
Abstract
The review discusses molecular and cellular mechanisms common to the temporal lobe epileptogenesis/epilepsy and depressive disorders. Comorbid temporal lobe epilepsy and depression are associated with dysfunction of the hypothalamic-pituitary-adrenocortical axis. Excessive glucocorticoids disrupt the function and impair the structure of the hippocampus, a brain region key to learning, memory, and emotions. Selective vulnerability of the hippocampus to stress, mediated by the reception of glucocorticoid hormones secreted during stress, is the price of the high functional plasticity and pleiotropy of this limbic structure. Common molecular and cellular mechanisms include the dysfunction of glucocorticoid receptors, neurotransmitters, and neurotrophic factors, development of neuroinflammation, leading to neurodegeneration and loss of hippocampal neurons, as well as disturbances in neurogenesis in the subgranular neurogenic niche and formation of aberrant neural networks. These glucocorticoid-dependent processes underlie altered stress response and the development of chronic stress-induced comorbid pathologies, in particular, temporal lobe epilepsy and depressive disorders.
Similar content being viewed by others


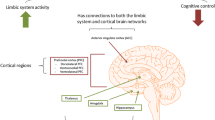
INTRODUCTION
Epilepsy is one of the most common neurological diseases, affecting ~70 million people worldwide. A heterogeneous disease associated with various etiological factors (genetic, infectious, traumatic, vascular, toxic, tumor, etc.), epilepsy is characterized by periodic spontaneous seizures caused by hyperexcitability and hypersynchronization of brain neurons. In almost a third of patients, epilepsy is pharmacoresistant. Epileptic brain is an example of chronic alterations of neuroplasticity (functional and structural changes in the brain allowing to adapt to environmental conditions, learn, remember information, and restore after damage), which is based on biochemical processes in synapses and other compartments of neurons [1].
According to the current definition, epilepsy it is not only a disorder accompanied by seizures, but also a disease that can often be associated with comorbid cognitive and mental disorders [2]. The prevalence of associated mental illness in patients with epilepsy is relatively high: one in three patients experiences a mental disorder (usually mood disorders and anxiety) during the lifetime. Comorbid mental diseases often precede the onset of seizures and affect the quality of life of patients and the course of epilepsy in several aspects. These include poorer tolerability of antiepileptic drug therapy, in particular, development of iatrogenic psychiatric symptoms after pharmacological and surgical treatment, increased mortality risk, and higher economic burden on the patient, family, and society as a whole. The problem seems so important that it is debated whether psychiatric comorbidities should be included in the classification of epilepsies [3].
Depression is one of the most disabling disease associated with epilepsy, and it has a profound negative impact on the quality of life of patients with epilepsy. Although known since the ancient times, the link between depression and epilepsy is still poorly understood. For a long time, depressive disorders were considered only as an expression of reactivity to the stressful factors in life of patients with epilepsy. However, epilepsy and associated mental illnesses have a complex relationship, which can be manifested by both their high comorbidity and bidirectional relationships between them. As a result, not only people with epilepsy are at a greater risk of developing mental disorders, but also patients with primary mental disorders are at a higher risk of developing epilepsy [4]. This relationship can only be explained by the existence of pathogenetic mechanisms shared by both diseases. Indeed, the data obtained over the past three decades have demonstrated biochemical, neuropathological and neurophysiological changes common to epilepsy and depression, confirmed by studies in animal models and involved in the development of mood disorders and seizures.
Among many different forms of epilepsy, the most common epileptic syndrome in adults is temporal lobe epilepsy (TLE). From 30 to 70% of patients with TLE have neuropsychiatric complications, most often depressive spectrum disorders. Comorbid mood disorders in patients with TLE include personality disorders (especially obsessive-compulsive disorders) and major depression, which are often manifested in drug-resistant (refractory, pharmacoresistant) TLE with the hippocampal (mesial temporal) sclerosis [5, 6]. The incidence of depression ranges from 20 to 55% in patients with refractory epilepsy, most of whom have TLE associated with the hippocampal sclerosis. Patients with refractory epilepsy are generally characterized by a frequent occurrence of dysfunctional personality patterns, these patterns differing depending on the epileptogenic zone [7, 8]. Interestingly, another risk factor for comorbid depression in TLE, in addition to pharmacoresistance, is gender (women suffer more, which is typical for depressive disorders in general) [9].
PATHOGENETIC CONCEPT OF COMORBIDITY OF EPILEPSY AND DEPRESSION
An existence of common pathogenetic mechanisms in primary psychiatric disorders and epilepsy has been postulated as one of the leading hypotheses to explain close and very complex relationship between these illnesses, and neurobiological characteristics of mood disorders and epilepsy have been analyzed to test this hypothesis [4]. The data obtained in animal models were used to suggest how the neurobiological aspects of mood disorders contribute to the epileptogenic process and to explain an increased risk of developing epilepsy, in particular, pharmacoresistant TLE, in patients with primary mood disorders. Analysis of these data confirmed the existence of common pathogenetic mechanisms functioning in both depressive (and, probably, anxiety) disorders and epilepsy, that could explain their relatively high comorbidity, their bidirectional relationship, and the worse course of convulsive disorder associated with a previous history of depression [2, 4]. The authors of this analysis rightly pointed out that the available data represent only a tip of the iceberg, and research in this area was just beginning.
Depressive-like behaviors, such as immobility in the forced swimming test as a manifestation of behavioral despair and anhedonia (lack of preference for sweetened water), were described in the absence epilepsy model (WAG/Rij rats) and in TLE models. The lithium-pilocarpine model of epileptic status followed by a period of epileptogenesis over several weeks is one of the most adequate TLE models, characterized by the brain (primarily, the hippocampus) damage and changes in neuroplasticity, similar to those observed in human TLE. Both epilepsy models cause the development of behavioral symptoms of despair, anhedonia, and changes in the serotonergic pathways of the hippocampus [10], as well as a number of other biochemical alterations. These changes directly or indirectly affect the hippocampus, including excessive activation of the hypothalamic-pituitary-adrenocortical system (HPACS) and pro-inflammatory processes.
EPILEPSY AS A MODEL OF CHRONIC STRESS
Depressive disorders are classified as stress-dependent diseases. The key role of various stress factors (chronic stress, early life stress) and alterations in stress reactivity in the pathogenesis of these diseases has been confirmed by numerous experimental [11-14] and clinical data [15-20] and is currently unquestionable. However, the role of stress factors in epileptogenesis and epilepsy has been studied to a much lesser extent and remains mostly obscure, as well as underestimated, both in scientific research and medicine. Neurobiological analysis of the clinical data, as well as the results of animal studies on the influence of various stress factors indicate that stress is fundamentally involved in the pathogenesis of epilepsy at all stages, at different levels, and through various mechanisms. In a sense, epilepsy itself can be considered as a model of chronic stress.
The stress hormones glucocorticoids (GCs, cortisol in humans, and corticosterone in laboratory rodents), are secreted into the blood as a result of the HPACS functioning and continuously affect the brain. Natural changes in their levels are mediated by the ultradian or circadian rhythms or can be caused by stressful situations [21]. Brain cells express 2 types of corticosteroid receptors (glucocorticoid, GR, and mineralocorticoid) that differ in regional distribution and affinity. These receptors mediate both fast non-genomic and slower genomic effects in neurons via membrane and cytoplasmic receptors, respectively. That is why natural, including stress-induced, changes in the level of GCs are associated with a complex mosaic of time-dependent and region-dependent modulations in brain neural activity. Such changes in the activity of brain cells and the corresponding neural networks lead to different shifts in cognitive and emotional patterns [22]. Immediately after stress (at the peak of GC concentration), the activity of the brain regions involved in emotional responses increases, followed activation of areas involved in higher cognitive functions. Since the hippocampus, a limbic structure with the maximum density of corticosteroid receptors, is essential for both cognitive function (dorsal/posterior hippocampus) and emotional response (ventral/anterior hippocampus), it is the regulation of the hippocampal function by stress hormones that becomes pivotal for the body’s response to stress [21, 23]. It is also important that unlike other brain structures, in the hippocampus, the basal membrane covers only 30% of the vascular surface [24], which facilitates hormone penetration into the neurons. Overcoming stress requires a balanced response during both phases of acute stress (at the peak GC concentration and during its subsequent decrease). However, chronic stress disturbs this balance, especially in genetically vulnerable individuals, which leads to more pronounced manifestation of the disease. This applies to mental disorders, as well as neurological diseases, in particular, epilepsy. Both stress response and seizure activity significantly depend on endogenous neurosteroids, which have an important neurophysiological activity. Neurosteroids interact with the glutamatergic, serotonergic, adrenergic, GABAergic systems, potential-dependent cationic and anionic channels, neurotrophin system, etc. Changes in neurosteroid levels may be associated with neuroinflammation and stress-related diseases, including depression, as well as with epilepsy [25].
It has become clear that stress factors are key components in the pathogenesis of early stages of epilepsy. Stress episodes in early pre-and postnatal ontogenesis are quite frequent and produce a range of negative effects on physical and cognitive status in later life. These clinical observations have been confirmed in experimental studies. One of the consequences of early stress is seizure activity. The underlying mechanisms remain poorly understood, although there is evidence of the involvement of the glutamatergic, GABAergic, and adrenergic systems [26].
Several approaches are used to investigate the association between epilepsy and stress in patients. When interviewed, patients with epilepsy often report triggering of seizures by stressful events, and many believe that it is stress that caused epilepsy. However, preclinical, epidemiological, and clinical studies of the association between stress and epilepsy provide rather contradictory results. The data of clinical studies are the most contradictory; however, the experimental results of preclinical studies mostly confirm that chronic stress exacerbates the manifestations of epilepsy, and, in some cases can cause it [27, 28]. The negative impact of depression on the control of convulsive disorders has been documented in various studies [29]. Most patients with epilepsy who report stress as an inducer of seizures have a history of anxiety, depression, or childhood trauma, as opposed to those who do not consider stress important for the onset of the attack. Most patients with stress-induced seizures who use self-reported stress reduction techniques find this approach effective for relieving seizures [30]. Despite the fact that the mechanisms behind the pro-epileptic effects of stress are still insufficiently studied, we believe that in order to formalize the analysis of biological mechanisms of epileptogenesis/epilepsy, this disease should be regarded as a model of chronic stress.
Stress factors cause the release of stress mediators, hormones and neurotransmitters that bind to receptors in various brain structures, primarily in the hippocampus, a region that is sensitive to stress and easily develops seizure activity. The interaction of stress mediators with receptors in the hippocampus affects the excitability of individual neurons and hippocampal neural circuits. It is important that, depending on many factors determining the functional status of the organism, mediators and their effects demonstrate unique spatiotemporal profiles determining the nature of complex combinatorial effects on the excitability of hippocampal networks and the generation of seizures [31]. Despite numerous clinical evidences that stress can be a triggering factor for the development of seizures in patients with epilepsy, the status of HPACS (which determines stress reactivity) in such patients, is poorly understood. Although the obtained data vary significantly (which may be partly due to the use of antiepileptic drugs), association of seizures with an increased stress load and elevated cortisol levels has been demonstrated in numerous studies [32]. At the same time, high frequency of seizures is associated with increased cortisol levels and lower functional connectivity of the brain. The severity of psychological distress, realized primarily in depressive and anxiety disorders, is significantly higher in epilepsy than in other diseases. Some researchers believe that these comorbid conditions contribute to quality of life decline in these patients even more than epilepsy itself [33]. It is obvious that the neurobiological basis of the relationship between psychiatric comorbidities and epilepsy is to a great extent mediated by stress, which is involved in both psychophysiological and pathophysiological mechanisms of these diseases.
As mentioned above, the most common comorbidity of epilepsy is depressive episodes. Depending on the cohorts studied, the occurrence of depressive episodes may exceed 50% [34]. Depression belongs to a group of diseases characterized by the patient’s inability to adequately cope with stressful loads. Manifestations of depression in epilepsy involve many interacting neurobiological and psychosocial determinants, including clinical manifestations of epilepsy (frequency and type of seizures, source and lateralization), neurochemical and iatrogenic mechanisms. The most obvious and reasonable (although, partial) explanation of depressive symptoms in epilepsy is that these symptoms stem from the chronic multifactorial stress. The uncertainty and unpredictability of seizures causes the feeling of despondency, loneliness, despair, low self-esteem and self-reproach, which can lead to social isolation, social stigmatization and powerlessness. Depression is often seen as a response to social stigma and the associated reduced quality of life; the incidence of depression and suicidality in patients with epilepsy is 4-5 times higher than in the healthy population [35]. Epilepsy is also often accompanied by the anxiety symptoms, which are frequently peri-ictal. These symptoms are classified according to the period of their manifestation (preictal, ictal, and postictal), although they are generally believed to occur independently of seizures. Similar to depression, there is a two-way pathophysiological link between anxiety disorder and epilepsy. In some patients, anxiety initiates seizures, which can be explained by the concepts of stress and arousal [36]. Seizures originating in limbic networks involving the amygdala may have a fear-related symptomatology.
Analysis of available data suggests that a wide variety of stress factors of different nature, acting in early ontogenesis or on adulthood, are epileptogenic and can initiate the development of epilepsy. These factors can be chronic and act through the course of epileptogenesis and the developed disease, or they can act once, but trigger molecular processes that become the basis for the pathogenesis of epilepsy and comorbid depression (these factors are discussed in detail in articles [27, 37, 38]). A patient with developed epilepsy is affected by a complex of chronic stress factors, psychological and physical. Indeed, comorbid depressive and anxiety disorders are the evidence for the chronization of persistent stress. The idea that epilepsy is a model of chronic stress is schematically represented in Fig. 1 that presents pro-epileptic stress factors acting during different periods of epilepsy development, and their potential relationship with the development of psychopathology, as well as shows groups of factors that determine predisposition to epilepsy, promote epileptogenesis, and precipitate seizures. At the same time, persistent disturbances of HPACS (excessive activation and subsequent depletion of HPACS at the stage associated with chronic depression [39]) are considered as key events that trigger the development of comorbid epilepsy and depression.

Shared factors of development of epilepsy and comorbid psychopathology (based on the data from [27, 29, 37, 40]). Groups of factors that determine predisposition to epileptogenesis, promote epileptogenesis, and precipitate seizures are indicated. Amy, amygdala; Hip, hippocampus; PFC, prefrontal cortex.
In uncontrolled chronic stress, hypercortisolemia (in animals, hypercorticosteronemia) becomes the most important component of stress-associated vicious circle of epilepsy/depression comorbidity based on stressful and neurohumoral factors [41]. A consequence of comorbid depression and TLE developing on the background of HPACS dysfunction is amplification of the effects of chronic stress factors mutually potentiating comorbid pathologies. The diseases themselves begin to act as stress factors (e.g., patient’s awareness of his/her condition as a result of the manifestation of negative symptoms of the diseases). Thus, a vicious stressor circle is formed, which contributes to the development and aggravation of both diseases as they develop (Fig. 2). This vicious circle is based on the fact that the HPACS controls both stress response and seizure activity, since stress hormones, primarily GCs, affect both the excitability of neurons and the sensitivity to seizures [42, 43].

The vicious circle of epilepsy/depression comorbidity based on stressful and neurohumoral factors. The consequence of comorbid depression and TLE associated with HPACS dysfunction is mutually reinforcing effects of stress factors that form a vicious circle contributing to the progress and aggravation of both diseases. Chronic stress (including stress associated with epilepsy) leads to HPACS dysfunction, which in turn, disturbs stress response, thus exacerbating this dysfunction and resulting in the development of depressive disorder. The developing depression further aggravates the dysfunction of HPACS, acting as an additional chronic stress factor for the patient. Stressful events induce seizure episodes and increase in their frequency via multiple mechanisms. In turn, more severe epilepsy promotes chronic stress and aggravates comorbid depression.
HIPPOCAMPAL MOLECULAR AND CELLULAR MECHANISMS UNDERLYING COMORBIDITY OF TEMPORAL LOBE EPILEPSY AND DEPRESSION
Stages of neuroplasticity changes in the hippocampus in TLE. Using as an example TLE, the most common and studied form of adult epilepsy affecting the structure and functioning of the hippocampus, it is possible to outline the sequence of changes in hippocampal neuroplasticity. The main stages which are characterized by the opposite changes in neuroplasticity, are active epileptogenesis and the stage of “mature” TLE [44-46]. Events occurring during epileptogenesis result in the formation of epileptic neural networks and the corresponding connectopathies: changes in glutamatergic and GABAergic neurons, increased aberrant neurogenesis, axonal sprouting and remodeling of dendrites, in particular due to excessive augmentation of the brain-derived neurotrophic factor (BDNF) in specific areas of the hippocampus. With the progress of epileptogenesis, this stage of aberrant superplasticity is replaced by the formation of an epileptogenic focus, the functioning of which is accompanied by opposite events leading to the restriction of plasticity: depletion of the BDNF system, neuroinflammation, axon damage, neurodegeneration, neuronal death, hippocampal sclerosis, and suppression of subgranular neurogenesis (Fig. 3).

Molecular and cellular hippocampal mechanisms at the main stages of TLE pathogenesis [44-47]. Progress of epileptogenesis is accompanied by BDNF excess, enhanced neurogenesis, “aberrant superplasticity” in the subgranular zone of the dentate gyrus, and maturation of epileptic neural networks. Subsequently, against the background of HPACS dysfunction (excess of GCs and impaired functioning of their receptors), hyperglutamatergic signaling and activation of neuroinflammation suppress neurogenesis and induce neurodegeneration and neuronal death. At this stage, hippocampal plasticity becomes significantly reduced, and this plasticity deficit is further aggravated by recurrent convulsive activity.
Central role of HPACS in comorbidity between epilepsy and depression. The rearrangement of synaptic connections and neural networks during epileptogenesis is largely initiated by the dysregulation of HPACS. The control of the majority of the key mechanisms mediating epileptogenesis and formation of mature epileptic networks in TLE is impaired in depressive disorders, which are primarily associated with HPACS dysfunction as well. This gives a basis to suggest a central role of HPACS in the comorbidity of these diseases. Other mechanisms common to epilepsy and depression, including BDNF depletion, activation of microglia, neuroinflammation, impaired neurogenesis, neurodegeneration [40, 48], and alterations in the functioning of neurotransmitter systems (glutamatergic, serotonergic, monoaminergic, and GABAergic) [49] are generally associated, either directly or indirectly, with changes in HPACS function.
Recent studies have confirmed HPACS hyperactivation and elevated GC levels in patients with TLE. It was found that an excess of GCs increases the excitability of neurons, epileptiform activity and predisposition to seizures. Patients with TLE can exhibit abnormal stress response which facilitate ictal discharges and increase vulnerability to the development of comorbid psychopathologies [37, 50]. Long-term exposure to GCs not only exacerbates pre-existing TLE, but also can increase the predisposition to associated mental illnesses. The hyperactivity of the HPACS negatively affects the structure and function of the brain temporal lobe, an area deeply involved in both TLE and mood disorders, such as anxiety and depression. GCs can disrupt glutamatergic transmission through their receptors on the membranes and in the cytoplasm of glutamatergic synapses (see review [51]). Excessive excitatory activity of neurons during seizures damages the structures of the temporal lobes, further disinhibiting HPACS and concluding the vicious circle of neuronal damage by increasing the susceptibility to subsequent seizures and psychiatric comorbidities [43]. The hippocampus is particularly sensitive to chronic stress and to GCs [41]. Along with many other changes, these factors cause dendrite retraction, a reversible form of plasticity that involves dendrite restructuring without irreversible cell death. Dendrite retraction, which can occur for weeks, months, or even years, is associated with the increased probability of damage to the hippocampus, and the existence of this time window is one of the postulates on the GC vulnerability of the hippocampus [52].
Elucidation of the role of HPACS dysfunction in epilepsy requires more detailed mechanistic studies, since it is essential for understanding the development and progression of TLE and comorbid pathologies, as well as for identification of potential therapy targets. The meta-analysis of the relationship between cortisol levels and seizures in patients with epilepsy was performed using the data of 38 articles that met the inclusion criteria. Fourteen of them analyzed the basal cortisol levels; 8 – the effects of antiepileptic drugs; 13 – the effects of seizures on the cortisol levels; and 3 – the effects of stress on these parameters [53]. Basal cortisol levels were found to be higher in the patients than in healthy individuals in studies with the most homogenous groups (45% of 38 studies). Despite the heterogeneity of the results in the studies on antiepileptic drugs, seizures were found to be associated with the increased cortisol levels in 77% of the 38 studies. The only study with the presentation of acute stress revealed a higher cortisol reactivity in epilepsy as compared to the control [32]. In studies with self-reported stress, a higher frequency of seizures was associated with increased cortisol levels and reduced brain functional connectivity. The diurnal rhythm of epileptic seizures in different types of epilepsy was found to be similar to the cortisol circadian rhythm, which supports the hypothesis that circadian fluctuations in GC levels affect the occurrence of epileptic seizures [53, 54].
In the hippocampus of patients with pharmacoresistant TLE and comorbid depression, a decrease in GR density was found [55]. GR expression was evaluated immunohistochemically in patients with TLE (with or without comorbid depression), who underwent neurosurgery for pharmacoresistant epilepsy and in the control group (autopsy). The control samples revealed a high density of GRs in the granular neurons of the dentate gyrus and pyramidal cells of CA1 region, while in patients with TLE the expression of GRs was reduced, especially in the presence of depression. The ratio between the number of GR+/NeuN+ cells was lower in patients with TLE accompanied by depression as compared to TLE alone and negatively correlated with the severity of depression assessed based on psychiatric history. GR expression was also lower in glial cells in patients with TLE and depression as compared to individuals with TLE alone and negatively correlated with the severity of depression. Reduced GR expression in the hippocampus, an important brain region for HPACS feedback regulation, may be implicated in the etiology of depression in patients with TLE [55].
Hence, clinical data suggest that the elevated neuroendocrine response to stress in patients with TLE is chronic and potentially contributes to both epilepsy progression and the development of associated psychopathology. It is not clear whether HPACS dysregulation reflects the direct effects of the epilepsy- associated pathology, or secondary effects of the disease burden, such as social segregation, stigmatization, and other types of chronic stress associated with epilepsy. Animal models provide an opportunity to distinguish these factors by evaluating whether rodents with epilepsy reproduce the neuroendocrine and behavioral changes characteristic for human epilepsy. Wulsin et al. [56] modeled TLE on male FVB mice using pilocarpine to induce the epileptic status with subsequent development of spontaneous recurrent seizures. Basal corticosterone levels were elevated in pilocarpine-treated mice 1, 7, and 10 weeks after epileptic status as compared to the control group. Mice with epilepsy had increased adrenal weight and corticosterone hypersecretion after acute immobilization stress. Animals with TLE showed more pronounced anhedonic behavior (an index of depressive-like phenotype), hyperactivity, and anxious behavior. Hence, modeling TLE in mice resulted in the development of HPACS hyperactivity and depressive-like behavior. The epileptic status induces rapid hyperactivation of the HPACS, and dysfunction of this axis can exacerbate seizure-induced brain damage. The same research group [57] showed that administration of the GR antagonist RU486 to TLE animals prevented the increase in the cortisol levels caused by pilocarpine epileptic status. RU486 also prevented the development of pathological changes in the hippocampus, including the loss of mossy cells in the hilus and abnormal cell proliferation in the fascia dentata. Interestingly, the loss of mossy cells and the accumulation of ectopic newly formed cells in the hilus positively correlated with the severity of seizures, suggesting a potential antiepileptogenic effect of GR antagonists [57]. Using another TLE model, the pentylenetetrazole kindling, we have shown that even a single convulsive attack causes a gradual deterioration of learning and memory in the hippocampus-dependent tests [58]. Apparently, the corticoid-dependent mechanism of hippocampal damage may also underlie cognitive impairments described in patients with TLE.
Changes in neurogenesis. In almost all mammals, including humans, neurogenesis in the hippocampus occurs throughout life. Numerous studies in animal models have shown that neurogenesis in the subgranular niche (the dentate gyrus of the hippocampus) is important for learning, memory, and emotion, with newly formed neurons potentially more excitable than “old” ones. Neurogenesis in the hippocampus of adult brain is very sensitive to physiological and pathological stimuli. The key mechanism underlying the impact of stressful events on neurogenesis is modulation by GCs [59, 60]. Convulsive seizures alter neurogenesis both quantitatively and qualitatively [61]. Acute seizures are usually associated with increased neurogenesis and migration of newborn neurons to ectopic regions, such as the hilus and molecular layer of the dentate gyrus Abnormally (ectopically) migrating newborn neurons formed as a result of acute seizures, epileptic status, or traumatic brain injury, participate in the formation of epileptogenic neural networks of the hippocampus [62-69]. On the contrary, recurrent spontaneous seizures in chronic TLE are associated with significantly reduced neurogenesis, abnormal reorganization of neural networks, and loss of functional inhibition in the hippocampus and extrahippocampal regions [70]. Such changes are also accompanied by learning and memory impairments and depression [71, 72]. This is not surprising, since normal neurogenesis is important both for learning/memory and maintenance of normal emotional state [73]. Suppression of neurogenesis in the hippocampus is considered as one of the key mechanisms in the development of depressive disorders [74-77], so neurogenesis reduction in TLE might be a basis for TLE comorbidity with depression. In patients with pharmacoresistant TLE, who have undergone surgery, comorbid depression may be associated with a loss of calbindin-expressing neurons in the dentate gyrus accompanied with a higher axonal sprouting in these neurons (which is different from the patients with non-depressive TLE) [78]. These changes may indicate disturbances in the maturation of granular cells associated with the phenomena of epileptic hyperexcitability.
Neuroinflammation. In recent years, the hypothesis on the key role of neuroinflammation in the epileptic brain (including the hippocampus in TLE) was postulated and gained experimental confirmation (see reviews [79-83]). At the same time, evidence from another pool of studies strongly suggests that neuroinflammation in the hippocampus is also important in the pathogenesis of depressive disorders (see reviews [84, 85]. Current understanding of the role of cytokines as neuromodulators in the epileptic and depressive brain is based on the results of studies in humans and experimental animals [86, 87]. Cytokine receptors are present on the populations of neurons and glial cells in specific brain areas. In the hippocampus, cytokines are integral participants in physiological processes such, as memory and learning, as well as in pathological conditions, e.g., depression and epilepsy. Normally, interleukin (IL)-1β is necessary to maintain long-term potentiation in the hippocampus, while IL-6 plays a negative regulatory role in the formation of long-term memory. Tumor necrosis factor (TNF) secreted by the astrocytes is involved in functioning of glutamatergic synapses by promoting the surface translocation of glutamate AMPA receptors. Chemokine CXCL12 can suppress tonic activity of Cajal–Retzius neurons in the hippocampus. The concentrations of IL-10, interferon-γ, ICAM1, CCL2, and CCL4 were found to be significantly increased in the hippocampus of patients with TLE [88].
Glial cells play a crucial role in epilepsy pathogenesis by modulating both inflammatory processes (primarily microglia, but also astrocytes) and neuronal excitability (astrocytes). The mechanisms behind the involvement of glial cells in these processes were discussed in detail in the comprehensive review [89]. The loss of hippocampal neurons induced by excitotoxicity and neuroinflammation, as well as reactive astrogliosis, are typical signs of TLE pathology. In patients with TLE, functional changes in hippocampal astrocytes are associated with mental disorders and expression of astrogliosis-related genes coding for glial fibrillary acid protein, metallothioneins I/II, and aquaporin 4 [90].
Glutamatergic system. Glutamatergic system is the main excitatory neurotransmission system of the temporal lobe. Changes in this system have been described in both epilepsies, in which they are essential for the development of excessive arousal [91-96], and depression [97-101]. Overactive glutamatergic neurotransmission (recently, the term hyperglutamatergic neurotransmission was suggested) in the hippocampus is a central factor in the pathogenesis of epilepsy, depression, and cognitive impairments [101]. It induces neurodegenerative changes and loss of hippocampal neurons. Aberrant signaling affects both ionotropic [102, 103] and metabotropic glutamate receptors [104] and induces the excitotoxicity and oxidative stress, which also contribute to the comorbidity of TLE and psychiatric diseases [93, 105]. It is important to note that the functioning of glutamatergic synapses carrying mineralocorticoid receptors and GRs on the membrane and in the cytoplasm is regulated by GCs via multiple pathways (see review [51]).
Experimental studies of the involvement of glutamate receptors in the comorbidity of epilepsy and depression in animal models are just beginning. For example, it was shown that chronic disturbances of the signal transduction through the NMDA receptors, including signaling via the ERK/CREB/ELK1/BDNF/CFOs pathway and CREB (cyclic AMP response element binding protein) phosphorylation, in the model of fluoxetine-resistant epilepsy associated with depressive-like behavior is an important factor in the development of comorbid pathologies [106]. Glutamate synapses also contain cannabinoid receptors, and disorders of the endocannabinoid system are involved in the epileptogenesis, pharmacoresistance, and psychiatric comorbidity in patients with TLE [107, 108]. Epilepsy and mood disorders also share common receptor and transduction mechanisms involved in the serotonin, dopamine, and gamma-aminobutyric acid (GABA) systems [109].
BDNF system. The role of the hippocampal BDNF system in the development of epileptogenesis in TLE [110, 111] (Fig. 3) and depressive disorders [112, 113] has been studied in detail. In both cases, BDNF deficiency is associated with the suppression of neurogenesis and activation of neuroinflammatory processes in the hippocampus. The BDNF system is closely involved in functioning of various types of neurons potentially important for the comorbidity of epilepsy and depression; it is primarily responsible for the multifactorial reciprocal regulation of the glutamatergic system [114].Molecular mechanisms common for stress response and epileptogenesis (an outline). It is important to emphasize once again that all the above-mentioned major molecular mechanisms involved in the hippocampal damage in response to chronic stress and associated with development of epileptogenesis/epilepsy and comorbid depressive disorders are interrelated and interdependent. The central integrating and modulating component of these mechanisms, both in the brain and in entire organism, is the HPACS. The dysregulation of the HPACS results in the excess of GCs and dysfunction of GRs (Fig. 4).

Main molecular mechanisms involved in the hippocampal damage in response to chronic stress and associated with epileptogenesis, epilepsy, and development of comorbid depressive disorders. See the text for details.
It is principally important that stress response and epileptogenesis share molecular mechanisms (Fig. 4), which explains the comorbidity of epilepsy and stress-induced diseases, including depressive/anxiety disorders. The central event in a complex system of interrelated molecular processes is the dysregulation of the HPACS, associated with disturbances in the functioning of corticosteroid receptors and hypercortisolemia. The excitatory glutamatergic system and the BDNF system are essential for the cellular and synaptic plasticity. They are connected by numerous and reciprocal links providing their mutual regulation. These intricate but well-coordinated links ensure the optimal cellular and synaptic plasticity in the normal brain [114]. Both systems are involved in the pathogenesis of affective disorders, and disturbances in the close and balanced relationships between them negatively affect brain neuroplasticity, underlying depression, anxiety, and other mental illnesses. Neuroinflammation, which occurs in response to the dysfunction of the HPACS (mainly via activation of the microglia), is also closely related to the above-mentioned systems. Neuroinflammation induces neurodegeneration and neuronal loss in the brain limbic system, as well as suppresses neurogenesis. Excessively secreted GCs in patients with TLE, especially in those with initially abnormal stress response due to the HPACS dysfunction, interact with the hippocampal GRs, inducing signaling pathways that stimulate neuroinflammation and other processes, such as neurodegeneration and changes in neurogenesis [21, 41, 60]. Functional and structural damages to the hippocampus, which is selectively vulnerable to extreme factors and responds to them by increasing cytokine secretion [115], form an individual basis for the development of both epilepsy and depressive/anxiety disorders in each particular patient.
CHANGES IN NEURAL NETWORKS INVOLVED IN THE COMORBIDITY OF EPILEPSY AND DEPRESSION
The above-described molecular and cellular mechanisms of epilepsy pathogenesis, which also participate in the development of depressive disorders and underlie the comorbidity of epilepsy and depression, should undoubtedly form the basis for the existence of shared mechanisms at higher (network and system) levels. Epilepsy is a disorder associated with recurrent seizures that are defined as “transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous activity of neurons in the brain” [116]. This hyperactivity is the result of abnormal activity in large neuronal groups or networks. The organization of aberrant neural networks and potential mechanisms for their regulation in TLE are discussed in detail in [116], both at the level of “microchips”, i.e., the connections between individual neurons or small groups of similar neurons, and at the level of macrosystems covering large areas of the brain. TLE is an appropriate biological model for establishing a common structural basis for depression and epilepsy. TLE and depression share similar neural network properties, including temporal lobes with the hippocampus, amygdala, and entorhinal cortex and neocortex; frontal lobes with the cingulate gyrus; subcortical structures such as the basal ganglia and thalamus; and connective pathways [117]. In other words, there is clinical and structural evidence that depression and TLE are epiphenomena based on similar properties of neural networks.
A common question in the studies of depression and epilepsy comorbidity is which one of them is primary. It is believed that depression and anxiety in epilepsy reflect the dysfunction of widely distributed limbic networks, which may be either a cause or consequence of epileptogenesis. In refractory epilepsy, seizures and/or certain anticonvulsants may disrupt central emotional homeostatic mechanisms that perpetually increase the risk of seizures [118]. In TLE, fear-related neural networks are associated with the symptoms of anxiety and ictal fear [6]. Despite that many researchers believe that changes in the neural networks in TLE form the basis for the development of mental disorders [5], the systemic review of neuroimaging studies of depression in adult patients with epilepsy [119] showed that the available data are quite contradictory. In particular, some authors suggest that depression, at least in TLE, is associated with a different pattern of brain changes as compared to major depression, which confirms an existing idea of the phenomenological features of depression in epilepsy [119].
Interestingly, neuroticism, the main personality trait characterized by a tendency to experience negative effects, is higher in patients with TLE as compared to healthy people. Neuroticism is a commonly recognized predictor of depression and anxiety, which are also more frequent in patients with TLE. According to [120], patients with increased neuroticism with depression and anxiety symptoms, demonstrated both unique and common patterns of abnormal functional connectivity of resting networks between the mesial temporal and frontal lobes. For example, increased neuroticism was significantly associated with hyposynchrony between the right hippocampus and the Brodmann area, and this unique relationship was independent of the symptoms of depression and anxiety.
CONCLUSION: HIPPOCAMPUS AS COUNTERPOINT TO STRESS, EPILEPSY, AND DEPRESSION
Epilepsy is a neurological condition associated with seizures, neurodegeneration, neural network rearrangement, and other structural and functional disorders which can ultimately lead to changes in behavior and emotional state (anxiety, depression), cognitive dysfunctions (learning disabilities, memory impairment), and accelerated aging. A growing number of studies have demonstrated that neurodegenerative diseases, depressive disorders, and epilepsy share common mechanisms [21, 121]. Cognitive impairments and emotional/personality disorders are regarded as symptoms, and the hippocampus is believed to be the principal affected area.
A significant pool of clinical and experimental data has been accumulated for evidence-based association of TLE and depression. Multiple approaches are used to study the pathogenesis of comorbid disorders at the levels from ultrastructural (expression of cellular receptors) to behavioral (studies of comorbid depression in animals with modeled epilepsy). Investigations of neurobiological aspects of primary mood disorders, mood disorders in TLE, and pathogenetic mechanisms of epilepsy in animal models and human subjects allows better understanding of complex relationships between these two conditions and provide an emerging picture of shared pathophysiological mechanisms.
When validating the concept of the neuroplasticity/neuropathology continuum [47], we emphasized the commonality and pleiotropy (involvement in both the implementation of normal adaptive plasticity and in the pathogenesis of brain disease) of mechanisms at the molecular, synaptic, cellular, and network levels. These mechanisms are associated with a high adaptive plasticity of the hippocampus, which is responsible for brain integrative function, including learning, memory and emotions. However, the price for this high plasticity is a selective sensitivity of the hippocampus to the development of pathological processes. Signaling via secretion of small molecules is characteristic of the nervous, endocrine, and immune systems. The scale and significance of interactions between these have become a subjects of scientific research only recently. Neurotransmitters, hormones, and cytokines act on the target cells via membrane or intracellular receptors that switch on various molecular cascades. There is no doubt that the hippocampus is one of the central structures regulating interactions between these systems.
There is a growing body of evidence that in both epilepsy and depression, the alterations in the interaction of serotonergic and noradrenergic neurons with the glutamatergic systems is associated with formation of abnormal neural networks and increased excitability. The hyperexcitability of neurons can cause convulsive activity, as well as disturb emotional responses. In addition, altered synaptic levels of neurotransmitters and high GC levels affect intracellular signaling pathways, causing dysfunction of the neurotrophin system and other neurotrophic factors [122]. Comorbid mechanisms may be associated with memory impairment and hippocampal atrophy observed in magnetic resonance imaging. Positron emission tomography studies revealed glucose hypometabolism in the temporal and frontal lobes. In both TLE and depression, astrocytes are involved in various processes, playing a role that goes far beyond their involvement in the hippocampal sclerosis. All these mechanisms are closely intertwined and probably bidirectional, so that structural and functional changes associated with a particular disease can increase the risk of developing the other one. The limbic system (primarily, the hippocampus) is a counterpoint to depression and epilepsy, in particular TLE.
Understanding the general neurobiological pathogenetic mechanisms that may play a role in the high comorbidity of epilepsy and depression has a high translational significance and potential clinical use. The impact of shared pathogenetic mechanisms is not limited to high comorbidity, but may be important in the disease exacerbation, as well as in the response to pharmacological and surgical treatment of seizures in patients with a history of depression. Individual stress reactivity of a patient is a principal factor that determines the development of epilepsy and comorbid mental disorders. Unfortunately, this concept has not been routinely adapted for patients with epilepsy; existing simple but efficient approaches to evaluate individual stress reactivity based on the assessment of stress markers are not used sufficiently. Such markers, commonly determined in the blood, can also be evaluated in biomaterials obtained noninvasively (hair, saliva, etc.) [39] and can be even more informative when using a moderate stress test [123]. The idea of the involvement of individual stress reactivity in the development of epilepsy and comorbid diseases can be experimentally tested and used in medical practice to develop new personalized strategies for correcting the functional state of patient and monitoring the efficacy of treatment.
Abbreviations
- BDNF:
-
brain-derived neurotrophic factor
- GC:
-
glucocorticoid
- GR:
-
glucocorticoid receptors
- HPACS:
-
hypothalamic-pituitary-adrenocortical system
- TLE:
-
temporal lobe epilepsy
References
Gulyaeva, N. V. (2017) Molecular mechanisms of neuroplasticity: an expanding universe, Biochemistry (Moscow), 82, 237-242, https://doi.org/10.1134/S0006297917030014.
Kanner, A. M. (2017) Psychiatric comorbidities in new onset epilepsy: should they be always investigated? Seizure, 49, 79-82, https://doi.org/10.1016/j.seizure.2017.04.007.
Kanner, A. M. (2016) Psychiatric comorbidities in epilepsy: should they be considered in the classification of epileptic disorders? Epilepsy Behav., 64 (Pt B), 306-308, https://doi.org/10.1016/j.yebeh.2016.06.040.
Ribot, R., and Kanner, A. M. (2019) Neurobiologic properties of mood disorders may have an impact on epilepsy: should this motivate neurologists to screen for this psychiatric comorbidity in these patients? Epilepsy Behav., 98 (Pt B), 298-301, https://doi.org/10.1016/j.yebeh.2019.01.026.
Sair, A., Şair, Y. B., Saracoğlu, İ., Sevincok, L., and Akyol, A. (2021) The relation of major depression, OCD, personality disorders and affective temperaments with temporal lobe epilepsy, Epilepsy Res., 171, 106565, https://doi.org/10.1016/j.eplepsyres.2021.106565.
Melo, H. M., Guarnieri, R., Vascouto, H. D., Formolo, D. A., de Carvalho, C. R., et al. (2020) Ictal fear is associated with anxiety symptoms and interictal dysphoric disorder in drug-resistant mesial temporal lobe epilepsy, Epilepsy Behav., 115, 107548, https://doi.org/10.1016/j.yebeh.2020.107548.
Kim, S. J., Lee, S. A., Ryu, H. U., Han, S. H., Lee, G. H., et al. (2020) Factors associated with obsessive-compulsive symptoms in people with epilepsy, Epilepsy Behav., 102, 106723, https://doi.org/10.1016/j.yebeh.2019.106723.
Novais, F., Franco, A., Loureiro, S., Andrea, M., Figueira, M. L., Pimentel, J., and Pestana, L. C. (2019) Personality patterns of people with medically refractory epilepsy – does the epileptogenic zone matter? Epilepsy Behav., 97, 130-134, https://doi.org/10.1016/j.yebeh.2019.05.049.
Ho, P. H., Leung, W. C. Y., Leung, I. Y. H., and Chang, R. S. K. (2020) Factors associated with depression in people with epilepsy: a retrospective case-control analysis, Hong Kong Med. J., 26, 311-317, https://doi.org/10.12809/hkmj198310.
Sankar, R., and Mazarati, A. (2012) Neurobiology of depression as a comorbidity of epilepsy, in Jasper’s Basic Mechanisms of the Epilepsies [Internet] (Noebels, J. L., Avoli, M., Rogawski, M. A., Olsen, R. W., and Delgado-Escueta, A. V., eds) 4th Edn., Bethesda (MD): National Center for Biotechnology Information (US).
Duric, V., Clayton, S., Leong, M. L., and Yuan, L. L. (2016) Comorbidity factors and brain mechanisms linking chronic stress and systemic illness, Neural Plast., 2016, 5460732, https://doi.org/10.1155/2016/5460732.
Du Preez, A., Eum, J., Eiben, I., Eiben, P., Zunszain, P. A., et al. (2020) Do different types of stress differentially alter behavioural and neurobiological outcomes associated with depression in rodent models? A systematic review, Front. Neuroendocrinol., 61, 100896, https://doi.org/10.1016/j.yfrne.2020.100896.
Khan, A. R., Geiger, L., Wiborg, O., and Czéh, B. (2020) Stress-induced morphological, cellular and molecular changes in the brain-lessons learned from the chronic mild stress model of depression, Cells, 9, 1026, https://doi.org/10.3390/cells9041026.
Li, M., Fu, X., Xie, W., Guo, W., Li, B., et al. (2020) Effect of early life stress on the epigenetic profiles in depression, Front. Cell. Dev. Biol., 8, 867, https://doi.org/10.3389/fcell.2020.00867.
Negrón-Oyarzo, I., Aboitiz, F., and Fuentealba, P. (2016) Impaired functional connectivity in the prefrontal cortex: a mechanism for chronic stress-induced neuropsychiatric disorder, Neural Plast., 2016, 753906, https://doi.org/10.1155/2016/7539065.
Ross, R. A., Foster, S. L., and Ionescu, D. F. (2017) The role of chronic stress in anxious depression, chronic stress (thousand oaks), Chronic Stress, 1, 2470547016689472, https://doi.org/10.1177/2470547016689472.
Larrieu, T., and Sandi, C. (2018) Stress-induced depression: is social rank a predictive risk factor? Bioessays, 40, e1800012, https://doi.org/10.1002/bies.201800012.
Slavich, G. M., and Sacher, J. (2019) Stress, sex hormones, inflammation, and major depressive disorder: extending social signal transduction theory of depression to account for sex differences in mood disorders, Psychopharmacology (Berl), 236, 3063-3079, https://doi.org/10.1007/s00213-019-05326-9.
Sotiropoulos, I., Silva, J. M., Gomes, P., Sousa, N., and Almeida, O. F. X. (2019) Stress and the etiopathogenesis of Alzheimer’s disease and depression, Adv. Exp. Med. Biol., 1184, 241-257, https://doi.org/10.1007/978-981-32-9358-8_20.
LeMoult, J., Humphreys, K. L., Tracy, A., Hoffmeister, J. A., Ip, E., and Gotlib, I. H. (2020) Meta-analysis: exposure to early life stress and risk for depression in childhood and adolescence, J. Am. Acad. Child. Adolesc. Psychiatry, 59, 842-855, https://doi.org/10.1016/j.jaac.2019.10.011.
Gulyaeva, N. V. (2019) Biochemical mechanisms and translational relevance of hippocampal vulnerability to distant focal brain injury: the price of stress response, Biochemistry (Moscow), 84, 1306-1328, https://doi.org/10.1134/S0006297919110087.
Joëls, M. (2018) Corticosteroids and the brain, J. Endocrinol., 238, R121-R130, https://doi.org/10.1530/JOE-18-0226.
Gulyaeva, N. V. (2015) Ventral hippocampus, stress and psychopathology: translational implications, Neurochem. J., 9, 85-94, https://doi.org/10.1134/S1819712415020075.
Licht, T., Sasson, E., Bell, B., Grunewald, M., Kumar, S., et al. (2020) Hippocampal neural stem cells facilitate access from circulation via apical cytoplasmic processes, Elife, 9, e52134, https://doi.org/10.7554/eLife.52134.
Tuem, K. B., and Atey, T. M. (2017) Neuroactive steroids: receptor interactions and responses, Front. Neurol., 8, 442, https://doi.org/10.3389/fneur.2017.00442.
Saboory, E., Mohammadi, S., Dindarian, S., and Mohammadi, H. (2019) Prenatal stress and elevated seizure susceptibility: molecular inheritable changes, Epilepsy Behav., 96, 122-131, https://doi.org/10.1016/j.yebeh.2019.04.046.
Galtrey, C. M., Mula, M., and Cock, H. R. (2016) Stress and epilepsy: fact or fiction, and what can we do about it? Pract. Neurol., 16, 270-278, https://doi.org/10.1136/practneurol-2015-001337.
Novakova, B., Harris, P. R., Ponnusamy, A., and Reuber, M. (2013) The role of stress as a trigger for epileptic seizures: a narrative review of evidence from human and animal studies, Epilepsia, 54, 1866-76, https://doi.org/10.1111/epi.12377.
Kanner, A. M., Schachter, S. C., Barry, J. J., Hesdorffer, D. C., Mula, M., et al. (2012) Depression and epilepsy: epidemiologic and neurobiologic perspectives that may explain their high comorbid occurrence, Epilepsy Behav., 24, 156-68, https://doi.org/10.1016/j.yebeh.2012.01.007.
McKee, H. R., and Privitera, M. D. (2017) Stress as a seizure precipitant: Identification, associated factors, and treatment options, Seizure, 44, 21-26, https://doi.org/10.1016/j.seizure.2016.12.009.
Gunn, B. G., and Baram, T. Z. (2017) Stress and seizures: space, time and hippocampal circuits, Trends Neurosci., 40, 667-679, https://doi.org/10.1016/j.tins.2017.08.004.
Cano-López, I., and González-Bono, E. (2019) Cortisol levels and seizures in adults with epilepsy: a systematic review, Neurosci. Biobehav. Rev., 103, 216-229, https://doi.org/10.1016/j.neubiorev.2019.05.023.
Kotwas, I., McGonigal, A., Bastien-Toniazzo, M., Bartolome, I. F., and Micoulaud-Franchi, J. A. (2017) Stress regulation in drug-resistant epilepsy, Epilepsy Behav., 71 (Pt A), 39-50, https://doi.org/10.1016/j.yebeh.2017.01.025.
Yang, Y., Yang, M., Shi, Q., Wang, T., and Jiang, M. (2020) Risk factors for depression in patients with epilepsy: a meta-analysis, Epilepsy Behav., 106, 107030, https://doi.org/10.1016/j.yebeh.2020.107030.
Błaszczyk, B., and Czuczwar, S. J. (2016) Epilepsy coexisting with depression, Pharmacol. Rep., 68, 1084-1092, https://doi.org/10.1016/j.pharep.2016.06.011.
Hingray, C., McGonigal, A., Kotwas, I., and Micoulaud-Franchi, J. A. (2019) The relationship between epilepsy and anxiety disorders, Curr. Psychiatry Rep., 21, 40, https://doi.org/10.1007/s11920-019-1029-9.
Wulsin, A. C., Solomon, M. B., Privitera, M. D., Danzer, S. C., and Herman, J. P. (2016) Hypothalamic-pituitary-adrenocortical axis dysfunction in epilepsy, Physiol. Behav., 166, 22-31, https://doi.org/10.1016/j.physbeh.2016.05.015.
Jones, N. C., and O’Brien, T. J. (2013) Stress, epilepsy, and psychiatric comorbidity: how can animal models inform the clinic? Epilepsy Behav., 26, 363-369, https://doi.org/10.1016/j.yebeh.2012.09.002.
Pochigaeva, K., Druzhkova, T., Yakovlev, A., Onufriev, M., Grishkina, M., et al. (2017) Hair cortisol as a marker of hypothalamic-pituitary-adrenal Axis activity in female patients with major depressive disorder, Metab. Brain Dis., 32, 577-583, https://doi.org/10.1007/s11011-017-9952-0.
Kanner, A. M. (2017) Can neurochemical changes of mood disorders explain the increase risk of epilepsy or its worse seizure control? Neurochem. Res., 42, 2071-2076, https://doi.org/10.1007/s11064-017-2331-8.
Gulyaeva, N. V. (2019) Functional neurochemistry of the ventral and dorsal hippocampus: Stress, depression, dementia and remote hippocampal damage, Neurochem. Res., 44, 1306-1322, https://doi.org/10.1007/s11064-018-2662-0.
Maguire, J., Salpekar, J. A. (2013) Stress, seizures, and hypothalamic-pituitary-adrenal axis targets for the treatment of epilepsy, Epilepsy Behav., 26, 352-362, https://doi.org/10.1016/j.yebeh.2012.09.040.
Basu, T., Maguire, J., Salpekar, J. A. (2021) Hypothalamic-pituitary-adrenal axis targets for the treatment of epilepsy, Neurosci. Lett., 746, 135618, https://doi.org/10.1016/j.neulet.2020.135618.
Gulyaeva, N. V. (2015) Neuronal plasticity and epilepsy: modern concepts and mechanisms of epilepsy and depression comorbidity, Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova, 115, 148-153, https://doi.org/10.17116/jnevro2015115112148-153.
Gulyaeva, N. V. (2016) Cerebral plasticity and connectopathies: mechanisms of comorbidity of neurological diseases and depression, Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova, 116, 157-162, https://doi.org/10.17116/jnevro2016116111157-162.
Gulyaeva, N. V. (2017) Staging of neuroplasticity alterations during epileptogenesis (temporal lobe epilepsy as an example), Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova, 117, 10-16, https://doi.org/10.17116/jnevro20171179210-16.
Gulyaeva, N. V. (2020) Physiological continuum of plasticity and pathology of the nervous system [in Russian], Integr. Physiol., 1, 294-302, https://doi.org/10.33910/2687-1270-2020-1-4-294-302.
Stepanichev, M., Dygalo, N. N., Grigoryan, G., Shishkina, G. T., and Gulyaeva, N. (2014) Rodent models of depression: neurotrophic and neuroinflammatory biomarkers, Biomed. Res. Int., 2014, 932757, https://doi.org/10.1155/2014/932757.
Kanner, A. M. (2008) Mood disorder and epilepsy: a neurobiologic perspective of their relationship, Dialogues Clin. Neurosci., 10, 39-45, https://doi.org/10.31887/DCNS.2008.10.1/amkanner.
Pauli, E., and Stefan, H. (2009) Emotional and affective disorders, anxiety and personality disorders in epilepsies, Nervenarzt, 80, 1440-1451, https://doi.org/10.1007/s00115-009-2740-2.
Gulyaeva, N. V. (2021) Glucocorticoid regulation of the glutamatergic synapse: Mechanisms of stress-dependent neuroplasticity, Ross. Fiziologicheskii Zhurnal im. I. M. Sechenova (Sechenov Physiology Journal), 107, 518-532, https://doi.org/10.31857/S0869813921040099.
Conrad, C. D. (2008) Chronic stress-induced hippocampal vulnerability: the glucocorticoid vulnerability hypothesis, Rev. Neurosci., 19, 395-411, https://doi.org/10.1515/revneuro.2008.19.6.395.
Van Campen, J. S., Valentijn, F.A., Jansen, F. E., Joels, M., and Braun, K. P. (2015) Seizure occurrence and the circadian rhythm of cortisol: a systematic review, Epilepsy Behav., 47, 132-137, https://doi.org/10.1016/j.yebeh.2015.04.071.
Van Campen, J. S., Hompe, E.L., Jansen, F. E., Velis, D.N., Otte, W.M., et al. (2016) Cortisol fluctuations relate to interictal epileptiform discharges in stress sensitive epilepsy, Brain, 139 (Pt 6), 1673-1679, https://doi.org/10.1093/brain/aww071.
D’Alessio, L., Mesarosova, L., Anink, J. J., Kochen, S., Solís, P., et al. (2020) Reduced expression of the glucocorticoid receptor in the hippocampus of patients with drug-resistant temporal lobe epilepsy and comorbid depression, Epilepsia, 61, 1595-1605, https://doi.org/10.1111/epi.16598.
Wulsin, A. C., Franco-Villanueva, A., Romancheck, C., Morano, R. L., Smith, B. L., et al. (2018) Functional disruption of stress modulatory circuits in a model of temporal lobe epilepsy, PLoS One, 13, e0197955, https://doi.org/10.1371/journal.pone.0197955.
Wulsin, A. C., Herman, J. P., and Danzer, S. C. (2016) RU486 mitigates hippocampal pathology following status epilepticusm, Front. Neurol., 7, 214, https://doi.org/10.3389/fneur.2016.00214.
Aniol, V. A., Ivanova-Dyatlova, A. Y., Keren, O., Guekht, A. B., Sarne, Y., and Gulyaeva, N. V. (2013) A single pentylenetetrazole-induced clonic-tonic seizure episode is accompanied by a slowly developing cognitive decline in rats, Epilepsy Behav., 26, 196-202, https://doi.org/10.1016/j.yebeh.2012.12.006.
Gulyaeva, N. V. (2010) Aberrant neurogenesis in adult epileptic brain: compensatory or pathologic, Neurochem. J., 4, 84-89, https://doi.org/10.1134/S1819712410020029.
Podgorny, O. V., and Gulyaeva, N. V. (2021) Glucocorticoid-mediated mechanisms of hippocampal damage: Contribution of subgranular neurogenesis, J. Neurochem., 157, 370-392, https://doi.org/10.1111/jnc.15265.
Kuruba, R., Hattiangady, B., and Shetty, A. K. (2009) Hippocampal neurogenesis and neural stem cells in temporal lobe epilepsy, Epilepsy Behav., 14 (Suppl 1), 65-73, https://doi.org/10.1016/j.yebeh.2008.08.020.
Parent, J. M., and Lowenstein, D. H. (2002) Seizure-induced neurogenesis: are more new neurons good for an adult brain? Prog. Brain. Res., 135, 121-131, https://doi.org/10.1016/S0079-6123(02)35012-X.
Parent, J. M. (2002) The role of seizure-induced neurogenesis in epileptogenesis and brain repair, Epilepsy Res., 50, 179-89, https://doi.org/10.1016/s0920-1211(02)00078-5.
Parent, J. M. (2007) Adult neurogenesis in the intact and epileptic dentate gyrus, Prog. Brain Res., 163, 529-40, https://doi.org/10.1016/S0079-6123(07)63028-3.
Kokaia, M. (2011) Seizure-induced neurogenesis in the adult brain, Eur. J. Neurosci., 33, 1133-1138, https://doi.org/10.1111/j.1460-9568.2011.07612.x.
Jessberger, S., and Parent, J. M. (2015) Epilepsy and adult neurogenesis, Cold Spring Harb Perspect. Biol., 7, a020677, https://doi.org/10.1101/cshperspect.a020677.
Scharfman, H. E. (2004) Functional implications of seizure-induced neurogenesis, Adv. Exp. Med. Biol., 548, 192-212, https://doi.org/10.1007/978-1-4757-6376-8_14.
Scharfman, H. E., and Gray, W. P. (2007) Relevance of seizure-induced neurogenesis in animal models of epilepsy to the etiology of temporal lobe epilepsy, Epilepsia, 48 (Suppl 2), 33-41, https://doi.org/10.1111/j.1528-1167.2007.01065.x.
Siebzehnrubl, F. A., and Blumcke, I. (2008) Neurogenesis in the human hippocampus and its relevance to temporal lobe epilepsies, Epilepsia, 49 (Suppl 5), 55-65, https://doi.org/10.1111/j.1528-1167.2008.01638.x.
Hattiangady, B., and Shetty, A. K. (2008) Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy, Epilepsia, 49 (Suppl 5), 26-41, https://doi.org/10.1111/j.1528-1167.2008.01635.x.
Danzer, S. C. (2012) Depression, stress, epilepsy and adult neurogenesis, Exp. Neurol., 233, 22-32, https://doi.org/10.1016/j.expneurol.2011.05.023.
Braun, S. M., and Jessberger, S. (2014) Adult neurogenesis and its role in neuropsychiatric disease, brain repair and normal brain function, Neuropathol. Appl. Neurobiol., 40, 3-12, https://doi.org/10.1111/nan.12107.
Abrous, D. N., Koehl, M., and Le Moal, M. (2005) Adult neurogenesis: from precursors to network and physiology, Physiol. Rev., 85, 523-569, https://doi.org/10.1152/physrev.00055.2003.
Hayashi, Y., Jinnou, H., Sawamoto, K., and Hitoshi, S. (2018) Adult neurogenesis and its role in brain injury and psychiatric diseases, J. Neurochem., 147, 584-594, https://doi.org/10.1111/jnc.14557.
Lino de Oliveira, C., Bolzan, J. A., Surget, A., and Belzung, C. (2020) Do antidepressants promote neurogenesis in adult hippocampus? A systematic review and meta-analysis on naive rodents, Pharmacol. Ther., 210, 107515, https://doi.org/10.1016/j.pharmthera.2020.107515.
Patel, D., Kas, M. J., Chattarji, S., and Buwalda, B. (2019) Rodent models of social stress and neuronal plasticity: relevance to depressive-like disorders, Behav. Brain Res., 369, 111900, https://doi.org/10.1016/j.bbr.2019.111900.
Planchez, B., Surget, A., and Belzung, C. (2020) Adult hippocampal neurogenesis and antidepressants effects, Curr. Opin. Pharmacol., 50, 88-95, https://doi.org/10.1016/j.coph.2019.11.009.
D’Alessio, L., Konopka, H., Solís, P., Scévola, L., Lima, M. F., et al. (2019) Depression and temporal lobe epilepsy: expression pattern of calbindin immunoreactivity in hippocampal dentate gyrus of patients who underwent epilepsy surgery with and without comorbid depression, Behav. Neurol., 2019, 7396793, https://doi.org/10.1155/2019/7396793.
Vezzani, A., Balosso, S., and Ravizza, T. (2019) Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy, Nat. Rev. Neurol., 15, 459-472, https://doi.org/10.1038/s41582-019-0217-x.
Vezzani, B., Carinci, M., Patergnani, S., Pasquin, M. P., Guarino, A., et al. (2020) The dichotomous role of inflammation in the CNS: a mitochondrial point of view, Biomolecules, 10, 1437, https://doi.org/10.3390/biom10101437.
Sharma, A. A., and Szaflarski, J. P. (2020) In vivo imaging of neuroinflammatory targets in treatment-resistant epilepsy, Curr. Neurol. Neurosci. Rep., 20, 5, https://doi.org/10.1007/s11910-020-1025-9.
Mukherjee, S., Arisi, G. M., Mims, K., Hollingsworth, G., O’Neil, K., and Shapiro, L. A. (2020) Neuroinflammatory mechanisms of post-traumatic epilepsy, J. Neuroinflammation, 17, 193, https://doi.org/10.1186/s12974-020-01854-w.
Sanz, P., and Garcia-Gimeno, M. A. (2020) Reactive glia inflammatory signaling pathways and epilepsy, Int. J. Mol. Sci., 21, 4096, https://doi.org/10.3390/ijms21114096.
Meyer, J. H., Cervenka, S., Kim, M. J., Kreisl, W. C., Henter, I. D., and Innis, R. B. (2020) Neuroinflammation in psychiatric disorders: PET imaging and promising new targets, Lancet Psychiatry, 7, 1064-1074, https://doi.org/10.1016/S2215-0366(20)30255-8.
Troubat, R., Barone, P., Leman, S., Desmidt, T., Cressant, A., et al. (2021) Neuroinflammation and depression: a review, Eur. J. Neurosci., 53, 151-171, https://doi.org/10.1111/ejn.14720.
Ting, E. Y., Yang, A. C., and Tsai, S. J. (2020) Role of interleukin-6 in depressive disorder, Int. J. Mol. Sci., 21, 2194, https://doi.org/10.3390/ijms21062194.
Tsai, S. J. (2021) Role of interleukin 8 in depression and other psychiatric disorders, Prog. Neuropsychopharmacol. Biol. Psychiatry, 106, 110173, https://doi.org/10.1016/j.pnpbp.2020.110173.
Arisi, G. M. (2014) Nervous and immune systems signals and connections: cytokines in hippocampus physiology and pathology, Epilepsy Behav., 38, 43-7, https://doi.org/10.1016/j.yebeh.2014.01.017.
Devinsky, O., Vezzani, A., Najjar, S., De Lanerolle, N. C., and Rogawski, M. A. (2013) Glia and epilepsy: excitability and inflammation, Trends Neurosci., 36, 174-84, https://doi.org/10.1016/j.tins.2012.11.008.
Lu, J., Huang, H., Zeng, Q., Zhang, X., Xu, M., et al. (2019) Hippocampal neuron loss and astrogliosis in medial temporal lobe epileptic patients with mental disorders, J. Integr. Neurosci., 18, 127-132, https://doi.org/10.31083/j.jin.2019.02.16.
Albrecht, J., and Zielińska, M. (2017) Mechanisms of excessive extracellular glutamate accumulation in and temporal lobe epilepsy, Neurochem. Res., 42, 1724-1734, https://doi.org/10.1007/s11064-016-2105-8.
Barker-Haliski, M., and White, H. S. (2015) Glutamatergic mechanisms associated with seizures and epilepsy, Cold Spring Harb. Perspect. Med., 5, a022863, https://doi.org/10.1101/cshperspect.a022863.
Eid, T., Gruenbaum, S. E., Dhaher, R., Lee, T. W., Zhou, Y., and Danbolt, N. C. (2016) The glutamate–glutamine cycle in epilepsy, Adv. Neurobiol., 13, 351-400, https://doi.org/10.1007/978-3-319-45096-4_14.
Nicolo, J. P., O’Brien, T. J., and Kwan, P. (2019) Role of cerebral glutamate in post-stroke epileptogenesis, Neuroimage Clin., 24, 102069, https://doi.org/10.1016/j.nicl.2019.102069.
O’Neil, D. A., Nicholas, M. A., Lajud, N., Kline, A. E., and Bondi, C. O. (2018) Preclinical models of traumatic brain injury: emerging role of glutamate in the pathophysiology of depression, Front. Pharmacol., 9, 579, https://doi.org/10.3389/fphar.2018.00579.
Cui, W., Ning, Y., Hong, W., Wang, J., Liu, Z., and Li, M. D. (2019) Crosstalk between inflammation and glutamate system in depression: signaling pathway and molecular biomarkers for ketamine’s antidepressant effect, Mol. Neurobiol., 56, 3484-3500, https://doi.org/10.1007/s12035-018-1306-3.
Fullana, N., Gasull-Camós, J., Tarrés-Gatius, M., Castañé, A., Bortolozzi, A., and Artigas, F. (2020) Astrocyte control of glutamatergic activity: downstream effects on serotonergic function and emotional behavior, Neuropharmacology, 166, 107914, https://doi.org/10.1016/j.neuropharm.2019.107914.
Henter, I. D., de Sousa, R. T., and Zarate, C. A., Jr. (2018) Glutamatergic modulators in depression, Harv. Rev. Psychiatry, 26, 307-319, https://doi.org/10.1097/HRP.0000000000000183.
Li, C. T., Yang, K. C., and Lin, W. C. (2019) Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies, Front. Psychiatry, 9, 767, https://doi.org/10.3389/fpsyt.2018.00767.
Reus, G. Z., de Moura, A. B., Silva, R. H., Resende, W. R., and Quevedo, J. (2018) Resilience dysregulation in major depressive disorder: focus on glutamatergic imbalance and microglial activation, Curr. Neuropharmacol., 16, 297-307, https://doi.org/10.2174/1570159X15666170630164715.
Gulyaeva, N. V. (2021) Hippocampal hyperglutamatergic signaling matters: early targeting glutamate neurotransmission as a preventive strategy in Alzheimer’s disease: an editorial highlight for “riluzole attenuates glutamatergic tone and cognitive decline in AβPP/PS1 mice” on https://doi.org/10.1111/jnc.15224, J. Neurochem., 156, 399-402, https://doi.org/10.1111/jnc.15238.
Hanada, T. (2020) Ionotropic glutamate receptors in epilepsy: a review focusing on AMPA and NMDA receptors, Biomolecules, 10, 464, https://doi.org/10.3390/biom10030464.
Mihály, A. (2019) The reactive plasticity of hippocampal ionotropic glutamate receptors in animal epilepsies, Int. J. Mol. Sci., 20, 1030, https://doi.org/10.3390/ijms20051030.
Qian, F., and Tang, F. R. (2016) Metabotropic glutamate receptors and interacting proteins in epileptogenesis, Curr. Neuropharmacol., 14, 551-562, https://doi.org/10.2174/1570159x14666160331142228.
Miladinovic, T., Nashed, M. G., and Singh, G. (2015) Overview of glutamatergic dysregulation in central pathologies, Biomolecules, 5, 3112-3141, https://doi.org/10.3390/biom5043112.
Sadeghi, M. A., Hemmati, S., Mohammadi, S., Yousefi-Manesh, H., Vafaei, A., et al. (2021) Chronically altered NMDAR signaling in epilepsy mediates comorbid depression, Acta Neuropathol. Commun., 9, 53, https://doi.org/10.1186/s40478-021-01153-2.
Scarante, F. F., Vila-Verde, C., Detoni, V. L., Ferreira-Junior, N. C., Guimarães, F. S., and Campos, A. C. (2017) Cannabinoid modulation of the stressed hippocampus, Front. Mol. Neurosci., 10, 411, https://doi.org/10.3389/fnmol.2017.00411.
Cheung, K. A. K., Peiris, H., Wallace, G., Holland, O. J., and Mitchell, M. D. (2019) The interplay between the endocannabinoid system, epilepsy and cannabinoids, Int. J. Mol. Sci., 20, 6079, https://doi.org/10.3390/ijms20236079.
Rocha, L., Alonso-Vanegas, M., Orozco-Suárez, S., Alcántara-González, D., Cruzblanca, H., and Castro, E. (2014) Do certain signal transduction mechanisms explain the comorbidity of epilepsy and mood disorders? Epilepsy Behav., 38, 25-31, https://doi.org/10.1016/j.yebeh.2014.01.001.
Koyama, R., and Ikegaya, Y. (2005) To BDNF or not to BDNF: that is the epileptic hippocampus, Neuroscientist, 11, 282-7, https://doi.org/10.1177/1073858405278266.
McNamara, J. O., and Scharfman, H. E. (2012) Temporal lobe epilepsy and the BDNF receptor, TrkB, in Jasper’s Basic Mechanisms of the Epilepsies [Internet] (Noebels, J. L., Avoli, M., Rogawski, M. A., Olsen, R. W., and Delgado-Escueta, A. V., eds.) 4th Edn., Bethesda (MD): National Center for Biotechnology Information (US).
Brunoni, A. R., Lopes, M., and Fregni, F. (2008) A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression, Int. J. Neuropsychopharmacol., 11, 1169-1180, https://doi.org/10.1017/S1461145708009309.
Martinowich, K., Manji, H., and Lu, B. (2007) New insights into BDNF function in depression and anxiety, Nat. Neurosci., 10, 1089-1093, https://doi.org/10.1038/nn1971.
Gulyaeva, N. V. (2017) Interplay between brain BDNF and glutamatergic systems: a brief state of the evidence and association with the pathogenesis of depression, Biochemistry (Moscow), 82, 301-307, https://doi.org/10.1134/S0006297917030087.
Bolshakov, A. P., Tret’yakova, L. V., Kvichansky, A. A., and Gulyaeva, N. V. (2021) Glucocorticoids: Dr. Jekyll and Mr. Hyde of hippocampal neuroinflammation, Biochemistry (Moscow), 86, 156-167, https://doi.org/10.1134/S0006297921020048.
Alexander, A., Maroso, M., and Soltesz, I. (2016) Organization and control of epileptic circuits in temporal lobe epilepsy, Prog. Brain Res., 226, 127-154, https://doi.org/10.1016/bs.pbr.2016.04.007.
Valente, K. D., and Busatto, F. G. (2013) Depression and temporal lobe epilepsy represent an epiphenomenon sharing similar neural networks: clinical and brain structural evidences, Arq. Neuropsiquiatr., 71, 183-190, https://doi.org/10.1590/s0004-282x2013000300011.
Krishnan, V. (2020) Depression and anxiety in the epilepsies: from bench to bedside, Curr. Neurol. Neurosci. Rep., 20, 41, https://doi.org/10.1007/s11910-020-01065-z.
Elkommos, S., and Mula, M. (2020) A systematic review of neuroimaging studies of depression in adults with epilepsy, Epilepsy Behav., 115, 107695, https://doi.org/10.1016/j.yebeh.2020.107695.
Rivera Bonet, C. N., Hwang, G., Hermann, B., Struck, A. F., Cook, C. J., et al. (2020) Neuroticism in temporal lobe epilepsy is associated with altered limbic-frontal lobe resting-state functional connectivity, Epilepsy Behav., 110, 107172, https://doi.org/10.1016/j.yebeh.2020.107172.
Szot, P. (2012) Common factors among Alzheimer’s disease, Parkinson’s disease, and epilepsy: possible role of the noradrenergic nervous system., Epilepsia, 53 (Suppl 1), 61-6, https://doi.org/10.1111/j.1528-1167.2012.03476.x.
Kondziella, D., Alvestad, S., Vaaler, A., and Sonnewald, U. (2007) Which clinical and experimental data link temporal lobe epilepsy with depression? J. Neurochem., 103, 2136-52, https://doi.org/10.1111/j.1471-4159.2007.04926.x.
Druzhkova, T., Pochigaeva, K., Yakovlev, A., Kazimirova, E., Grishkina, M., et al. (2019) Acute stress response to a cognitive task in patients with major depressive disorder: potential metabolic and proinflammatory biomarkers, Metab. Brain. Dis., 34, 621-629, https://doi.org/10.1007/s11011-018-0367-3.
Funding
The work was supported by the Russian Foundation for Basic Research (grant no. 20-115-50019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares that there is no conflict of interest. This article does not contain experimental studies performed by the author involving humans or animals as objects.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gulyaeva, N.V. Stress-Associated Molecular and Cellular Hippocampal Mechanisms Common for Epilepsy and Comorbid Depressive Disorders. Biochemistry Moscow 86, 641–656 (2021). https://doi.org/10.1134/S0006297921060031
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297921060031