggo 2013). These studies illustrate how underestimated is viral diversity associated with mosquitoes and outline a way to bridge the gap between our current state of knowledge and the vast viral biodiversity occurring in nature.
Here we report the screening of 22 Illumina transcriptomes of wild individuals of Culex pipiens, C. hortensis, and C. torrentium species and the subsequent discovery and genetic characterization of a new mosquito virus species associated with C. pipiens. Results of genome annotation and phylogenies of Culex pipiens associated tunisia virus (CpATV) were used to decipher its evolutionary relationships to a new group of viruses recently found in insects.
2. Materials and methods
2.1 Mosquito sampling
Twenty-two transcriptomes were obtained from single adult female mosquitoes, belonging to the species C.pipiens collected in France, Algeria, Tunisia, Burkina Faso, Israel, Reunion Island, Philippine, China, Costa Rica, and the USA, C. hortensis, collected in France and C. torrentium collected in France and Sweden (Supplementary Table S1). All mosquitoes were sampled as larvae in fresh water puddles and grown in laboratory conditions. After emergence, females were conserved in liquid nitrogen, or kept under laboratory conditions before they had the opportunity to take any blood meal. All animals were caught in the wild and preserved after emergence, except from C. pipiens from USA, for which a laboratory strain was maintained since 1950 and the transcriptomes sequenced in 2011.
2.2 RNA extraction for transcriptome sequencing and assembly
Total RNA isolation was performed using RNeasy Mini Spin Columns (Qiagen, Chatsworth, CA, USA) on individual mosquitoes as previously described (Gayral et al. 2011). RNA quality was assessed on Agilent Bioanalyzer 2100 system and a RNA 6000 Nano Lab-Chip (Agilent). Then, 5 µg total RNA was reverse-transcribed using the SMART cDNA library Construction kit (Clontech, Mountain View, USA). An oligo(dT)-primed first-strand synthesis followed by a cap-primed second-strand synthesis was performed. Eight libraries were sequenced per lane using an Illumina HiSeq 2000 sequencer to produce 50 bp single-end reads. All 22 transcriptomes were de novo assembled using a previously developed bioinformatics pipeline (Romiguier et al. 2014): a first assembly with ABYSS V 1.2.0 (Birol et al. 2009; Simpson et al. 2009) with Kmer set at 40 (Cahais et al. 2012) was followed by contig re-assembly with CAP3 (Huang and Madan 1999). Complete and 5′- and/or 3′-truncated ORFs were detected and translated with standard genetic code using Prodigal V2_60 software for metagenomic data (Hyatt et al. 2010, 2012). ORFs displaying undetermined nucleotides were not discarded in subsequent analyses.
2.3 Virus discovery
Protein homology searches were performed on all translated ORFs of the 22 transcriptomes using the accurate and sensitive HHblits program implemented in the HHSuite package (Söding 2005; Remmert et al. 2011). The Nr20 NCBI protein database, a clustered version of the protein nonredundant database from NCBI down to a maximum pairwise sequence identity of 20% protein, was used as a search database as recommended (Söding 2005; Remmert et al. 2011). To minimize false-positive results, only ORFs displaying homology e-value <10−5 and probability >95% were kept. When a positive hit was detected, NCBI taxonomic identifier (TaxID; ftp://ftp.ncbi.nih.gov/pub/taxonomy) of the corresponding Nr protein was retrieved using the BLAST+ program (Camacho et al. 2009) and assigned to the predicted ORFs. Only ORFs identified as ‘viruses’ in the superkingdom taxonomic rank were kept for further analysis. Then, to reduce false-discovery rate and ensure the detection of functional infectious viruses, isolated virus-like ORFs or single ORFs of dubious viral homology were discarded and only putative full-length viral genomes were further analyzed.
To verify the accuracy of viral contigs assembly, Illumina reads were mapped on the assembled viral genome using BWA (Li and Durbin 2009) with default parameters. Mapping results (SAM files) were used to calculate the coverage at each nucleotide position along the viral genomes using Geneious 8.1.7 (http://www.geneious.com; Kearse et al. 2012). The distribution of the quality score of mapped reads was plotted using the fastx_quality_stats and fastq_quality_boxplot programs implemented in FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/).
Lastly, nucleotide polymorphism at each genomic position (pi) was estimated on the mapping results using PoPoolation (version 1.2.2) with default parameters (Kofler et al. 2011). Three nucleotides were trimmed from both 5′ and 3′ ends of all viral reads to ensure that sequencing errors would not bias the estimation of intra-host virus diversity as such errors are more frequent at the extremities of Illumina reads.
2.4 Prevalence in other hosts
We screened several public databases to evaluate if the virus described in this study might be widespread or infecting other hosts. First BLASTN searches were performed using the entire CpATV genome (e-value threshold=10−5) on the Culex genome (C. quinquefasciatus) and on the 15 available mosquito transcriptomes (Supplementary Table S2). In addition, Culex reads produced in this study were mapped on the CpATV genome using BWA software with defaults parameters (Li and Durbin 2009).
Further BLASTN and BLASTX searches (e-value threshold=1) were performed against the Transcriptome Shotgun Assembly (TSA), Whole-genome Shotgun contigs (WGS) and Metagenomic proteins (env-nr) databases (October 2016), as well as the 60 Arachnida, Gastropoda and Insecta genomes available in VectorBase (September 2016, https://www.vectorbase.org/).
2.5 Genome annotation
Viral genomes were annotated based on conserved protein domains searched using InterProScan (version 5) (Jones et al. 2014; Mitchell et al. 2015), NCBI Conserved Domain and Conserved Domain Database (v3.14) (Marchler-Bauer et al. 2015), as well as SMART (version 7) (Schultz et al. 1998; Letunic, Doerks, and Bork 2012). Signal peptides were detected using SignalP (version 4.0) (Petersen et al. 2011) and TargetP (version 1.1) (Emanuelsson et al. 2000). The detection of potential poly(A) tail was performed using PolyApred (Ahmed, Kumar, and Raghava 2009). RNA motifs and cis-regulatory elements such as tRNA-like structures were searched using RegRNA 2.0 (Chang et al. 2013). Internal ribosomal entry sites (IRES) were predicted using Viral IRES Prediction System (VIPS) with default parameters (Hong et al. 2013).
BLAST searches (e-value threshold=1) were performed against Plasmodium genomes available in PlasmoDB (Aurrecoechea et al. 2009) (release 2015/07/23) to detect homologies with 13 available Plasmodium genomes (Supplementary Table S3) as well as the unpublished P. relictum genome (Ana Rivero, personal communication).
2.6 Phylogenetic analyses
Amino acid sequences alignment of each conserved protein domain were performed with MAFFT (Katoh et al. 2002) using default parameters and curated manually. Nonhomologous sites located in ORFs extremities were discarded from alignments. The best substitution model was selected using ProtTest v3.2 (Abascal, Zardoya, and Posada 2005). Maximum Likelihood (ML) phylogenetic trees were inferred for each alignment using PhyML (Guindon, Gascuel, and Rannala 2003) and robustness of nodes was assessed with aLRT statistics (SH-like branch supports) (Anisimova and Gascuel 2006). Shimodaira–Hasegawa (SH) (Shimodaira and Hasegawa 1999) and a one-sided Kishino-Hasegawa (1sKH) (Kishino and Hasegawa 1989) tests were performed using TREE-PUZZLE (version 5.6.rc16) (Schmidt et al. 2002) to detect possible phylogenetic incongruence between different domains of the same ORF.
2.7 Molecular evolutionary analyses
PAL2NAL program (Suyama, Torrents, and Bork 2006) was used to obtain relevant codon alignments guided by protein alignments. Only the most closely related sequences to the taxon of interest were kept to ensure genuine homology of aligned sites. Selective pressures acting on viral coding sequences were assessed using branch-models which estimated the ratio of nonsynonymous (dN)/synonymous (dS) substitution rates (Nei and Gojobori 1986; Kryazhimskiy and Plotkin 2008) in a chosen branch or subtree, in codeml (Yang 1998; Yang and Bielawski 2000) implemented in PAML version 4.9c (Yang 2007). Likelihood ratio tests (LRTs) were employed using the χ2 tests statistics = 2ΔLnL (i.e. twice the difference of the Log-Likelihood of each model) and a type I error = 0.05, df = 1 (i.e. the difference of number of parameters between two models).
2.8 Virus detection by RT-PCR
Culex pipiens mosquitos conserved at −80 °C and originating from the same populations Ayed and Jedaida studied for CpATV discovery using bioinformatics approach were used to confirm the presence of the virus. Insects were ground in 200 µl TRI Reagent BD (Molecular Research Center, USA) using a plastic pestle and a final volume of 1 ml of TRI Reagent per sample was used. Following procedure was modified from the manufacturer’s recommendations. Samples were incubated at room temperature for 5 min in TRI Reagent and centrifuged at 12,000g at 4 °C for 10 min. After removal of the supernatant, 200 µl of chloroform was added, shacked for 15 s and incubated at room temperature for 3 min. After centrifugation at 12,000g at 4 °C for 15 min, 500 µl isopropanol was added to the supernatant to precipitate RNA for 10 min at room temperature. Tubes were centrifuged at 12,000g at 4 °C for 10 min. After removal of the supernatant, the RNA precipitate was washed once with 1 ml 75% alcohol. Tubes were shacked and centrifuged at 7500g at 4 °C for 5 min. After removal of the supernatant, the RNA was dried at 37 °C for 10 min. The RNA was dissolved with 30 µl of DEPC-treated water. After stirring, tubes were stored at 55–60 °C for 10 min to promote dissolution. A DNAse treatment was performed with the TURBO DNA-free™ Kit (Life technologies) in a 30-µl volume following the manufacturer’s instructions. Absence of residual genomic DNA was confirmed by an absence of PCR amplification from RNA samples (data not shown) using primers Intron2dir (GCGCGAGCATATCCATAGCAC) and CpEx3rev (GACTTGCGACACGGTACTGCA) (Osta et al. 2012) targeting 500 pb from ace-1 gene of C. pipiens (see below for PCR conditions).
RNA from each sample were reverse transcribed using the SuperSript III Reverse transcriptase Kit (Invitrogen, Life technologies) from 400 ng to 1.5 µg total RNA, 30 ng of random primers (RP-10) and 1 µl of dNTP mixture (10 mM each). The remaining procedure was performed following the manufacturer's instructions.
The quality of the cDNA was tested by PCR with primers Intron2dir and CpEx3rev targeting ace-1 gene of C. pipiens. The PCR was performed in a 40 µl volume containing 2 µl of cDNA and using GoTaq G2 polymerase (Promega, USA) following manufacturer's instructions. An initial denaturing of the template at 94 °C for 2 min was followed by 35 cycles: 94 °C, 30 s; 59 °C, 45 s; 72 °C, 1 min and by a final extension period at 72 °C for 10 min. CpATV detection targeting 843 nucleotides of ORF1 was performed using primers CpATV_1815F (TGGGGCTGGTAGAAGACGTA) and CpATV_2657R (ACGGCAGAGTATTCGTAAGGTG) in a 40 µl volume containing 2 µl of cDNA and using GoTaq G2 polymerase following manufacturer's instructions. These primers were designed to amplify both CpATV strains. Same PCR cycle than for ace-1 gene (see above) was used for CpATV detection. Amplified products were visualized by electrophoresis in 2% agarose gels with ethidium bromide. PCR products were purified using EZ 10 Spin Column PCR Purification Kit (BioBasic) and sequenced in forward and reverse using SANGER technology. CpATV sequence of samples Ayed_15 and Ayed_19 were deposited in GenBank under accession number MG557616 and MG557617.
3. Results
3.1 Virus detection in C. pipiens GA35C from Tunisia
Twenty-two individual mosquito transcriptomes, including eight new, were assembled from a total of 687 million Illumina reads. Average contig size (N50) was 230 bp (range 164–318 bp; Supplementary Table S4). Out of 69,756 ORFs predicted in this dataset, the virus detection pipeline identified only one 6,818 bp contig presenting high homology with viral sequences in the C.pipiens individual GA35C, sampled in 2005 in Tunisia.
This contig contained 4 ORFs of sizes 3,240, 414, 2,118, and 483 bp, consistent with a putative full-length viral genome (Fig. 1a). Initial protein homology search results showed ORF1 and ORF4 were strongly homologous to structural proteins of Tobamovirus, a ssRNA(+) plant virus belonging to the Virgaviridae family (e-value = 8.40E−210 and 4.70E−65, P = 8.00E−210 and 4.20E−70 for ORF1 and ORF4, respectively). The coding region was surrounded by 2 noncoding sequences of 333 and 99 bp at the 5′ and 3′ extremities respectively, possibly representing two UTRs. Advanced analyses of genomic motifs did not reveal any regulatory sequences, such as poly(A) tail, IRES signals or tRNA-like structures.
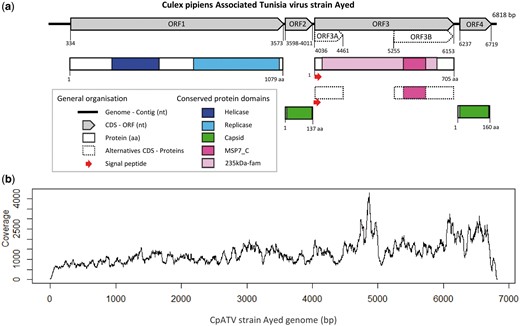
Culex pipiens associated tunisia virus (CpATV) characteristics. (a) Genome organization of CpATV with conserved domains and predicted ORFs. (b) Read coverage of CpATV_Ayed genome.
To ensure this viral contig did not result from chimeric assembly, several verification steps were undertaken. First, read mapping quality was assessed. A total of 176,684 very good quality reads (median quality scores >36 at each position of the reads; Supplementary Fig. S1a) from C. pipiens GA35C mapped on the viral genome (Fig. 1b). This indicated that at least 99.9% of bases were accurately called. Second, the viral contig was evenly covered along its entire length, without any coverage drop that could have indicated a chimera (Fig. 1b). Third, the viral contig was very abundant in the mosquito transcriptome as its mean sequence coverage was 1,322× compared with the 227× mean coverage obtained for C. pipiens GA35C. Such difference in transcript abundance may indicate the presence of a replicating infectious virus. As our contig bared all the hallmarks of a viral genome, we named this new virus CpATV strain Ayed (CpATV_Ayed).
3.2 CpATV_Ayed displays typical viral ORFs
The ORF1 of CpATV_Ayed contained a viral helicase domain (pfam01443, e-value 8.82E−35) and an RNA-dependent RNA polymerase domain (RdRp_2 pfam00978, e-value 4.74E−110) (Fig. 1a). Interestingly, both domains possessed all amino acids conserved within ssRNA(+) viruses of closely related genera in the seven conserved domains of helicase (Supplementary Fig. S2) as well as in the eight RdRp domain (Supplementary Fig. S3) as defined by Koonin and Dolja (1993). RdRp catalyzes RNA replication while helicase facilitates RdRp initiation and elongation by unfolding secondary structures of ssRNA and unwinding dsRNA templates. Thus, CpATV ORF1 is putatively a functional viral replicase.
ORF2 and ORF4 both contained a complete capsid domain similar to the TMV capsid-like domain (pfam00721) found in Tobacco Mosaic Virus (Tobamovirus, Virgaviridae). This result was supported by strong homologies of protein domains: e-value =5.45E−3 and 1.43E−11 for ORF2 and ORF4, respectively. Protein alignment also showed the two amino acids identified as functionally important to form an R(+)–E(−) salt bridge (Koonin and Dolja 1993) were present in both CpATV capsid sequences (Supplementary Fig. S4).
3.3 CpATV_Ayed ORF3 has similarities with Plasmodium domains
Two conserved protein domains were discovered in ORF3. First, homology was detected for protein 235kDa-fam (TIGR01612; 634 aa, e-value = 2.25E−4), which encodes a reticulocyte binding protein or Rhoptry protein. This Plasmodium protein plays a role in the red blood cell invasion process (Counihan et al. 2013). Second, ORF3 displayed homology with a complete MSP7_C domain (Pfam id = pfam12948; 114 aa, e-value =6.76E−3); which is the C-terminal part of the Merozoite Surface Protein 7 of Plasmodium falciparum (Pachebat et al. 2001). However, phylogenetic analyses did not reveal any direct relationships between the CpATV sequence and the Plasmodium genes (data not shown). Further BLASTN and BLASTX searches of the entire ORF3 against the Plasmodium genomes did not produce any significant hits towards more closely related sequences. The N-terminal region of ORF3 harbored a 36-bp signal peptide (secretory pathway score 0.764, reliability class 3/5 where 5 is the least reliable class, see Emanuelsson et al. 2000), indicating that the translated protein is likely secreted.
3.4 Comparative viral genomic analyses
The CpATV genome content was compared with genomes of closely related to Virgaviridae and ‘Negeviruses’. Additionally, fourteen new viral sequences associated with diverse insects (including many fruit flies, Diptera) (Webster et al. 2015,, 2016) were annotated for the first time in the present study (Fig. 2). In all virus genomes, the large ORF1 contained a viral helicase domain adjacent to an RdRp domain. The 5′ end of ORF1 CpATV did not harbor any methyltransferase domain (Fig. 2), which plays a role in the biosynthesis of the 5′ cap of RNA viral genomes (Rozanov, Koonin, and Gorbalenya 1992; Byszewska et al. 2014).

Comparative genomics of Culex pipiens associated tunisia virus (CpATV) genome and related virus genomes. The fourteen new insect viruses are from Webster et al. (2015, 2016) and the outgroups are Citrus leprosis virus C RNA1 (Cilevirus), Negev virus (Nelorpivirus), tobacco mosaic virus (Tobamovirus, Virgaviridae). Sequences details and GenBank accessions numbers are available in Supplementary Table S6.
The remaining ORFs were variable both in number (1–5, depending on the virus genome) and size (67–681 aa) and contained structural and virulence domains (Fig. 2). Capsid domains (TMV-like coat protein—IPR001337) similar to those of CpATV were found in the Boutonnet virus and the TSA Musca domestica viral sequence (both displayed two capsid ORFs), as well as in the TSA Argochrysis armilla, TSA Cotesia vestalis and TSA Latrodectus hesperus venom viral sequences (all three displayed one ORF) (Fig. 2). The Blackford virus contained a putative F-Box protein (domain PF00646), which, in Poxviruses, is involved in the ubiquitination of proteins targeted for degradation in the proteasome and deregulation of host cells (Mercer, Fleming, and Ueda 2005; Barry et al. 2010). The TSA Monomorium pharaonis sequence contained a putative envelope glycoprotein and a putative virion membrane protein of plant and insect viruses (η and ν in Fig. 2). Both genes were recently described in the RNA2 of Cilevirus and in Negev virus (Nelorpivirus) as well as in the unrelated Chronic Bee paralysis virus (Kuchibhatla et al. 2014). The other ORFs could not be associated to specific functions in public databases. However, reciprocal BLASTP analyses (e-value threshold = 10−3) within the dataset revealed thirty-nine ORFs could be clustered into twelve homologous groups (detailed by Greek alphabet in Fig. 2), the ten remaining ORFs were ORFans. Signal peptides towards the secretory pathway were observed at the 5′ end of accessory ORFs of eleven viruses including CpATV as well as in ORF2 of the TSA Bactocera dorsalis viral sequence targeting the mitochondrion compartment.
The Boutonnet virus (GenBank KU754539) and a virus-like sequence from TSA Musca domestica comp20588_c0 transcribed RNA sequence (GenBank GARN01041480) have similar genomic organization to CpATV (Fig. 2). However, their ORF3 did not possess the MSP7_C domain, or the 235kDa-fam domain similarities found in CpATV. The genome of CpATV, lacking the methyltransferase domain, is shorter than those of these two viruses. This likely represents the true biological size of CpATV, as the absence of the methyltransferase domain in the GA35C and GA35E mosquito transcriptomes was confirmed by BLASTN using the 5′ end of the Boutonnet virus ORF1 as query against mosquito transcriptomes.
3.5 Replicase phylogeny
Phylogenetic analyses were performed to determine the relationships between CpATV and other viruses close to the Virgaviridae (King et al. 2012a,b), including fifteen new insect viruses recently discovered (Webster et al. 2015,, 2016; Li et al. 2017). Tymovirales (Gammaflexiviridae, Betaflexiviridae, Alphaflexiviridae, and Tymoviridae) and Benyviridae were used as outgroup in order to resolve the relationships between the new viruses and unassigned genera such as Cilevirus, Higrevirus, Nelorpivirus, and Sandewavirus as well as the Virgaviridae and the Bromoviridae. As the helicase and RdRp domains had congruent phylogenetic signals (Helicase: p-SH = 0.115; p-1sKH = 0.097; RdRp: p-SH = 0.499; p-1sKH = 0.181), their alignments were concatenated into a 979 aa alignment for increased resolving power.
Maximum likelihood phylogenetic analyses, using beet necrotic yellow vein virus (Benyviridae) sequence as outgroup, showed the Tymovirales formed an early-diverging clade at the root of the tree (Fig. 3a). All remaining sequences formed a highly supported monophyletic group (aLRT support =1, here called superclade), which based on branch length is as genetically diversified as the order Tymovirales. At the base of the superclade, the sixteen new virus sequences, including CpATV formed eight early-diverging clades. All these viruses are associated with insects and none are closely related to characterized plant viruses (Virgaviridae, Bromoviridae, Closteroviridae, Idaeovirus, as well as Cilevirus and Higrevirus) or described mosquito-infecting viruses (Sandewavirus and Nelorpivirus, grouped under the term negeviruses), which are not monophyletic unlike previously observed (Kallies et al. 2014). CpATV clustered with the Boutonnet virus, the TSA Musca domestica sequence and ASV1 (Adelphocoris suturalis-associated virus 1) into clade 8 with high support 0.94. The use of appropriate outgroups in the phylogenetic analyses clearly showed the sixteen new insect viruses did not evolved from a recent common ancestor with Nelorpivirus and Sandewavirus clades, and thus belong to different viral genera and families, even though they also infect mainly dipteran hosts.
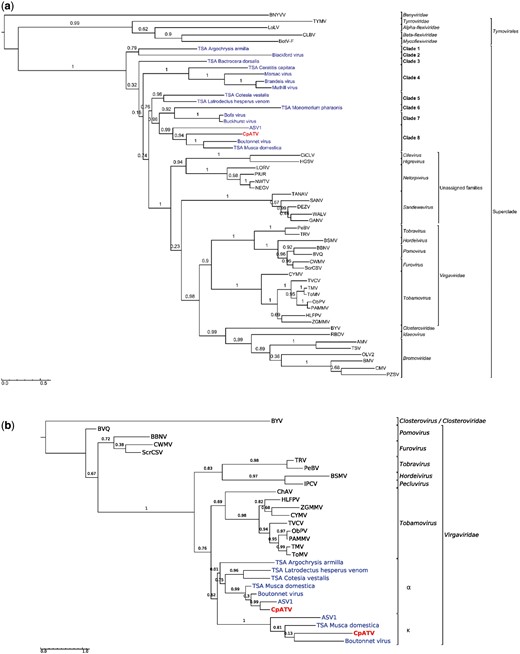
Maximum likelihood phylogenies of conserved protein domains in the Culex pipiens associated tunisia virus. (a) Concatenated helicase and RNA-dependent RNA polymerase domains from ORF1 (979 aa, substitution model LG + G). (b) Capsid domains from ORF2 and ORF4 (203 aa, substitution model LG + I+G). Taxon names represent virus acronyms. In red: CpATV; in blue: new viruses from Webster et al (2016). The scale bar represents the substitutions rate per site. aLRT statistics are indicated above nodes. Sequences details and GenBank accessions numbers are available in Supplementary Table S6.
3.6 Phylogeny of CpATV capsids
Both CpATV TMV coat capsids, as well as the seven other homologous capsids identified in the new insect virus dataset were aligned to TMV coat capsid proteins from the Virgaviridae and Closterovirus. The capsid phylogeny (Fig. 3b) was incongruent with the RdRp tree (Fig. 3a) as all nine insect-virus capsids fell inside the Virgaviridae clade instead of forming the ancestral lineages of the superclade (Fig. 3b). The insect virus capsids formed two distinct evolutionary lineages. The first group of sequences corresponded to homologs of CpATV ORF4 (α in Fig. 2), and encompassed ORF4 of the three closely related viruses Boutonnet virus, TSA Musca domestica and ASV1, as well as ORF2 of TSA Cotesia vestalis, and ORF3 of TSA Latrodectus hesperus venom and TSA Argochrysis armilla. The topology of this group reflected that of RdRp tree, suggesting a single capsid acquisition event from a Virgaviridae ancestor. The second group of sequence comprised the homologs of CpATV ORF2 (κ in Fig. 2), and only included CpATV, Boutonnet virus, TSA Musca domestica and ASV1. This second lineage also derived from a Virgaviridae ancestor, and the topology suggested that this four κ sequences derived from a single acquisition event. Altogether, our results suggest at least that two independent capsid acquisition events (α and κ) occurred in some of the new insect viruses. As the Virgaviridae, like most ancestral virus of the superclade (TSA Argochrysis armilla, TSA Cotesia vestalis, and TSA Lactrodectum herpesum venom) possess a single TMV coat capsid (King et al. 2012a,b), this might represent the ancestral genome content and indicate that a second capsid acquisition (ORF κ) occurred later in the lineage of CpATV.
3.7 Molecular evolution of the CpATV_Ayed genome
Ratios of nonsynonymous over synonymous mutations (dN/dS) were used to estimate the selective pressures acting on the helicase, RdRp, and capsid conserved domains. All conserved domains identified in CpATV evolved under strong purifying selection with dN/dS values <0.1 (Table 1). Similar values were obtained for the entire clade of new insect viruses (clades 5, 6, 7, 8) as well as for the closely related infectious viruses’ clades. LRT test showed that all the conserved viral domains of CpATV have the same molecular evolutionary rates as closely related infectious viruses (Table 1, p-value >0.40). Altogether, these results showed that CpATV and the other new viruses discovered in silico are probably functional since they harbor the molecular evolution hallmarks of known infectious viruses.
dN/dS ratio estimations and LRT comparisons between CpATV and closely related viruses.
| Branch or subtree of interest | ORF | |||
|---|---|---|---|---|
| Helicase | RdRp | Capsids | Capsids | |
| ORF1 | ORF1 | α and κ (shared estimation) | α and κ (separate estimation) | |
| CpATV | 0.004 | 0.003 | 0.10 | ORF2 (κ): 0.10 |
| ORF4 (α): 0.08 | ||||
| New insect viruses | Clades 5, 6, 7, 8 | All sequences with α and κ domains | ||
| 0.01 | 0.008 | 0.01 | 0.01 | |
| Closely-related infectious viruses | Nelorpivirus, Sandewavirus, Higrevirus, Cilevirus | Virgaviridae | ||
| 0.004 | 0.006 | 0.02 | 0.02 | |
| p-valuea | 0.93 | 0.56 | 0.40 | 0.95 |
| Branch or subtree of interest | ORF | |||
|---|---|---|---|---|
| Helicase | RdRp | Capsids | Capsids | |
| ORF1 | ORF1 | α and κ (shared estimation) | α and κ (separate estimation) | |
| CpATV | 0.004 | 0.003 | 0.10 | ORF2 (κ): 0.10 |
| ORF4 (α): 0.08 | ||||
| New insect viruses | Clades 5, 6, 7, 8 | All sequences with α and κ domains | ||
| 0.01 | 0.008 | 0.01 | 0.01 | |
| Closely-related infectious viruses | Nelorpivirus, Sandewavirus, Higrevirus, Cilevirus | Virgaviridae | ||
| 0.004 | 0.006 | 0.02 | 0.02 | |
| p-valuea | 0.93 | 0.56 | 0.40 | 0.95 |
aLikelihood ratio test comparison with a null model assuming a common rate between the CpATV branch and branches of closely related infectious viruses and an alternative model assuming independent rates for CpATV and closely related infectious viruses. New insect viruses were excluded of the comparison.
dN/dS ratio estimations and LRT comparisons between CpATV and closely related viruses.
| Branch or subtree of interest | ORF | |||
|---|---|---|---|---|
| Helicase | RdRp | Capsids | Capsids | |
| ORF1 | ORF1 | α and κ (shared estimation) | α and κ (separate estimation) | |
| CpATV | 0.004 | 0.003 | 0.10 | ORF2 (κ): 0.10 |
| ORF4 (α): 0.08 | ||||
| New insect viruses | Clades 5, 6, 7, 8 | All sequences with α and κ domains | ||
| 0.01 | 0.008 | 0.01 | 0.01 | |
| Closely-related infectious viruses | Nelorpivirus, Sandewavirus, Higrevirus, Cilevirus | Virgaviridae | ||
| 0.004 | 0.006 | 0.02 | 0.02 | |
| p-valuea | 0.93 | 0.56 | 0.40 | 0.95 |
| Branch or subtree of interest | ORF | |||
|---|---|---|---|---|
| Helicase | RdRp | Capsids | Capsids | |
| ORF1 | ORF1 | α and κ (shared estimation) | α and κ (separate estimation) | |
| CpATV | 0.004 | 0.003 | 0.10 | ORF2 (κ): 0.10 |
| ORF4 (α): 0.08 | ||||
| New insect viruses | Clades 5, 6, 7, 8 | All sequences with α and κ domains | ||
| 0.01 | 0.008 | 0.01 | 0.01 | |
| Closely-related infectious viruses | Nelorpivirus, Sandewavirus, Higrevirus, Cilevirus | Virgaviridae | ||
| 0.004 | 0.006 | 0.02 | 0.02 | |
| p-valuea | 0.93 | 0.56 | 0.40 | 0.95 |
aLikelihood ratio test comparison with a null model assuming a common rate between the CpATV branch and branches of closely related infectious viruses and an alternative model assuming independent rates for CpATV and closely related infectious viruses. New insect viruses were excluded of the comparison.
3.8 Detection of CpATV in other NGS datasets
The CpATV sequence was screened by BLASTX search in our remaining twenty-one Culex transcriptomes, as well as in sixty arthropod and gastropod NGS datasets publicly available. Only one dataset (Culex pipiens individual GA35E) returned a significant hit and no other homology was found in the sixty arthropod and gastropod sequence dataset at the time of analysis. A complete viral sequence was found in the transcriptome of the Culex pipiens individual GA35E. This mosquito had been sampled in Jedaida, Tunisia (160 km from CpATV_Ayed) and six years later than the one in which CpATV_Ayed was detected (Supplementary Table S1). A mapping step on the CpATV_Ayed sequence as reference genome allowed the reconstruction of a 6,816 bp full-length genome. This second CpATV genome of the strain named Jedaida (CpATV_Jedaida) was covered by 42,924 good quality reads for a mean coverage of 320× (Supplementary Fig. 1b), whereas the mean coverage of GA35E individual transcriptome was 205×.
3.9 Comparison of the CpATV Ayed and Jedaida strains
Comparison of the CpATV_Ayed and CpATV_Jedaida consensus sequences showed twenty-seven mutations; four indels occurring in intergenic regions and twenty-three single-nucleotide polymorphism (SNPs) (Supplementary Table S5). One SNP occurred in intergenic regions and the remaining twenty-two were dispersed in ORFs 1, 3, and 4. Only six SNPs were nonsynonymous substitutions, but they did not occur in any of the identified functional domains (helicase, RdRp, capsids). This suggests that both strains may have the same activity. The genetic divergence between the Ayed and Jedaida strains excludes the possibility that cross-contamination from a single biological sample happened during RNA extraction, library construction or sequencing, and confirmed two independent CpATV discoveries in distinct mosquitoes sampled at different times and places.
3.10 Intra-host diversity
Deep sequence coverage of 1,322× for CpATV_Ayed and 320× for CpATV_Jedaida allowed the analysis of viral genetic variations within each mosquito host (ID GA35C and GA35E). Both CpATV strains displayed similar intra host genetic diversity with mean pi values of 1.0× 10−3 and 0.96× 10−3. Interestingly, two high frequency variants were observed.
A first SNP (83% G–17% A) at genome position 2021 was found only in CpATV_Ayed. In the Jedaida strain the Adenine was fixed at this position (100% A). This SNP, which is located within ORF1 outside the helicase and RdRp domains, caused an Asn-Ser amino acid replacement. Both amino acids share similar biochemical properties (polar, hydrophilic, neutral and relatively small), suggesting no major functional change associated with this SNP.
The second high frequency polymorphism was detected in both CpATV_Ayed and CpATV_Jedaida. The variants presented the insertion of a seventh Adenine in ORF3 between the positions 4449 and 4454 (CpATV_Ayed). This indel caused a frameshift resulting in a premature stop codon at position 4461 for CpATV_Ayed (Fig. 1a). As a result, two additional ORFs (ORF3A and ORF3B) could be predicted in the variants. ORF3A contained the full signal peptide, and ORF3B the full-length MSP7_C domain (Fig. 1a). Remarkably, this indel was observed at very similar frequencies of 19.2 and 21.5% mapped reads in CpATV_Ayed and CpATV_Jedaida, respectively.
3.11 RT-PCR detection of CpATV in wild mosquitoes
To confirm the real biological existence of CpATV in Culex pipiens, an RT-PCR detection of ORF1 of CpATV was conducted on ten individual mosquitoes from Jedaida population and twenty mosquitoes from Ayed population (Supplementary Fig. S5). After verification of the good cDNA quality by PCR amplification of the genomic ace-1 locus, PCR detection of CpATV indicated that three samples from Ayed population were positive for CpATV but none from Jedaida population. The specificity of the RT-PCR was confirmed by sequencing 2 PCR products from samples Ayed_15 and Ayed_19. After removing primer sequences, 801 nucleotides were aligned with CpATV_Ayed genome. Pairwise identity between Ayed_15 or Ayed_19 sequences and CpATV_Ayed was 99.9%. Both sequences displayed a clear double pick in the electrophoregram (A and G) at position 2,021 (Ayed genome as a reference), and CpATV Ayed-15 sequence showed a supplementary double pick (A and T) at position 2,079, reflecting intra-host polymorphism. This RT-PCR detection demonstrated that CpATV virus first discovered using bioinformatic tools only has a genuine biological existence.
4. Discussion
High throughput sequencing accelerates virus discovery (Paez-Espino et al. 2016). Recent metaviromic approaches on invertebrates, based on pools of several species, have shown the virosphere extends far beyond the current taxonomy of viruses (Webster et al. 2015; Shi et al. 2016). Here, we focused our analyses on the transcriptomes of individual mosquitoes to warrant virus–host interactions.
We discovered a new mosquito virus, named CpATV, through systematic bioinformatic search of individual transcriptomes. The 6.8-kb genome of CpATV contains four ORFs, which encode a RNA replicase with helicase and RdRp domains (ORF1), two capsids (ORF2 and 4), and an accessory gene (ORF3, λ) with similarities to Plasmodium domains. This genomic organization shows some similarities but also differences with that of Virgaviridae, ssRNA(+) viruses, to which CpATV is related. Within ORF1, the synteny between the helicase and replicase domains is highly conserved in all viruses investigated. However in contrast to all other genera, CpATV lacked a 5′ methyltransferase, protease or movement protein domains (King et al. 2012a,b; Vasilakis et al. 2013). This could indicate that the CpATV genome does not possess a covering 5′ cap, which is mainly synthetized by this protein (Decroly et al. 2011). Other ssRNA(+) viruses such as Picornaviridae or Calicivirus use other 5′-end protection such as IRES and/or a viral genome-linked protein (VPg) recruitment (Goodfellow et al. 2005), however, no such elements could be detected in CpATV. Regulatory elements normally found in Virgaviridae, such as IRES, poly(A) tail, and tRNA-like cis-regulatory elements (i.e. hairpin-type pseudoknots) could not be identified in CpATV. These features appear facultative in several unassigned genera. For example, the mosquito infecting Negev virus (Nelorpivirus) has a 30−34 nt Poly(A) tail (Vasilakis et al. 2013), a putative IRES structure in the 5′-UTR (Gorchakov et al. 2014) but not hairpin-type pseudoknot.
The accessory genome of CpATV is distinguishable from those of other described viruses. It contains two capsid genes (α and κ) as well as ORF λ, with similarities to rhoptry proteins and Plasmodium MSP7 domain. We found this accessory genome composition in only three others recently described insect virus sequences: Boutonnet virus, TSA Musca domestica virus and ASV1, which together with CpATV form clade 8 (Webster et al. 2015,, 2016; Li et al. 2017). Both capsid genes derive from Virgaviridae, which only contains one capsid gene.
The capsid tree topology shows distinct phylogenetic origins for α and κ that apparently derive from two independent acquisitions rather than a unique ancestral acquisition followed by a duplication event producing two paralogs. Both horizontal gene transfer events occurred well after the initial diversification of the Virgaviridae ancestors, but not recently since ORF α was acquired before the divergence of clade 1, 7, and 8. Of note, not all viruses within this large clade possess a TMV-like coat protein. This suggests frequent capsid losses occurred in this group, potentially linked to adaption to new hosts, as we observed several host switches between plants and insects in the superclade. Viral capsid proteins are known source of evolutionary innovations in ssRNA(+) virus as they are implicated in multiple functions such as virus infectivity, pathogenicity, virus movement, and transmission (Weber and Bujarski 2015).
ORF λ was found in all viruses from clade 8. It shows high similarities with Rhoptry proteins (reticulocyte-binding proteins) involved in the process of invasion of the red blood cells by Plasmodium (Counihan et al. 2013). Interestingly, only ORF λ of CpATV, the mosquito virus in the group, displayed similarities to the Merozoite Surface Protein 7 domain, which is involved in vertebrate red blood cells invasion by the malaria agent (merozoite) during the Plasmodium life cycle (Pachebat et al. 2001). The MSP7 domain is not normally associated with Rhoptry proteins. It could derive from horizontal gene transfer of Plasmodium MSP7 sequence within a mosquito host. Culex pipiens, in which CpATV was found, is a natural vector of the avian malaria Plasmodium relictum (Tate and Vincent 1934; Melrose 2002; Turell 2012). However BLAST search in the P. relictum genome and in other Plasmodium species did not reveal any MSP7 homologs. Alternatively, the CpATV MSP7 domain could result from convergent evolution. At the molecular level, this evolutionary process occurs when two proteins, coded by unrelated genes in unrelated organisms, are facing similar specific environmental conditions. Evolution can retain a similar tertiary structure in unrelated proteins, which fulfill the same function (e.g. catalytic activity of an enzyme). For example, the convergent orientation of the active site of serine and cysteine proteases independently retained the same catalytic triad in twenty enzyme superfamilies (Buller and Townsend 2013). The hypothesis of interaction of the MSP7 domain in CpATV ORF λ with vertebrate host cells should now be addressed through functional studies.
Together with the other sequences from clade 8, CpATV does not bare the hallmarks of any viral group currently recognized by the ICTV. Comparative genomics and phylogenetic analysis indicate these sequences should belong to a new viral family. In support of this proposal, there are several lines of evidence indicating CpATV is a genuine infectious virus. First CpATV was sampled two times in 2005 and 2011 in C. pipiens collected as larvae in different regions of Tunisia. In both cases, the larvae were reared in the lab to adulthood, so the infection was not lethal to the larvae. Both mosquitoes were part of a larger effort to sample different Culex populations, which were not infected. This indicates that the CpATV sequence is not fixed in Culex genomes and that CpATV infections were initiated in the field rather than in the lab. Furthermore, as the sequence coverage of the virus is six times higher than the average transcriptome of the host in the Ayed sample, this points towards ongoing CpATV replication in the mosquito.
As the CpATV genome includes complete ORFs with ‘missing ends’ and is covered by over one hundred reads, it matches the definition of a ‘coding complete’ (CC) viral genome, which fits the recommendations for the description of novel viruses as it allows proper identifications and characterizations of the viral proteome and accurate phylogenetic analyzes (Ladner et al. 2014). This virus seemed to have a restricted local distribution so far, since no traces of CpATV were found in other sequence database or in broader environmental samples databases. Over the thirty mosquitoes from screened by PR-PCR Ayed and Jedaida populations, three from the Ayed locality were positive to CpATV. Altogether, these results suggest that CpATV prevalence is relatively low, but larger targeted field sampling of mosquitoes could reveal its full extent and species host range. Both CpATV_Ayed and CpATV_Jedaida share 99.6% of nucleotide identity and display low intra-host nucleotide polymorphism (pi = 1.0× 10−3 and 0.96×10−3, respectively). Low polymorphism appears as a biological characteristic of CpATV since both estimations were independent and yet very similar. Taking the high mutation rate expected by error-prone RNA-dependent RNA polymerase of RNA viruses into account (Drake and Holland 1999), it is therefore likely that CpATV may have small population size or suffered from large, recent or recurrent bottlenecks during vertical transmission (Zwart and Elena 2015; Elena et al. 2001). It is thus possible that the two strain-specific SNPs observed at high frequency in the genomes of CpATV_Ayed and CpATV_Jedaida originated by genetic drift only. However, the biological meaning of maintaining the indel polymorphism found in ORF3 seems incompatible with genetic drift, as the same pattern was observed twice. The high frequency (circa 20%) of Adenine insertion observed in ORF3 is in addition not compatible with known Illumina error rates in seven bases homopolymers were 0.02–0.002% error rate is expected (Minoche, Dohm, and Himmelbauer 2011). This indel polymorphism rather seems the consequence of natural selection. The finer evaluation of the strength of selection pressure using either dN/dS estimations as a proxy of selective pressures or the positions of nonsynonymous polymorphisms on functional domains on the CpATV genome shows that all conserved domains are under strong purifying selection. The conservation of important protein sites suggests that both CpATV strains are fully functional and adapted to their hosts. These results strongly suggest that both Ayed and Jedaida CpATV strains are infectious viruses.
Phylogenetic analyses retraced the evolution of CpATV among other recently discovered ssRNA(+) insect viruses (Webster et al. 2016). CpATV does not cluster with the clade comprising the mosquito specific Negev virus (Nelorpivirus). It is most closely related to the Boutonnet virus within a larger group of new insect viruses, several of which are associated with Diptera. These viruses do not form a monophyletic group and are early-diverging in the phylogeny. They do not belong to the same clade as Virgaviridae/Bromoviridae/Closteroviridae/Idaeovirus and Nelorpivirus/Sandewavirus/Higrevirus/Cilevirus, but clearly form distinct lineages. Overall, phylogenetic relationships show frequent host shifts between insect and plant shaped the evolution of these viruses, as recently pointed out (Shi et al. 2016; Nunes et al. 2017). A more exhaustive screening using degenerated PCR primers on a broad insect sample is now required to gain knowledge of host range, prevalence and biodiversity of these viruses. It would also be interesting to look at the real replication competence of these new viruses in plants and insects, to determine if the hosts in which they were first found are vectors or final hosts. Both Sandewavirus and Nelorpivirus have been shown to replicate efficiently in mosquitoes, and are true dipteran viruses (Vasilakis et al. 2013). Similar analyzes should be done for the newly discovered viruses, but this implies lifting technical issues on virus isolation. Thanks to NGS sequencing and combined efforts of scientists to analyze sequence datasets, viral phylogenies will certainly gain in accuracy and the knowledge of the whole virosphere has never been so close.
It was recently proposed to integrate in the taxonomic classification viruses discovered solely through metagenomics (Simmonds et al. 2017). The current classification of RNA viruses is mostly based on the replicase phylogeny (Koonin 1991). The analyses showed that CpATV represents a new viral species that does not belong to any currently described virus families or unassigned genera. The other three sequences from clade 8 (Boutonnet virus, TSA Musca domestica, and ASV1) found in different insect species, also represent different viral species based on phylogenetic distances (from 46.1 to 30.2% nucleotide identity between each pair of viruses). Together these four viruses form a monophyletic group that corresponds to higher classification rank at least at the level of genus within a new family. Furthermore, our study enabled the distinction of seven other clades of insect viruses, also representing new families or genera. To sum up this study described a powerful methodology for high-throughput virus discovery using transcriptomic analysis, which completed by comparative analyses accelerates viral taxonomy inference.
Despite the peculiarities resulting in exploring individual transcriptomes for virus discovery (i.e. focus on RNA viruses only and a lower discovery rate compared with pooled hosts), this work demonstrated that transcriptomic sequence datasets are good material for new virus discovery when the aim is to understand genome evolution and phylogenetic relationships. However, quantification of virus biodiversity in a complex environment for comparative purposes would certainly require a more exhaustive detection method such as the deep sequencing of virus metagenomes.
Data availability
Reads used for this analysis originated from a previous study (Romiguier et al. 2014) on Culex hortensis (SRA accession nos SRX565078 and SRX565079), Culex torrentium (SRA accession no. SRX565090 and SRX565091), Culex pipiens (SRA accession no. SRX565080 to SRX565089) and from this study for Culex pipiens (SRA accession SRX1453901, SRX1453908, SRX1457280 to SRX1457282, SRX1457496, SRX1457498, and SRX1457500). Genome sequences of CpATV strains Ayed and Jedaida were deposited on GenBank under accession numbers MG457154 and MG457155, respectively. Partial ORF1 sequences of CpATV Ayed_15 and Ayed_19 were deposited on GenBank under accession numbers MG557616 and MG557617, respectively.
Supplementary data
Supplementary data are available at Virus Evolution online.
Authors’ contributions
P.G. and E.A.H. performed the design and the coordination of the study. C.A., M.W., and F.J. carried out the sample collection, performed RNA isolation, RT-PCR detection and sequencing. D.B. carried out the bioinformatics analysis. D.B., E.A.H., and P.G. analyzed the result and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by European Research Council (ERC) grants E.A.H. (ERC GENOVIR 205206). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. This work has been supported by a European Research Council (ERC) grant to Nicolas Galtier (ERC PopPhyl 232971).
Conflict of interest: None declared.
Acknowledgements
Data used in this work were partly produced through molecular genetic analysis technical facilities of the SFR ‘Montpellier Environnement Biodiversite’, thanks to Dr P. Clair (UM2-Montpellier GenomiX) and the ‘Plateau de Génotypage-séquençage’ (Institut des Sciences de l'Evolution – Centre Méditerranéen Environnement et Biodiversité). Analyses largely benefited from the ISEM computing cluster platform. We are also grateful to the Genotoul Bioinformatics Platform Toulouse Midi-Pyrenees for providing computing and storage resources. We would like to thank J. Thézé and A. Bézier for fruitful discussions, A. Rivero for sharing unpublished data of P. relictum and N. Galtier, M. Ballenghien, J. Romiguier, K. Belkhir, R. Dernat, J. Veyssier for help in data accessibility, M. Rivera for English improvement and two anonymous reviewers for their helpful comments.
References
Author notes
Present address: Université de La Réunion, UMR PIMIT (Processus Infectieux en Milieu Insulaire Tropical), INSERM U1187, CNRS 9192, IRD 249, Sainte-Clotilde, Ile de La Réunion, France.
Elisabeth A. Herniou, Philippe Gayral contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}