
Abstract
The WT1 gene encodes a transcription factor implicated in normal and neoplastic development. The purpose of this study was to evaluate the diagnostic utility of a commercial WT1 antibody on a variety of pediatric small round blue cell tumors (SRBCT). A mouse monoclonal antibody (clone: 6F-H2, DAKO) raised against the N-terminal amino acids 1–181 of the human WT1 protein was tested. Microscopic sections from 66 specimens were stained using an antigen retrieval protocol with trypsin. The tumors included peripheral neuroectodermal tumors (PNET/Ewing's), neuroblastomas, desmoplastic small round cell tumors (DSRCT), lymphomas, Wilms' tumors, and rhabdomyosarcomas (RMS). One RMS case was investigated by Western blot analysis and RT-PCR to confirm the antibody specificity. A strong cytoplasmic staining was demonstrated in all RMS (11/11). The Western blot analysis confirmed the WT1 protein in the tissue, and the RT-PCR confirmed the presence of WT1 mRNA in the peripheral blood and tissue of one RMS patient. The Wilms' tumors had a variable nuclear and/or cytoplasmic positivity in most (17/24) cases. All PNET/Ewing's were negative. The nuclei of two lymphoblastic lymphomas stained strongly. A weak nuclear or cytoplasmic staining was reported in a few DSRCT (3/5), lymphomas (2/10), and neuroblastomas (2/8). This is a useful antibody in the differentiation of RMS from other SRBCTs. A strong cytoplasmic staining favors an RMS, and a strong nuclear staining is suggestive of a Wilms' tumor. A role for WT1 in the pathogenesis of rhabdomyosarcomas is raised. The limited sampling precludes any conclusions regarding the value of tissue or peripheral blood analysis for WT1 mRNA in patients with rhabdomyosarcoma.
Similar content being viewed by others

INTRODUCTION
The WT1 gene (1) encodes a protein with four zinc fingers of the Kruppel-type in the C-terminal region that recognizes a guanidine-cytidine (GC)–rich “EGR1” consensus sequence (2) required in tissue differentiation and proliferation (2, 3, 4). The N-terminal half contains a large proline-glutamine–rich domain important for inhibition of transcriptional activation (5, 6). There are at least eight protein isoforms ranging between 52 and 62 kDa in mammals produced by a combination of alternative splicing and RNA editing (4, 7).
The WT1 proteins are normally expressed in the nuclei of glomerular podocytes and mesothelial cells. It has also been demonstrated in stem cells bearing the CD34+ phenotype (8). The role of WT1 in normal human development also extends to a diversity of mammalian mesodermal tissues (9), including the body-wall musculature in a 13.5-days postconception (dpc) mouse embryo (43–49 dpc human). Embryologic studies of wt1-null mice reveal a failure to develop kidney and gonads (10). Mutations and splicing disruptions of WT1 have been described in Denys-Drash (11, 12, 13, 14), WAGR (15), and Frasier (13, 16) syndromes.
These proteins were first recognized as tumor suppressors. However, an activator or oncogenic behavior may be acquired by missense mutations. WT1 expression has been demonstrated in hematological malignancies (17, 18, 19, 20, 21, 22), mesothelial-derived neoplasms (23, 24, 25, 26, 27), breast cancer (28, 29), genitourinary tumors (30, 31), and small round blue cell tumors (SRBCT; 24, 27, 32, 33, 34). Recent studies have evaluated the possible role of peripheral blood RNA (18, 19), serum antibodies (35), and immunotherapy (36, 37) in the diagnosis, monitoring, and treatment of WT1-positive tumors.
The microscopic evaluation of an SRBCT can be difficult, especially in a small biopsy specimen. The diagnosis often relies on clinical history, laboratory analysis, and ancillary studies on the tissue (38, 39, 40). Immunohistochemistry has become one of the most important techniques in the interpretation of these biopsies, and the diagnostic work-up may include antibodies against mesenchymal, epithelial, neuronal, lymphoid, and myogenic antigens.
The WT1 antibodies have become recently available for immunohistochemical use on paraffin-embedded tissues, and a few reports have addressed the diagnostic utility in the differentiation of tumors (23, 24, 27, 30, 32). It has been our experience that the WT1 N-terminal antibody (clone 6F-H2) produces a different pattern of staining in SRBCT when compared with previous studies using the C-terminal antibody (24, 27, 32). This observation led us to evaluate the utility of the WT1 N-terminal antibody (6F-H2) in a cohort of pediatric patients diagnosed with SRBCT.
MATERIALS AND METHODS
Specimens
Sixty-six formalin-fixed and paraffin-embedded tumor biopsy specimens were retrieved from the files at the Children's Hospital of Philadelphia. These specimens included 8 peripheral neuroectodermal tumors (PNET/Ewing's), 8 neuroblastomas, 5 desmoplastic small round blue cell tumors (DSRCTs), 10 lymphomas, 24 Wilms' tumors, and 11 rhabdomyosarcomas (RMS). The lymphomas included seven lymphoblastic, three Burkitt's, and two large cell (one anaplastic). Five Wilms' tumors contained areas of heterologous differentiation (muscle or bone). The rhabdomyosarcomas included four embryonal, six alveolar, and one paratesticular spindle cell tumor. Supporting immunohistochemical stains included the following: MIC2, O13, NSE, CD56, AE1, AE3, desmin, LCA, CD20, UCHL-1, CD30, ALK-1, EMA, and muscle-specific actin. Molecular translocation studies (PAX3/PAX7-FKHR, EWS-FLI1/ERG, and EWS-WT1) were performed as indicated to confirm the diagnosis.
Immunohistochemistry
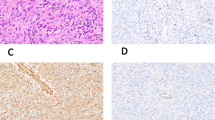
The WT1 antibody was a mouse monoclonal (clone 6F-H2; DAKO Corporation, Carpinteria, CA) antibody raised against the N-terminal 1–181 amino acids of human WT1 (41). The antibody was freshly diluted at 1:50 in phosphate buffered saline (PBS) every time that it was used. The antigen retrieval protocol required trypsin digestion for 20 minutes in a 37° C oven. The antibody was detected by a standard avidin–biotin method. Normal kidney tissue (Fig. 1A) and an 8-week-old human fetus (Fig. 1, B–D) were used as controls. The results were graded (Table 1) as 0 if negative or as weak (+1) or strong (+2) if positive in a nuclear (N) or cytoplasmic (C) pattern.

WT1 antibody (6F-H2) immunohistochemistry. A, normal kidney positive podocytes; B, 8-week-fetus intestinal wall with positive mesothelial cells; C, 8-week-fetus hand negative skeletal muscle staining; D,Wilms' tumor with epithelial and blastemal nuclear positivity; E, Wilms' tumor with muscle differentiation showing strong cytoplasmic reactivity; F, rhabdomyosarcoma with strong cytoplasmic expression.
Western Blot Analysis
The cell line 293T composed of transformed kidney epithelial cells were transiently transfected with an expression construct encoding human WT-1, isoform B (pCDNA3-WT1B, kindly provided by Dr. Daniel Haber, Massachusetts General Hospital Cancer Center) using calcium phosphate precipitation. After 48–72 hours, cells were rinsed one time with cold PBS and harvested by scraping. Whole-cell lysates were prepared from 293T cells, a Wilms' tumor, and a rhabdomyosarcoma sample using lysis buffer (1% NP-40, 150 mm NaCL, 50 mm Tris, pH 7.4) containing protease inhibitors. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose. After blocking in 5% bovine serum albumin–TBST for 1 hour at room temperature, Western blot analysis was performed using either anti-WT1 (clone 6F-H2; DAKO, Carpinteria, CA) or anti-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies, according to standard protocols.
Reverse Transcription Polymerase Chain Reaction
Tumor tissue and peripheral blood from a patient with RMS was snap-frozen in liquid nitrogen and stored at −70° C until the time of testing. The tissue was homogenized and washed twice in PBS. Approximately 10 million cells were lysed with 1 mL of Trizol reagent, and RNA was isolated according to the manufacturer's protocol. One microgram of total RNA from each sample was reverse transcribed into cDNA according to a standard protocol (Perkin Elmer Biosystems, Santa Clara, CA). Reverse transcription (RT) polymerase chain reaction (PCR) amplification of WT1 was performed using the following primers: 5′- GGCATCTGAGACCAG TGAGAA-3′ (outer sense), 5′ GAGAGTCAG ACTTGAAAGCAGT-3′ (outer antisense), 5′-GCT GTCCCACTTACAGATGCA-3′(inner sense), and 5′TCAAAGCGCCAGCTGGAGTTT-3′ (inner antisense). The first round of PCR was carried out with 30 amplification cycles, followed by a second round of 30 cycles with a DNA thermal cycler (R480, Perkin Elmer). PCR products were separated in 1.5% of agarose gel. The RNA obtained from K562 cell line and from a known patient with acute lymphoblastic leukemia were used as positive controls. The ratio of WT1 and human glyceraldehyde 3-phosphate dehydrogenase mRNA was used as an indication of the integrity of mRNA in the samples.
RESULTS
Positive and negative controls stained as expected with the WT-1 N-terminus antibody. Mature glomerular podocytes (Fig. 1A) and fetal mesothelial cells (Fig. 1B) nuclei were positive. Skeletal muscle from an 8-weeks' gestation fetus did not stain (Fig. 1C).
All (11/11) rhabdomyosarcomas revealed a strong cytoplasmic staining (Fig. 1F). This staining specificity was confirmed by Western blot analysis (Fig. 2). A band of 52 kDa from the RMS cell lysate comigrated with the transfected WT-1 (293/WT-1B) and the endogenous WT-1 (293, Wilms' tumor). Also, the RT-PCR (Fig. 3) from the RMS tissue (1:50 and 1:100 dilutions) and from the nondiluted peripheral blood (1:1) obtained from the same patient revealed a distinct and strong band that comigrated with the known controls (K562 and acute lymphoblastic leukemia). The diluted peripheral blood (1:100) revealed a very weak band.

Western blot gel of WT-1 in a rhabdomyosarcoma (RMS) tumor specimen. A band of 52 kDa that comigrates with either transfected WT-1 (293/WT-1B) or endogenous WT-1 (293, Wilms' tumor) is detected in the RMS sample. Equal amounts of protein are loaded as indicated by similar levels of tubulin expression.

WT1 RT-PCR from a patient with rhabdomyosarcoma (RMS): peripheral blood (PB; at 1:1 and 1:100 dilution) and RMS tissue (1:50 and 1:100) comigrating with controls (acute lymphoblastic leukemia patient and K562 cell line).
The Wilms' tumors stained in the nucleus and/or the cytoplasm of the epithelial, blastemal, and/or stromal cells (Table 2). The nuclear positivity (Fig. 1D) was always confined to the epithelial–blastemal elements and was noted in 54% (13/24) of the cases. The cytoplasmic pattern was seen in 75% (18/24) of cases and was almost exclusively stromal and weak (+1). All (5/5) Wilms tumors with heterologous (muscle) differentiation were strongly (+2) positive in a cytoplasmic pattern in the differentiated areas (Fig. 1E).
The remaining SRBCTs had a variable and mostly weak or negative staining (Table 2). The nuclei of two lymphoblastic lymphomas (20%) stained strongly. The weakly positive tumors included two neuroblastomas (25%), three DSRCTs (60%), and two lymphomas (20%). The latter included the cytoplasm of one Burkitt's lymphoma and the cytoplasm of one anaplastic large cell lymphoma. All (8/8) PNET/Ewing's lymphomas were negative.
DISCUSSION
This represents the first report on the diagnostic utility of a WT1 antibody (6F-H2) raised against the N-terminal portion of this protein in a large cohort of pediatric SRBCTs. The most intriguing finding of this analysis was the detection of WT1 protein in the cytoplasm of rhabdomyosarcoma cells supporting a possible linkage between Wilms' tumors, myogenic differentiation, and rhabdomyosarcomas (42, 43, 44, 45, 46, 47, 48, 49). Interestingly, rhabdomyogenesis is not uncommon in Wilms' tumors and correlates with a younger age and more favorable outcome. The literature also suggests that WT1 protein may be required for inhibition of skeletal muscle differentiation. However, the exact underlying molecular pathways for this association are still obscure.
A recent investigation (50) using cell lines derived from a panel of Wilms' tumors demonstrated that complete loss of WT1 function due to mutations could lead to muscle differentiation. In contrast, the larger cohort of Wilms' tumors from this same study was lacking WT1 gene mutations and rarely showed MyoD1, myosin heavy polypeptide 3 (MYH3), or myogenin (MYOG) expression. These findings in conjunction with in situ studies led to their conclusion that WT1 expression in the metanephric–mesenchymal stem cells of chick and mice kidneys probably inhibited skeletal muscle differentiation. Two other studies have correlated WT1 mutations (51, 52) with stromal-predominant Wilms' tumors. Also, lower levels of WT1 RNA transcripts have been described in Wilms' tumors with heterologous elements (53). Our molecular analysis of one RMS patient suggests that WT1 protein is not truncated.
The cytoplasmic staining in the rhabdomyosarcomas appears to be a true positive finding based on our molecular results. In fact, a similar strong cytoplasmic staining pattern has been previously described in two nephroblastomas within areas of desmin positivity using the same antibody (31). In contrast, previous immunohistochemical reports also described a weak cytoplasmic positivity with WT1 (C-terminus) antibodies in mesotheliomas (23, 25), leukemias (20), and SRBCT (24) raising the possibility of an artifact of fixation (24, 31, 54). Some of the tumors we examined (neuroblastomas, DSRCT, lymphomas, and Wilms' tumors) had a weak cytoplasmic reactivity and nonspecific staining could not be excluded. The lack of any positivity in the 8-week-old fetal skeletal muscle (Fig. 1C) and the expected nuclear pattern in mesothelial cells and podocytes further supports the specificity of the antibody (6F-H2) used in our study.
The cytoplasmic detection of WT1 is not surprising because many transcription factors are synthesized and reside in the cytoplasm in an inactive form. Activation of these factors by phosphorylation may be an essential mechanism for the nuclear translocation from the cytoplasm. Interestingly, WT1 appears to have potential phosphorylation sites in the C-terminal domain of the protein inhibiting its ability to bind DNA, but not RNA transcripts. In one study (55), a full-length isoform of the WT1 protein was phosphorylated by kinase A (PKA) leading to its translocation from the nucleus to the cytoplasm of 197/200 C2-transfected cells. The latter was documented by immunofluorescence with a C19 (C-terminal) polyclonal antibody. Another study (56) suggested that WT1 could be sequestered along with p53 as a cytoplasmic body in adenovirus-transformed kidney cells. However, the degree of expression noted in our tumors appeared to be quite different from the one described in the later report.
Another interesting aspect of this study was the weak positivity (+1) noted in the DSRCT contrasting with the previous literature (24, 32, 33). However, this apparent paradox may be explained by the underlying molecular pathology and the specificity of the antibodies used in previous reports. The DSRCT are characterized by EWS–WT1 translocation produced by the fusion of the first (7, 8, 9) exons of EWS and the last three exons of WT1. This fusion produces a protein containing the zinc finger region of WT1 that is recognized by antibodies directed to the C-terminal portion. In contrast, we used an antibody with specificity to the N-terminal region of the WT1 protein that is probably not or only partially expressed in the fusion protein of DSRCT. This would lead us to conclude that the mildly positive results (+1) noted in three of our DSRCT cases could be spurious. The reason and significance for the variable-staining pattern of WT1 in the Wilms tumors and lymphomas is not completely clear.
CONCLUSION
This study suggests that the WT1 antibody (6F-H2), when applied for immunohistochemistry studies in paraffin-embedded tissues, may be useful in the differentiation of rhabdomyosarcomas from other SRBCT in children, especially PNET/Ewing's tumors. Although the antibody may not clearly discriminate between the stromal cells of Wilms' tumors and RMS, this is a very uncommon diagnostic problem. These preliminary results also suggest that this antibody is useful across the spectrum of RMS and may not be limited to a rhabdomyosarcoma subtype or by the degree of differentiation, like myogenin and MyoD1 (57, 58, 59). The strong cytoplasmic expression appears to correlate with muscle differentiation and supports the idea that WT1 is deregulated in tumors showing this phenotype. The immunohistochemical findings support the hypothesis that the protein is stabilized in the cytoplasm and not functioning as a nuclear transcription factor in these tumors. Future molecular analysis may be of value to clarify this hypothesis. The limited sampling precluded any conclusions regarding the value of tissue or peripheral blood analysis for WT1 mRNA in patients with rhabdomyosarcoma.
References
Gessler M, Konig A, Bruns GAP . The genomic organization and expression of the WT1 gene. Genomics 1992; 12: 807–813.
Haber DA, Englert C, Maheswaran S . Functional properties of WT1. Med Pediatr Oncol 1996; 27: 453–455.
Moore AW, Schedl A, McInnes L, Doyle M, Hecksher-Sorensen J, Hastie ND . YAC transgenic analysis reveals Wilms' tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech Dev 1998; 79: 169–184.
Scharnhorst V, Dekker P, van der Eb AJ, Jochemsen AG . Internal translation initiation generates novel WT1 protein isoforms with distinct biological properties. J Biol Chem 1999; 274: 23456–23462.
Englert C . WT1—more than a transcription factor? Trends Biochem Sci 1998; 23: 389–393.
Reddy JC, Morris JC, Wang J, English MA, Haber DA, Shi Y, et al. WT1-mediated transcriptional activation is inhibited by dominant negative mutant proteins. J Biol Chem 1995; 270: 10878–10884.
Hastie ND . The genetics of Wilms' tumor—a case of disrupted development. Annu Rev Genet 1994; 28: 523–558.
Baird PN, Simmons PJ . Expression of the Wilms' tumor gene (WT1) in normal hemopoiesis. Exp Hematol 1997; 25: 312–320.
Armstrong JF, Pritchard-Jones K, Bickmore WA, Hastie ND, Bard JB . The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech Dev 1992; 40: 85–97.
Kreidberg JA . Gene targeting in kidney development. Med Pediatr Oncol 1996; 27: 445–452.
Denamur E, Bocquet N, Mougenot B, Da Silva F, Martinat L, Loirat C, et al. Mother-to-child transmitted WT1 splice-site mutation is responsible for distinct glomerular diseases. J Am Soc Nephrol 1999; 10: 2219–2223.
Machin GA . Atypical presentation of Denys-Drash syndrome in a female with a novel Wt1 gene mutation. Birth Defects Orig Artic Ser 1996; 30: 269–286.
Poulat F, Morin D, Konig A, Brun P, Giltay J, Sultan C, et al. Distinct molecular origins for Denys-Drash and Frasier syndromes. Hum Genet 1993; 91: 285–286.
Yang Y, Jeanpierre C, Dressler GR, Lacoste M, Niaudet P, Gubler MC . WT1 and PAX-2 podocyte expression in Denys-Drash syndrome and isolated diffuse mesangial sclerosis. Am J Pathol 1999; 154: 181–192.
Pelletier J, Bruening W, Li FP, Haber DA, Glaser T, Housman DE . WT1 mutations contribute to abnormal genital system development and hereditary Wilms' tumour. Nature 1991; 353: 431–434.
Klamt B, Koziell A, Poulat F, Wieacker P, Scambler P, Berta P, et al. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/−KTS splice isoforms. Hum Mol Genet 1998; 7: 709–714.
Im HJ, Kong G, Lee H . Expression of Wilms' tumor gene (WT1) in children with acute leukemia. Pediatr Hematol Oncol 1999; 169: 109–118.
Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, et al. Aberrant overexpression of the Wilms' tumor gene (WT1) in human leukemia. Blood 1997; 89: 1405–1412.
Menssen HD, Renkl HJ, Rodeck U, Maurer J, Notter M, Schwartz S, et al. Presence of Wilms' tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 1995; 9: 1060–1067.
Menssen HD, Renkl HJ, Rodeck U, Kari C, Schwartz S, Thiel E . Detection by monoclonal antibodies of the Wilms' tumor (WT1) nuclear protein in patients with acute leukemia. Int J Cancer 1997; 70: 518–523.
Miyagawa K, Hayashi Y, Fukuda T, Mitani K, Hirai H, Kamiya K . Mutations of the WT1 gene in childhood nonlymphoid hematological malignancies. Genes Chromosomes Cancer 1999; 25: 176–183.
Niegemann E, Wehner S, Kornhuber B, Schwabe D, Ebener U . wt1 gene expression in childhood leukemias. Acta Haematol 1999; 102: 72–76.
Amin KM, Litzky LA, Smythe WR, Mooney AM, Morris JM, Mews DJ, et al. Wilms' tumor 1 susceptibility (WT1) gene products are selectively expressed in malignant mesothelioma. Am J Pathol 1995; 146: 344–356.
Barnoud R, Sabourin JC, Pasquier D, Ranchere D, Bailly C, Terrier-Lacombe MJ, et al. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol 2000; 24: 830–836.
Kumar-Singh S, Segers K, Rodeck U, Backhovens H, Bogers J, Weyler J, et al. WT1 mutation in malignant mesothelioma and WT1 immunoreactivity in relation to p53 and growth factor receptor expression, cell-type transition, and prognosis. J Pathol 1997; 181: 67–74.
Park S, Schalling M, Bernard A, Maheswaran S, Shipley GC, Roberts D, et al. The Wilms tumour gene WT1 is expressed in murine mesoderm-derived tissues and mutated in a human mesothelioma. Nat Genet 1993; 4: 415–420.
Thorner P, Squire J, Plavsic N, Jong R, Greenberg M, Zielenska M . Expression of WT1 in pediatric small cell tumors: report of two cases with a possible mesothelial origin. Pediatr Dev Pathol 1999; 2: 33–41.
Loeb DM, Evron E, Patel CB, Sharma PM, Niranjan B, Buluwela L, et al. Wilms' tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res 2001; 61: 921–925.
Silberstein GB, Van Horn K, Strickland P, Roberts CT Jr, Daniel CW . Altered expression of the WT1 Wilms tumor suppressor gene in human breast cancer. Proc Natl Acad Sci U S A 1997; 94: 8132–8137.
Goldstein NS, Bassi D, Uzieblo A . WT1 is an integral component of an antibody panel to distinguish pancreatobiliary and some ovarian epithelial neoplasms. Am J Clin Pathol 2001; 116: 246–252.
Satoh F, Tsutsumi Y, Yokoyama S, Osamura RY . Comparative immunohistochemical analysis of developing kidneys, nephroblastomas and related tumors: considerations on their histogenesis. Pathol Int 2000; 50: 458–471.
Hill DA, Pfeifer JD, Marley EF, Dehner LP, Humphrey PA, Zhu X, et al. WT1 staining reliably differentiates desmoplastic small round blue cell tumor from Ewing sarcoma/primitive neuroectodermal tumor. An immunohistochemical and molecular diagnostic study. Am J Clin Pathol 2000; 114: 345–353.
Charles AK, Moore IE, Berry PJ . Immunohistochemical detection of the Wilms' tumour gene WT1 in desmoplastic small round cell tumor. Histopathology 1997; 30: 312–314.
Ramani P . Cowell JK . The expression pattern of Wilms' tumour gene (WT1) product in normal tissues and paediatric renal tumours. J Pathol 1996; 179: 162–168.
Gaiger A, Carter L, Greinix H, Carter D, McNeill PD, Houghton RL, et al. WT1-specific serum antibodies in patients with leukemia. Clin Cancer Res 2001; 7(3 Suppl): 761s–765s.
Sugiyama H . Wilms' tumor gene WT1: its oncogenic function and clinical application. Int J Hematol 2001; 73: 177–187.
Tsuboi A, Oka Y, Ogawa H, Elisseeva OA, Li H, Kawasaki K, et al. Cytotoxic T-lymphocyte responses elicited to Wilms' tumor gene WT1 product by DNA vaccination. J Clin Immunol 2000; 20: 195–202.
Bayani J, Thorner P, Zielenska M, Pandita A, Beatty B, Squire JA . Application of a simplified comparative genomic hybridization technique to screen for gene amplification in pediatric solid tumors. Pediatr Pathol Lab Med 1995; 15: 831–844.
Peter M, Gilbert E, Delattre O . A multiplex real-time PCR assay for the detection of gene fusions observed in solid tumors. Lab Invest 2001; 81: 905–912.
Thorner PS, Squire JA . Molecular genetics in the diagnosis and prognosis of solid pediatric tumors. Pediatr Dev Pathol 1998; 1: 337–365.
Rauscher FJ III, Morris JF, Fredericks WJ, Lopez-Guisa J, Balakrishnan C, Jost M, et al. Characterization of monoclonal antibodies directed to the amino-terminus of the WT1, Wilms' tumor suppressor protein. Hybridoma 1998; 17: 191–198.
Besnard-Guerin C, Newsham I, Winqvist R, Cavenee WK . A common region of loss of heterozygosity in Wilms' tumor and embryonal rhabdomyosarcoma distal to the D11S988 locus on chromosome 11p15.5 Hum Genet 1996; 97: 163–170.
Folpe AL, Patterson K, Gown AM . Antibodies to desmin identify the blastemal component of nephroblastoma. Mod Pathol 1997; 10: 895–900.
Garvin AJ, Surrete F, Hintz DS, Rudisill MT, Sens MA, Sens DA . The in vitro growth and characterization of the skeletal muscle component of Wilms' tumor. Am J Pathol 1985; 121: 298–310.
Gonzalez-Crussi F, Hsueh W, Ugarte N . Rhabdomyogenesis in renal neoplasia of childhood. Am J Surg Pathol 1981; 5: 525–532.
Gough DC . Wilms' tumour and genitourinary rhabdomyosarcoma. Br J Urol 1989; 63: 109–116.
Marcus R, Perez-Atayde AR . Unique dermal and subcutaneous botryoid rhabdomyosarcoma associated with mature renal tissue: is this an extrarenal Wilms' tumor? Pediatr Pathol 1994; 14: 617–625.
Steenman M, Tomlinson G, Westerveld A, Mannens M . Comparative genomic hybridization analysis of hepatoblastomas: additional evidence for a genetic link with Wilms tumor and rhabdomyosarcoma. Cytogenet Cell Genet 1999; 86: 157–161.
Vawter GF . Anaplastic Wilms' tumor with rhabdomyosarcomatous differentiation. Pediatr Pathol 1991; 11: 177–182.
Miyagawa K, Kent J, Moore A, Charlieu JP, Little MH, Williamson KA, et al. Loss of WT1 function leads to ectopic myogenesis in Wilms' tumour. Nat Genet 1998; 18: 15–17.
Pritchard-Jones K, Fleming S . Cell types expressing the Wilms' tumour gene (WT1) in Wilms' tumours: implications for tumour histogenesis. Oncogene 1991; 6: 2211–2220.
Schumacker V, Schneider S, Figge A, Wildhardt G, Harms D, Schimdt D, et al. Correlation of germ-line mutations and two-hit inactivation of the WT1 gene with Wilms tumors of stromal-predominant histology. Proc Natl Acad Sci USA 1997; 94: 3972–3977.
Gerald WL, Gramling S, Sens DA, Garvin J . Expression of the 11p13 Wilms' tumor gene, WT1, correlates with histologic category of Wilms' tumor. Am J Pathol 1992; 140: 1031–1037.
Loke SL, Neckers LM, Schwab G, Jaffe ES . c-myc protein in normal tissue. Effects of fixation on its apparent subcellular distribution. Am J Pathol 1988; 131: 29–37.
Ye Y, Raychaudhuri B, Gurney A, Campbell CE, Williams BR . Regulation of WT1 by phosphorylation: inhibition of DNA binding, alteration of transcriptional activity and cellular translocation. EMBO J 1996; 15: 5606–5615.
Maheswaran S, Englert C, Lee SB, Ezzel RM, Settleman J, Haber DA . E1B 55K sequesters WT1 along with p53 within a cytoplasmic body in adenovirus-transformed kidney cells. Oncogene 1998; 16: 2041–2050.
Cessna MH, Zhou H, Perkins SL, Tripp SR, Layfield L, Daines C, et al. Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am J Surg Pathol 2001; 25: 1150–1157.
Kumar S, Perlman E, Harris CA, Raffeld M, Tsokos M . Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues. Mod Pathol 2000; 13: 988–993.
Cui S, Hano H, Harada T, Takai S, Masui F, Ushigome S . Evaluation of new monoclonal anti-MyoD1 and anti-myogenin antibodies for the diagnosis of rhabdomyosarcoma. Pathol Int 1999; 49: 62–68.
Acknowledgements
The authors gratefully acknowledge the assistance of Dr. Daniel Haber from the Massachusetts General Hospital Cancer Center and the technical support from the laboratory staff of the Children's Hospital of Philadelphia and the Chicago's Children's Memorial Hospital.
Author information
Authors and Affiliations
Corresponding author
Additional information
The preliminary findings of this study were presented at the Society for Pediatric Pathology meeting in New Orleans, Louisiana in the Spring of 2000 and thus published previously in abstract form (Carpentieri et al. Mod Pathol 2000;13:6P).
Rights and permissions
About this article
Cite this article
Carpentieri, D., Nichols, K., Chou, P. et al. The Expression of WT1 in the Differentiation of Rhabdomyosarcoma from Other Pediatric Small Round Blue Cell Tumors. Mod Pathol 15, 1080–1086 (2002). https://doi.org/10.1097/01.MP.0000028646.03760.6B
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000028646.03760.6B
Keywords
This article is cited by
-
WT1 Alternative Splicing: Role of Its Isoforms in Neuroblastoma
Journal of Molecular Neuroscience (2017)
