
Abstract
The review describes the application of small-angle scattering (SAS) of neutrons and complementary methods to study the structures of biomacromolecules. Here we cover SAS techniques, such as the contrast variation, the neutron spin-echo, and the solution of direct and inverse problems of three-dimensional reconstruction of the structures of macromolecules from SAS spectra by means of molecular modeling. A special section is devoted to specific objects of research, such as supramolecular complexes, influenza virus nucleoprotein, and chromatin.
Similar content being viewed by others


CONTENT
Introduction
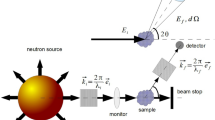
1. SANS and Complementary Methods
2. Contrast Variation
3. Time-Resolved SAS
4. Neutron Spin-Echo
5. Molecular Modeling and SAS
5.1. Debye Equation
5.2. Spherical Harmonic Decomposition
5.3. Direct Calculations of SANS and SAXS Spectra from the Scattering Length Density
5.4. Analysis of NSE Results
6. Unique Applications
6.1. Supramolecular Complexes
6.2. Influenza Virus Nucleoprotein
6.3. Chromatin
7. Future Biological Research at the PIK Reactor at the NRC “Kurchatov Institute”–PNPI
Conclusions
INTRODUCTION
Solving the most pressing problems in medicine and biology requires the investigation of the structures and the structural and dynamic organization of native proteins, their functional domains, synthetic polypeptides with defined peptide sequences, and supramolecular biological complexes at all levels of organization by physical methods, which are applicable to a wide range of objects. The development of comprehensive experimental approaches using high-flux neutron sources, other physical methods of research, molecular modeling, and molecular dynamics simulations provides new opportunities for investigating interactions of biomacromolecules, resulting in the formation, dissociation, and reorganization of macromolecular complexes and large supramolecular structures, and allows the understanding of the structural basis and molecular mechanisms of functioning of molecular and supramolecular biological complexes.
Neutron scattering is a unique tool for obtaining structural data on the functional states of biological macromolecular complexes in their native aqueous solution. Neutron scattering, unlike X-ray techniques, does not cause any radiation damage to biological macromolecules. Neutron techniques can be used to investigate the conformations of macromolecules and complexes incapable of crystallization, study labile supramolecular complexes, the native structure of which cannot be retained in atomic-force and electron-microscopy experiments, observe the dynamics of macromolecules, provide the structural description of the enzyme kinetics, and characterize complex hierarchical systems, in particular, in living tissues ex vivo, on scales from several to hundred nanometers [1]. An important advantage of small-angle neutron scattering (SANS) is the relative simplicity of the use of the method of contrast variation, which makes it possible to separate the scattering contributions from components of the system with different scattering length density, such as proteins, lipids, nucleic acids, and sugars, and to investigate the solvent accessibility of different compartments of the sample.
The development of the whole range of neutron scattering techniques is required to address important issues in studying biological structures. These techniques provide structural data on biological samples, characteristic sizes, the degree of polydispersity of the sample, the short-range structural order, in some cases, the internal structure of species, and integral characteristics of the large-scale structure, such as the fractal dimension in polymer and complex hierarchical systems. The currently available and planned facilities at the PIK reactor complex can be used to study such systems in the size range extremely wide for a single physical method, from 6–7 Å (SANS) to 15 µm (ultra-small-angle neutron scattering, USANS) and tens of micrometers (spin-echo small-angle neutron scattering, SESANS [2]).
Modern high-flux reactors and spectrometers for SANS and USANS experiments enable the reduction of the time required to measure the scattering signal to a few minutes or shorter. Hence, it becomes possible to study the kinetics of processes with a time resolution sufficient for the observation of structural changes associated with the biological function of the objects of research, as well as to search for and characterize intermediate states of the system. For this purpose, it is necessary to develop methods of analysis and interpretation of time-resolved SANS data, including dimensionality reduction methods (principal component analysis, singular value decomposition, etc.).
1 SANS AND COMPLEMENTARY METHODS
The self-organization is one of the key properties inherent in living systems. Specific interactions between biomacromolecules constitute the basis of all processes in living organisms. Historically, molecular biology has progressed from investigations of functions inherent in individual molecules to functions of biomolecular assemblies [3]. Parameters of protein–protein interactions and the structures of individual macromolecules can be studied by such methods as X-ray crystallography [4, 5] and nuclear magnetic resonance (NMR) spectroscopy [6], providing data on the all-atom structures of protein complexes and the whole range of biochemical processes. Hierarchically higher levels of organization of biomacromolecules (20–200 nm), from macromolecular complexes to organelle components, were inaccessible to studies by these methods because of the impossibility to crystallize these objects and the presence of a large number of overlapping signals in their NMR spectra. Electron microscopy (EM) was the only available method for structural studies [7]. However, the sample preparation for EM and experimental requirements, in particular, the necessity of drying and performing measurements in vacuum, did not allow one to state with certainty that the observed structures correspond to the functional structures existing in native conditions. Moreover, the EM method cannot be used to investigate the effect of the temperature on the structure of the object. Atomic force microscopy (AFM) [8] and laser correlation spectroscopy (LCS) [9], which were elaborated later, with the progress in the laser technique, also have drawbacks. Despite the possibility of performing measurements in liquids, the application of AFM under these conditions leads to a decrease in the resolution; meanwhile, this method does not obviate the need to attach biomacromolecular complexes to a solid substrate and take into account the probe shape in the image processing. Although the benefit of the LCS method is that the hydrodynamic radius of macromolecular complexes can be measured in the native environment at a specified temperature, this method produces images with a very low resolution, interfering with the conclusions about the morphology of the complexes.
The application of small-angle scattering (SAS) to study biological objects, which was first described in [10], has a number of advantages over the above methods. Although SAS belongs to low-resolution techniques, it can be used to characterize the size, shape, global conformational transitions of the protein, in particular, those occurring during folding, interactions between biomacromolecules, and their mutual arrangement in the complex [11]. However, the inverse problem of the determination of the structural parameters of biomacromolecular complexes from the scattering spectrum is mathematically incorrect. Thus, the same SAS spectra can be obtained for structurally different objects. Hence, to discriminate the solutions inconsistent with the structures, the SAS method requires addition data on the full or partial structure determined by complementary methods, e.g., by X-ray crystallography, EM, or AFM. The method, which allows a comparison of the solutions of the direct problem of obtaining the scattering spectrum from molecular modeling and the solution of the inverse problem, deserves separate consideration. Since the spectra can be measured at different temperatures, this method is promising for studying the temperature dependence of the structure of macromolecular complexes, which is necessary for the solution of a wide range of fundamental and applied problems [12, 13]. Also worthy of special attention is the application of SAS methods to study supramolecular complexes in solution. Weak non-covalent interactions responsible for the key properties of these complexes, such as the ability to form multicenter bonds [14, 15], are sensitive to environmental changes that occur during EM sample preparation or the separation of complexes by analytical gel filtration. It should be noted that cryo-electron microscopy (cryo-EM) [16], like SAS, can be used to determine the structures of biomacromolecules in the native environment, take into account the temperature dependence of the structure, and measure the kinetic changes [17, 18] but considerably outperforms all SAS techniques in resolution. However, like in conventional EM methods, the 3D reconstruction of biomacromolecular complexes by means of cryo-EM is performed taking into account only the images of the complexes capable of existing under cryo-EM conditions. Meanwhile, the ratio of complexes in different conformations in solution may significantly differ from that observed in cryo-EM experiments due to the different adsorption and aggregation behavior of the complexes [19]. In this case, SAS can be used as the complementary method, and the structures determined by cryo-EM can serve as the starting models for the solution of the direct problem. This allows the comparison with the SAS spectra and the verification of the models derived from the cryo-EM experiment. Therefore, SAS techniques can be employed to study the structure of macromolecular protein complexes in the native environment, although they require complementary methods for obtaining the structural data because of the incorrect solution of the inverse problem and the impossibility of determining the structures at high resolution.
2 CONTRAST VARIATION
Small-angle scattering measurements can be performed using either neutron (small-angle neutron scattering, SANS) or X-ray (small-angle X-ray scattering, SAXS) scattering from the object. Noteworthy is a significant advantage of SANS over SAXS in structural studies of biomacromolecular complexes, since it can be applied to multicomponent mixtures containing different classes of biomacromolecules. The difference in the scattering length density of biomacromolecules (proteins, nucleic acids, lipids) provides contrast between individual components, when varying the scattering length density of the buffer due to a change in the H2O : D2O ratio. The scattering length density of the buffer at the contrast match point corresponds to the scattering length density of the macromolecular component, and the component gives no more contribution to the scattering intensity. The mutual arrangement of macromolecules of different nature in complicated complexes can be studied by varying the scattering length density. This technique is called the contrast variation [20].
A classic example of the use of the contrast variation technique is the investigation of the structure of the ribosome. Initially, the mutual arrangement of the structural elements of the ribosome (ribosomal RNA and proteins) in its three-dimensional organization was established. It was demonstrated that, first, ribosomal proteins are compact globular macromolecules [21] and, second, ribosomal RNA is located mainly at the central part of the particle [22]. Selectively deuterated proteins were used for the mapping of the proteins in the small ribosomal subunit [23]. The translocation process was studied using a set of ribosomes with different deuteration levels (the degree of deuteration was different for the 30S and 50S subunits in the structure of one ribosome). It was demonstrated that during protein biosynthesis, the ribosome oscillates between two major states, which differ in the hydrodynamic radius due to a change in the position of the 30S subunit relative to 50S [24]. Much later, the functional states of the translocating ribosome were associated with conformational transitions in the ribosomal complex by means of cryo-EM [25] and time-resolved cryo-EM [18].
The contrast variation method can also be applied to study the organization of chromatin composed of nucleic-acid and protein components [26] and to investigate glycosylated proteins [27].
3 TIME-RESOLVED SAS
To understand the structural and functional properties of biological objects at all levels of organization, it is necessary not only to determine their structural characteristics but also to reveal the structural changes in time series. The functionality of most proteins is related to the large-scale conformational mobility of the protein by itself and the dynamics of components of the complexes and supramolecular systems involving the protein. Neutron scattering methods offer considerable possibilities for studying the functional dynamics of macromolecules in wide time and size ranges. A modification of the SAS technique can be applied to investigate the evolution of the structure with time (time-resolved small-angle scattering, TR-SAS) [28, 29]. This method using stopped-flow equipment installed at high-brilliance synchrotron beamlines allows the observation of structural changes of macromolecules that occur during a chemical reaction with a time resolution of 30 ms and higher [30, 31]. Time-resolved SAS data can be analyzed using both standard methods for the SAS data processing (the determination of integral characteristics of the object, the fitting with simple models, ab initio modeling) and dimensionality reduction methods (principal component analysis [32]), which provide structural information about both the main and intermediate conformational states of macromolecules that are present in the reaction mixture in the course of the reaction [28, 33].
The use of neutrons for time-resolved measurements is limited by a much lower flux compared to synchrotron sources and, as a consequence, a significantly lower time resolution (from tens and hundreds of milliseconds to tens of minutes). Meanwhile, considerable advantages of TR-SANS, like of all SANS techniques, are the complete absence of radiation damage of the sample and the ease of application of the contrast variation method. Despite the limited use in biology because of high requirements for the sample volume, the method was successfully employed to study conformational changes in proteins [34, 35] and structural changes in viral particles [36] and lipid vesicles [37, 38]. Stopped-flow rapid-mixing devices (e.g., Stopped-flowSFM-300, Bio-Logic, Seyssinet-Pariset, France) currently become standard instruments for small-angle spectrometers, such as D22 (ILL), KWS-2, and KWS-3 (MLZ) [39, 40].
4 NEUTRON SPIN-ECHO
Neutron spin-echo (NSE) is a unique technique for obtaining data on the structure and dynamics of macromolecular systems, which is also widely used in polymer physics. The application of this method in biology allows the determination of the spatial and temporal characteristics of conformational mobility of components of different protein and nucleoprotein complexes. The characteristic times of conformational changes recorded on modern NSE spectrometers vary from fractions of picoseconds to hundreds of nanoseconds, and the characteristic sizes and in the range from a few to tens of nanometers, which allows the observation of such functional conformational changes, as the motion of protein subdomains and internal diffusive motion in large macromolecular complexes.
Neutron spin-echo is based on the detection of very small (down to a few nanoelectronvolts) energy changes of neutrons interacting with the sample by measuring the Larmor precession phase difference between incident and scattered neutron spins in a magnetic field [41]. The intermediate scattering function S(q,t) measured by this method is a Fourier transform of the spatial and temporal correlation function of the scattering length density in the sample.
The ranges of the characteristic times of motion (from tens of picoseconds to hundreds of nanoseconds) and the characteristic sizes, where these motions occur (from a few to tens of nanometers), available for NSE measurements make this technique a unique direct method for the observation of the dynamics of biological objects at the molecular level. Neutron spin-echo is extensively used mainly to study the character of internal diffusive motion and intermolecular interactions in polymers and protein solutions at high concentrations [42–44]. The investigation of conformational mobility of the protein molecule by this method requires a thorough analysis of the data, the separation of the contributions of translational, rotational, and internal diffusive motion to the overall relaxation, and the interpretation of the contribution of the internal domain motion by modeling conformational transitions at different levels of complexity and reliability [17, 45–47]. Since the first use of NSE for the description of internal dynamics of the immunoglobulin molecule [48], this method has been applied to characterize the functional motion of domains in phosphoglycerate kinase [49], alcohol dehydrogenase [50], mercury reductase [51], Taq polymerase [52], the NHERF1 protein [53], and human lactoferrin [54]. It should also be noted that this method can be used to investigate changes in the physicochemical and structural properties of proteins under macromolecular crowding conditions [55] and the properties of lipid structures upon interaction with polypeptide molecules [56].
The further development of NSE instrumental facilities aimed at increasing the sensitivity due to the employment of high-flux neutron sources and extending the time scale may not only allow the determination of the spatial and temporal characteristics of conformational mobility of small individual enzymes but also will provide data on the functional movements in large protein oligomers and nucleoprotein complexes. The characteristic relaxation times in such systems can be as long as tens and hundreds of nanoseconds. In modern spectrometers, these times are achieved using superconducting magnets, providing the precessing magnetic field integral of up to 1.5 T m, and cold neutron sources that produce a sufficient flux at wavelengths of 15–25 Å. Thus, the upgraded J-NSE spectrometer at the FRM-2 reactor (Munich, Germany) has the upper limit of measured relaxation times of 500 ns at a neutron wavelength of 16 Å [57]; the spectrometer under development at the NIST Center for Neutron Research (Maryland USA), up to 700 ns (20 Å [58]); the upgraded IN-15 spectrometer (Grenoble, France), of up to 1000 ns (25 Å [59]). This method can potentially be used to study functional ribosomal particles, chromatin-remodeling complexes, protein–DNA systems, in particular, DNA recombination and repair complexes, membrane proteins, and proteins interacting with cell membranes.
5 MOLECULAR MODELING AND SAS
As mentioned above, SANS and SAXS can be successfully used in structural biology in combination with molecular modeling methods. In some cases, the combined interpretation of the data obtained by molecular modeling and SAS makes it possible to generate all-atoms models of biomacromolecular complexes [60–63]. The inverse problem of SAS can be solved using molecular modeling as a complementary method, which allows the discrimination of some structures, i.e., the exclusion from consideration of the structures, the spectra of which are identical to the experimental spectra but which have no physical and biological sense [61, 64, 65]. On the other hand, the solution of the direct problem of SAS provides the verification of the structures determined by molecular modeling [61]. The comparison of the data obtained by these two methods requires the solution of the direct problem: the calculation of SAXS or SANS spectra from the structure determined by molecular modeling. The three approaches considered below are most often applied to solve the direct problem.
5.1 Debye Equation
The oldest calculation method, which uses the Debye equation to solve the direct problem, was described back in 1915 [66]. A simplified version of the complete equation, which describes the scattering cross-sections on the object taking into account the averaging over all orientations, is employed in this approach. The spectrum can be generated by calculating pair-distribution functions, which is a rather time-consuming procedure. As a consequence, the Debye equation can be used to describe the scattering from molecules in solution only provided that there are predominant orientations. Another drawback of this method is that it is impossible to correctly take into account the solvent effect on the scattering spectrum, i.e., the method does not allow the modeling of contrast variation. Nevertheless, the Debye equation is currently widely employed to simulate SAXS and SANS spectra for the structures determined by molecular modeling [60, 67].
5.2 Spherical Harmonic Decomposition
This method takes into account the scattering contributions from the solvent and hydration shells of proteins and nucleic acids in aqueous solutions [68, 69]. The method is based on the assumption that biomacromolecules have a uniform scattering length density regardless of the type of the molecule (protein, nucleic acid, or lipid). The SAXS or SANS spectrum can be calculated from the molecular shape and the hydration-shell size using a model of the molecule with a hydration shell, which is constructed in terms of spherical harmonic functions. Generally, this description does not require a large number of spherical harmonics and, consequently, the calculations are not time-consuming. This method has some drawbacks. In particular, it is suitable for calculations of the scattering spectra of globular proteins; however, in the case of elongated structures, such as filaments, or flattened structures, this method can give rise to artifacts.
5.3 Direct Calculations of SANS and SAXS Spectra from the Scattering Length Density
In this method, the SAXS or SANS spectra are generated by calculating the scattering length density function for the overall system. It directly accounts for the effects associated with the contrast in SANS and the effects associated with the hydration shells of the molecules and solvent-density fluctuations in the vicinity of the macromolecule. A drawback of this method is that it is time-consuming, particularly, in calculations of the scattering length density. As applied to calculations of SANS and SAXS spectra, it is more versatile compared to the above two methods. In particular, depending on the system under study, either the presence of the specified orientation of the molecules can be taken into account or the averaging over all orientations can be performed.
All the above methods are applicable when using the static structure of the molecule, derived by modeling, as the starting data for the simulation of SANS and SAXS spectra. However, a significant part of biological systems studied in aqueous media are dynamical. As a consequence, in many cases none of the structures of biomacromolecules, which represent an individual static conformation and serve as the starting models in these methods, does not give a satisfactory fit of the simulated spectra to the experimental SAXS or SANS spectra, because the scattering occurs on an ensemble of molecules adopting similar but not identical conformations. In these cases, it is necessary to use data obtained by molecular dynamics simulation in aqueous solutions to take into account the conformational mobility of the system. For a series of systems, the consideration of the internal dynamics is a critical factor in calculations of the SANS and SAXS spectra. Examples of such systems are RecA protein filaments [61, 70], multidomain proteins, and multicomponent complexes [27].
In some cases, when simulating the SANS spectra of multicomponent systems containing glycosylated proteins, the dynamics of hydrogen–deuterium exchange during the contrast variation experiment should be taken into account.
It is worth noting that in the past years, a number of methods were developed, in which the experimental SAXS or SANS spectra are directly taken into account in molecular dynamics simulations. For this purpose, a special term, which takes into account the correlation between the experimental SAS spectrum and the spectrum generated by molecular dynamics simulations, is introduced into the energy functions [62, 63, 71, 72]. Despite the fact that this approach potentially allows the structure determination at atomic resolution, it considers the only structure in calculations, while biomacromolecules in solution are rather labile and the only structure is not representative.
5.4 Analysis of NSE Results
The NSE method enables direct measurements of the parameters related to the dynamics of macromolecular systems. The main difficulty of the application of this technique is the interpretation of the results. For this purpose, both simplified dynamic-system models and the results of all-atom molecular dynamics simulations can be used and the whole range of methods of molecular dynamics trajectory analysis can be employed. Both approaches require the solution of the direct problem: the molecular dynamics simulation of NSE data. The generalized description of the motion of the components of the system can serve as the simplified model in studies of large-scale motions, which is often used to describe non-biological polymers and elongated filaments (Rose model, reptation model, [73]). Simplified methods are employed also for the interpretation of NSE by means of all-atom molecular dynamics simulations. For example, the normal mode decomposition [45] or the principal component decomposition of the system dynamics [52, 74, 75] are employed. In this case, rather long molecular dynamics trajectories can be used for the interpretation of NSE. Alternatively, NSE spectra can be directly calculated from molecular dynamics trajectories. This may be performed either by employing the Debye equation or by direct calculations of the spectra from the scattering length density [76].
6 UNIQUE APPLICATIONS
6.1 Supramolecular Complexes
Small-angle scattering methods hold promise for studying the properties of supramolecular complexes in solution [77]. A characteristic feature of most of these complexes is that they exist only in solution in dynamic equilibrium and, as a consequence, they undergo dissociation when using such methods as analytical gel filtration, electrophoretic separation, etc. In view of such facts as the interaction with the substrate and local changes in the concentration, resulting in the structural changes during sample preparation for atomic force and electron microscopy, the molecular weight insufficient for cryo-EM (lower than 100 kDa), a large number of identical elements, interfering with the interpretation of NMR signals, and the interaction energy much lower than the crystal lattice energy, the SAS and dynamic light scattering methods are unique tools that can be applied to study the structural features of such systems [78]. It should be emphasized that only SAS methods provide structural data at sufficient resolution and, in combination with molecular modeling techniques, allow the construction of all-atom models of these systems [79]. The knowledge of supramolecular systems is required for both investigation of the pathogenesis of a number of diseases [80] and the development of new drugs [81–83]. It is worth noting that SAS, unlike many other methods, can be used to perform kinetic measurements and probe the real-time structural evolution of biomacromolecular complexes [84] on short time scales by means of SAXS and on large time scales by SANS due to the difference in the count rate because of the difference in the scattered beam intensity. The use of a high-flux neutron reactor [85] will make it possible to improve the time scales for SANS.
Investigation of large systems with sizes of tens and hundreds of nanometers, in particular, oligomeric complexes of proteins and nucleic acids, isolated cell nuclei, and amyloid-like fibrils, is an important and unique field of application of SAS to study the structure and dynamics of biomacromolecular complexes.
6.2 Influenza Virus Nucleoprotein
A combination of SANS methods and molecular dynamics simulations can be used to study large-scale structural changes of biomacromolecular complexes associated with minimal changes in the primary structure, for example, as single amino-acid substitutions in proteins. An example of this application is the study of the effect of a single-point mutation in influenza virus nucleoprotein on the appearance of new functional properties. In the oligomeric state (about 100 monomers), influenza virus nucleoprotein encapsidates the viral RNA, and the resulting ribonucleoprotein (RNP) complex is arranged in space as a long helical filament about 100 nm in length [86]. It was demonstrated that the E292G mutation is associated with the appearance of the cold-adapted properties of the influenza virus strain (i.e., the influenza virus acquires the ability to replicate at low temperature) [87]. The molecular dynamics simulation of the wild-type and mutant RNPs and the trajectory analysis demonstrated that the most significant differences in their three-dimensional structures are observed for the distances between the chains of the nucleoprotein complexes. These differences are up to a few Angströms. Small-angle neutron scattering studies of supramolecular structures of isolated RNPs at low and ambient temperatures showed that the structures of the complexes at high temperature differ from the model of the rod-like particles and are differently affected by changes in the temperature. A comparison of the data obtained by molecular modeling and SANS demonstrated that the mechanism of cold adaptation of the strain bearing the E292G substitution is related to a weakening of the interaction between the RNP chains and, as a consequence, the lability of linkages between the chains required for the functioning of the complex at low temperature [13].
6.3 Chromatin
Chromatin is an example of a more complex structural organization of biological macromolecules. Chromatin is a nucleoprotein complex bearing cellular DNA. It is a hierarchical structure containing nucleosome as the constitutive basic element, which consists of ~ 150 bp of DNA wrapped around an heterooctamer of histone proteins. The structure of nucleosomes was determined at atomic resolution by X-ray crystallography [88], and their biological function was extensively studied in the past thirty years by molecular biology and biophysical methods. Much less is known about higher levels of chromatin organization, which are still debated. The regular structures of components of the chromatin, isolated from moderate-ionic-strength solutions (30-nm fibrils), were determined for the first time by electron microscopy [89]. However, as opposed to the nucleosome, the existence of these structures in vivo was not established, and all observations can be interpreted using a model an irregular folding of the 10-nm chromatin fiber [90–92].
Since chromatin elements are involved in such essential cellular processes as the transcription, replication, recombination, and repair of the genome, the knowledge of the structural and dynamic organization of chromatin in eukaryotic cell nuclei under physiological conditions is required for a detailed understanding of all these processes at the molecular level.
In recent years, increasing evidence for the fractal model of chromatin architecture at the supranucleosomal level has been reported [93]. Let us mention the study concerned with models of DNA folding [94]. In terms of polymer theory, it was demonstrated that DNA can be folded into a structure with fractal properties. The in vivo study of the diffusion of fluorescent probes in rat kidney cell nuclei and mouse fibroblasts also supported the hypothesis of the fractal chromatin organization [95]. The data on the diffusion of green fluorescent protein oligomers obtained by fluorescence correlation spectroscopy and the direct observation of the movement of a single quantum dot and local photoactivation of chromatin-interacting proteins were interpreted in terms of anomalous diffusion theory, which describes the behavior of particles in a fractal environment. The fractal dimensions calculated by this method turned out to be different for euchromatin (2.6) and heterochromatin (2.2). The use of the experimental conformation capture assay (the Hi-C method) in human lymphoblastoid cell nuclei [96] resulted in the creation of a model of chromatin architecture as a fractal globule [97, 98], which predicts the fractal dimension of 3, exhibits self-similar behavior, and has territorial organization, which are not observed in the random folding of the polymer chain. Despite a wide recognition of this model, it is worth noting that some high-resolution experimental data obtained, in particular, by the Hi-C technique require further refinement of this model [99, 100].
Therefore, on the one hand, systematic and quantitative comparative studies of the fractal organization of DNA folding in chromatin of animal cells, in particular human cells, have been extensively performed; however, on the other hand, the available studies are just the beginning of a long line of research on the structure of native chromatin. As can be seen in the review [101], a lot remains unclear in terms of polymer physics.
Non-destructive techniques in reciprocal space (in pulse space), which are based on the analysis of Fourier-transform images of fractal objects obtained by light, X-ray, and neutron scattering techniques [102, 103] are the most informative and precise methods for investigation of fractal structures. The choice of the type of irradiation depends on both the size of the objects and the nature of the material under study. The best results are obtained by the complementary use of different types of radiation (e.g., X-ray and neutron radiation), which significantly expands and completes the informative value of the data. Small-angle scattering experiments on fractal structures showed the power dependence of the scattering intensity on the transmitted pulse I(q) ≈ q–n (n ≤ 6) in a certain range of transmitted pulses q > 1/R, where R is the characteristic scale of the scattering system. The fractal structure of the system can be evaluated from the value of n, more precisely from its deviation from Porod’s law (n = 4) [103]. For the volume and mass fractals, n coincides with the fractal dimension DV; 1 < DV < 3. For the scattering from three-dimensional objects with fractal surfaces, 3 < n ≤ 4; n = 6 – DS, where DS is the fractal dimension of the surface, 2 < DS < 3. Therefore, the SAS method not only allows the direct measurement of the fractal dimension of the object but also can be appliedused to determine whether its fractal properties are due to the distribution of inhomogeneities within the object or due to the shape of its surface.
Currently, SANS seems to be the most powerful tool to determine the fractal properties of native chromatin. Modern SANS spectrometers allow researchers to measure the data in a wide range of characteristic sizes, from nucleosome sizes up to 1 µm and larger. The upper limit of observed inhomogeneity sizes achieved with unique ultra-small-angle scattering instruments [40] is up to 10 µm and larger, i.e., it reaches the cell nucleus size. Additional advantages of the SANS method are the possibility of the investigation of the chromatin structure under nearly native conditions and the ease of application of the contrast variation tecnhique, which, as mentioned above, makes it possible to separate the scattering contributions from the protein and nucleic-acid components of chromatin. It should be noted that this method requires the accurate selection of the conditions for the sample preparation, which ensure the maintenance of chromatin integrity and provide long-term measurements required for recording the spectra in a wide range of transmitted pulse amplitudes. Recent studies demonstrated that the exposure of chromatin to high shear forces may have a significant additional effect on the large-scale chromatin structure, resulting in the formation of degenerate logarithmic-fractal structures of chromatin [104]. The conditions of choice for the preparation and purification of cell nuclei are such, under which the mechanical effects on chromatin are reduced, combined with mild conditions of the fixation of chromatin with glutaraldehyde at a concentration of up to 0.5%. These conditions ensure long-term measurements with the maintenance of the supranucleosomal chromatin structure [26, 105].
In 2005, the study of the chromatin organization in chicken erythrocyte nuclei by small-angle and ultra-small-angle scattering methods combined with the contrast variation technique [24] showed that both the protein structure of chromatin and the nucleic-acid architecture exhibit the mass-fractal properties, the nucleic-acid component having two levels of organization or two phases with different fractal dimensions and the crossover point in the region corresponding to the sizes of scattering inhomogeneities of about 300–400 nm. More recently, SANS and SAXS methods were used for the direct observation of the fractal structure of rat lymphocyte nuclei [105], the rat glioma cell line (C6), the human adenocarcinoma cell line (HeLa), and drosophila embryonic cells [106, 107]. In total, the SAS data allow the conclusion that the fractal structure of chromatin with two modes of chromatin organization, including the small-scale organization as a mass fractal and the large-scale organization (the chromosomal-scale region) as a fractal globule or a surface fractal, is a universal chromatin architecture in eukaryotic cells. It should be noted that the observed fractal dimensions and crossover points in different types of cells may be significantly different depending on the genome size, the degree of genomic rearrangements, and transcriptional activity. The chromatin organization in cells containing inactive chromatin can be described as a fractal globule on a large scale, whereas active chromatin, particularly in tumor cell lines, behaves as a surface fractal with a dimension of 2.2–2.4 [106, 108].
Mathematical models of the chromatin organization in the cell nucleus can be used to relate the integral characteristics of the density distribution of chromatin to elements of supranucleosomal structure observed by other methods. One of such models based on the randomized algorithm of the construction of the Koch curve was employed to obtain a system of nucleosomes within a given volume with a given fractal dimension or two fractal dimensions (structures with a crossover point) [109]. In order to validate the agreement between the model and the experimental small-angle scattering data, a method was developed for the calculation of SANS spectra [97], and the fractal model of the supranucleosomal chromatin structure of the genomic size, which has two fractal dimensions and describes the two-level fractal chromatin organization, was analyzed [109]. Although the model rather well describes the available experimental data on a scale larger than the nucleosome size and, in general, reflects the properties of the fractal globule, it has a limited application because the chromatin dynamics cannot be adequately taken into account both at the chromosome and nucleosome levels. Currently, it is possible to more adequately simulate the spatial genome organization based on contact frequencies mapped by the Hi-C method [110], which opens new possibilities for the interpretation of the experimental results using SANS.
The elucidation of the functional role of the structural and dynamic characteristics of chromatin is an important issue in studies of the chromatin organization. The integral properties of the chromatin organization as a polymer chain are of great importance for an understanding of the mechanism of interaction between distant chromatin sites, the looping, and, finally the formation and dynamics of topologically associated chromatin domains: genome regions associated in space and having similar characteristics of transcriptional activity [111, 112]. The self-organization and evolution of these domains are mediated by biochemical (histone post-translational modification, nucleosome dynamics, interaction of chromatin with specific nuclear proteins) [113, 114] and physicochemical (charge–charge interactions, macromolecular crowding) [115, 116] factors and, under particular conditions, are apparently also induced by a number of non-nuclear proteins and protein complexes [108].
Small-angle scattering methods hold promise for both the elucidation of the mechanisms of action of different factors on the epigenetic regulation in cells and the investigations of structural changes in chromatin and their relationship with the changes in the expression profile in cell differentiation, malignant transformation, and programmed cell death.
7 FUTURE BIOLOGICAL RESEARCH AT THE PIK REACTOR AT THE NRC “KURCHATOV INSTITUTE”–PNPI
Apart from the obvious advantages of neutron scattering methods used to investigate the structure and dynamics of macromolecular complexes, the most essential drawback is a worldwide shortage of neutron sources, resulting in their heavy workload. As a consequence, research groups have to complete for the beam time, and certain proposals highly rated by experts are rejected. The large-scale user research center “PIK neutron research facility” at the NRC “Kurchatov Institute”–PNPI (Gatchina) is the unique experimental complex, which will be included in experimental facilities in the near future. The Russian scientific community pins particular hope on the installation of new planned experimental beamilnes. They include SAS facilities similar in parameters to the state-of-the-art instruments (such as D22 and D33, ILL, Grenoble) and a neutron spin-echo spectrometer with superconducting magnets, which will provide longer characteristic times (about 2 µs at 25 Å) and, therefore, will become the best in the world, outperforming the J-NSE PHOENIX spectrometers installed at the FRM-2 reactor (Heinz Maier-Leibnitz Zentrum, Germany) [57] and the IN15 spectrometer (ILL, Grenoble). This will open a possibility to study the structural dynamics of domain motion in protein structures and the large-scale conformational motion of protein and nucleoprotein complexes. The design of such hi-tech facilities will be the unique breakthrough in the development of neutron scattering methods for biological studies in the Russian Federation and will expand the range of applications in fundamental molecular biophysics, modern medicine, pharmaceutics, and biotechnology.
CONCLUSIONS
The small-angle scattering method can be used to solve a wide range of problems, investigation of the structural organization of macromolecules and biomacromolecular complexes to the determination of their dynamic characteristics.
Small-angle scattering methods have a relatively low spatial resolution and, in the general case, it is impossible to solve ab initio the inverse problem of the structure determination from the SAS data. Hence, the development of approaches based on neutron scattering and their application in combination with various methods of structural biology, including X-ray crystallography, cryo-electron microscopy, nuclear magnetic resonance spectroscopy, atomic force and fluorescence microscopy, and molecular modeling, is of great importance for the successful use of neutron scattering techniques.
REFERENCES
KWS-3. Very Small Angle Scattering Diffractometer with Focusing Mirror. https://mlz-garching.de/kws-3
M. T. Rekveldt, Nucl. Instrum. Methods Phys. Res. B 114 (3–4), 366 (1996). https://doi.org/10.1016/0168-583X(96)00213-3
S. E. Bresler, Introduction to Molecular Biology (Nauka, Moscow, 1966) [in Russian].
J. C. Kendrew, G. Bodo, H. M. Dintzis, et al., Nature 181 (4610), 662 (1958). https://doi.org/10.1038/181662a0
J. D. Watson and F. H. C. Crick, Cold Spring Harb. Symp. Quantum Biol. 18, 123 (1953). https://doi.org/10.1101/SQB.1953.018.01.020
M. Saunders, A. Wishnia, and J. G. Kirkwood, J. Am. Chem. Soc. 79 (12), 3289 (1957). https://doi.org/10.1021/ja01569a083
S. E. Luria and T. F. Anderson, Proc. Natl. Acad. Sci. 28 (4), 127 (1942). https://doi.org/10.1073/pnas.28.4.127
G. Binnig, C. F. Quate, and C. Gerber, Phys. Rev. Lett. 56 (9), 930 (1986). https://doi.org/10.1103/PhysRevLett.56.930
B. J. Berne and R. Pécora, Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics (Wiley, New York, 1976).
R. Schneider, A. Mayer, W. Schmatz, et al., J. Mol. Biol. 41 (2), 231 (1969). https://doi.org/10.1016/0022-2836(69)90388-X
H. D. T. Mertens and D. I. Svergun, J. Struct. Biol. 172 (1), 128 (2010). https://doi.org/10.1016/j.jsb.2010.06.012
V. V. Egorov, A. A. Shaldzhyan, A. N. Gorshkov, et al., J. Surf. Invest. X-ray, Synchrotron Neutron Tech. 10 (2), 322 (2016). https://doi.org/10.1134/S1027451016020063
A. V. Shvetsov, D. V. Lebedev, Y. A. Zabrodskaya, et al., J. Biomol. Struct. Dyn. (2020). https://doi.org/10.1080/07391102.2020.1776636
V. V. Egorov, Y. A. Zabrodskaya, D. V. Lebedev, and A. N. Gorshkov, J. Phys. Conf. Ser. 848 (1), 012022 (2017). https://doi.org/10.1088/1742-6596/848/1/012022
A. V. Shvetsov, Y. A. Zabrodskaya, P. A. Nekrasov, and V. V. Egorov, J. Biomol. Struct. Dyn. 36 (10), 2794 (2018). https://doi.org/10.1080/07391102.2017.1367329
R. Henderson and G. McMullan, Microscopy 62 (1), 43 (2013). https://doi.org/10.1093/jmicro/dfs094
R. Biehl and D. Richter, J. Phys. Condens. Matter 26 (50), 503103 (2014). https://doi.org/10.1088/0953-8984/26/50/503103
N. Fischer, A. L. Konevega, W. Wintermeyer, et al., Nature 466 (7304), 329 (2010). https://doi.org/10.1038/nature09206
A. Shvetsov, E. Pichkur, T. Shtam, et al., Int. J. Biomed. 9 (Suppl. 1), S32 (2019). https://doi.org/10.21103/IJBM.9.Suppl_1.P36
B. Jacrot, Rep. Prog. Phys. 39 (10), 911 (1976). https://doi.org/10.1088/0034-4885/39/10/001
I. N. Serdyuk, G. Zaccai, and A. S. Spirin, FEBS Lett. 94 (2), 349 (1978). https://doi.org/10.1016/0014-5793(78)80974-0
I. N. Serdyuk, A. K. Grenader, and G. Zaccaï, J. Mol. Biol. 135 (3), 691 (1979). https://doi.org/10.1016/0022-2836(79)90172-4
M. S. Capel, D. M. Engelman, B. R. Freeborn, et al., Science 238 (4832), 1403 (1987). https://doi.org/10.1126/science.3317832
I. Serdyuk, V. Baranov, T. Tsalkova, et al., Biochimie 74 (4), 299 (1992). https://doi.org/10.1016/0300-9084(92)90107-P
P. Julián, A. L. Konevega, S. H. W. Scheres, et al., Proc. Natl. Acad. Sci. 105 (44), 16924 (2008). https://doi.org/10.1073/pnas.0809587105
D. V. Lebedev, M. V. Filatov, A. I. Kuklin, et al., FEBS Lett. 579 (6), 1465 (2005). https://doi.org/10.1016/j.febslet.2005.01.052
A. E. Schmidt, A. V. Shvetsov, A. I. Kuklin, et al., Crystallogr. Rep. 61 (1), 149 (2016). https://doi.org/10.1134/S1063774516010223
A. G. Fowler, A. M. Foote, M. F. Moody, et al., J. Biochem. Biophys. Methods 7 (4), 317 (1983). https://doi.org/10.1016/0165-022X(83)90057-X
N. M. Kirby and N. P. Cowieson, Curr. Opin. Struct. Biol. 28 (1), 41 (2014).
C. E. Blanchet, A. Spilotros, F. Schwemmer, et al., J. Appl. Crystallogr. 48 (2), 431 (2015). https://doi.org/10.1016/j.sbi.2014.07.007
T. Narayanan, M. Sztucki, P. Van Vaerenbergh, et al., J. Appl. Crystallogr. 51 (6), 1511 (2018). https://doi.org/10.1107/S1600576718012748
G. H. Golub and C. Reinsch, Singular Value Decomposition and Least Squares Solutions. Handbook for Automatic Computation (Springer, Berlin, 1971), p. 134. https://doi.org/10.1007/978-3-642-86940-2_10
Y. Chen, J. M. Tokuda, T. Topping, et al., Nucleic Acids Res. 42 (13), 8767 (2014). https://doi.org/10.1093/nar/gku562
R. Stegmann, E. Manakova, M. Rößle, et al., J. Struct. Biol. 121 (1), 30 (1998). https://doi.org/10.1006/jsbi.1997.3938
Z. Ibrahim, A. Martel, M. Moulin, et al., Sci. Rep. 7 (1), 40948 (2017). https://doi.org/10.1038/srep40948
R. Aramayo, C. Mérigoux, E. Larquet, et al., Biochim. Biophys. Acta: Gen. Subj. 1724 (3), 345 (2005). https://doi.org/10.1016/j.bbagen.2005.05.020
S. Maric, T. K. Lind, M. R. Raida, et al., Sci. Rep. 9 (1), 7591 (2019). https://doi.org/10.1038/s41598-019-43713-6
S. Mahabir, D. Small, M. Li, et al., Biochim. Biophys. Acta: Biomembr. 1828 (3), 1025 (2013). https://doi.org/10.1016/j.bbamem.2012.11.002
D22: Large Dynamic Range Small-Angle Diffractometer. ILL Neutrons for Society: Instrument Layout. https://www.ill.eu/users/instruments/instruments-list/d22/description/instrument-layout/ (accessed: 28.05.2020).
V. Pipich and Z. Fu, J. Large-Scale Res. Facil. JLSRF 1, A31 (2015). https://doi.org/10.17815/jlsrf-1-28
F. Mezei, Z. Phys. A: Hadron. Nucl. 255 (2), 146 (1972). https://doi.org/10.1007/BF01394523
S. Longeville, W. Doster, and G. Kali, Chem. Phys. 292 (2–3), 413 (2003). https://doi.org/10.1016/S0301-0104(03)00292-1
L. Porcar, P. Falus, W.-R. Chen, et al., J. Phys. Chem. Lett. Am. Chem. Soc. 1 (1), 126 (2010). https://doi.org/10.1021/jz900127c
E. J. Yearley, P. D. Godfrin, T. Perevozchikova, et al., Biophys. J. 106 (8), 1763 (2014). https://doi.org/10.1016/j.bpj.2014.02.036
N. Smolin, R. Biehl, G. R. Kneller, et al., Biophys. J. 102 (5), 1108 (2012). https://doi.org/10.1016/j.bpj.2012.01.002
D. J. Callaway and Z. Bu, Curr. Opin. Struct. Biol. 42, 1 (2017). https://doi.org/10.1016/j.sbi.2016.10.001
D. J. E. Callaway, B. Farago, and Z. Bu, Eur. Phys. J. E 36 (7), 76 (2013). https://doi.org/10.1140/epje/i2013-13076-1
Y. Alpert, L. Cser, B. Faragó, et al., Biopolymers 24 (9), 1769 (1985). https://doi.org/10.1002/bip.360240908
R. Inoue, R. Biehl, T. Rosenkranz, et al., Biophys. J. 99 (7), 2309 (2010). https://doi.org/10.1016/j.bpj.2010.08.017
R. Biehl, B. Hoffmann, M. Monkenbusch, et al., Phys. Rev. Lett. 101 (13), 138102 (2008). https://doi.org/10.1103/PhysRevLett.101.138102
L. Hong, M. A. Sharp, S. Poblete, et al., Biophys. J. 107 (2), 393 (2014). https://doi.org/10.1016/j.bpj.2014.06.013
Z. Bu, R. Biehl, M. Monkenbusch, et al., Proc. Natl. Acad. Sci. 102 (49), 17646 (2005). https://doi.org/10.1073/pnas.0503388102
B. Farago, J. Li, G. Cornilescu, et al., Biophys. J. 99 (10), 3473 (2010). https://doi.org/10.1016/j.bpj.2010.09.058
C. Sill, R. Biehl, B. Hoffmann, et al., BMC Biophys. 9 (1), 7 (2016). https://doi.org/10.1186/s13628-016-0032-3
K. Ciepluch, A. Radulescu, I. Hoffmann, et al., Bioconjug. Chem. 29 (6), 1950 (2018). https://doi.org/10.1021/acs.bioconjchem.8b00203
C. Ricci, M. Maccarini, P. Falus, et al., J. Phys. Chem. B 123 (3), 631 (2019). https://doi.org/10.1021/acs.jpcb.8b11719
S. Pasini, O. Holderer, T. Kozielewski, et al., Rev. Sci. Instrum. 90 (4), 043107 (2019). https://doi.org/10.1063/1.5084303
A New Neutron Spin Echo Spectrometer on NG-A (NIST). https://www.nist.gov/ncnr/installation-upgraded-cold-source/new-neutron-spin-echo-spectrometer-ng
IN15 Spin-Echo Spectrometer with Time-Of-Flight Option and Focusing Option. ILL Neutrons for Society: Characteristics. https://www.ill.eu/users/instruments/instruments-list/in15/characteristics
A. V. Shvetsov, A. E. Shmidt, D. V. Lebedev, and V. V. Isaev-Ivanov, J. Surf. Investig. X-ray, Synchrotron Neutron Tech. 7 (6), 1124 (2013). https://doi.org/10.1134/S1027451013060372
A. V. Shvetsov, D. V. Lebedev, D. B. Chervyakova, et al., FEBS Lett. 588 (6), 948 (2014). https://doi.org/10.1016/j.febslet.2014.01.053
A. Björling, S. Niebling, M. Marcellini, et al., J. Chem. Theory Comput. 11 (2), 780 (2015). https://doi.org/10.1021/ct5009735
S. Niebling, A. Björling, and S. Westenhoff, J. Appl. Crystallogr. 47 (4), 1190 (2014). https://doi.org/10.1107/S1600576714009959
C. Olsson and J. Swenson, Mol. Phys. 117 (22), 3408 (2019). https://doi.org/10.1080/00268976.2019.1640400
E. V. Shtykova, E. N. Bogacheva, L. A. Dadinova, et al., Crystallogr. Rep. 62 (6), 894 (2017). https://doi.org/10.1134/S1063774517060220
P. Debye, Ann. Phys. 351 (6), 809 (1915). https://doi.org/10.1002/andp.19153510606
P. Chen and J. S. Hub, Biophys. J. 107 (2), 435 (2014). https://doi.org/10.1016/j.bpj.2014.06.006
D. I. Svergun, S. Richard, M. H. J. Koch, et al., Proc. Natl. Acad. Sci. 95 (5), 2267 (1998). https://doi.org/10.1073/pnas.95.5.2267
F. Merzel and J. C. Smith, Acta Crystallogr. D 58 (2), 242 (2002). https://doi.org/10.1107/S0907444901019576
Y. Garmay, A. Shvetsov, D. Karelov, et al., J. Phys. Conf. Ser. 340, 012094 (2012). https://doi.org/10.1088/1742-6596/340/1/012094
D. Kimanius, I. Pettersson, G. Schluckebier, et al., J. Chem. Theory Comput. Am. Chem. Soc. 11 (7), 3491 (2015). https://doi.org/10.1021/acs.jctc.5b00299
P. Chen and J. S. Hub, Biophys. J. 108 (10), 2573 (2015). https://doi.org/10.1016/j.bpj.2015.03.062
D. Richter, Hyperfine Interact. 106 (1–4), 3 (1997). https://doi.org/10.1023/A:1012624631098
J. Lal, P. Fouquet, M. Maccarini, and L. Makowski, J. Mol. Biol. 397 (2), 423 (2010). https://doi.org/10.1016/j.jmb.2010.01.029
R. Biehl, M. Monkenbusch, and D. Richter, Soft Matter 7 (4), 1299 (2011). https://doi.org/10.1039/C0SM00683A
B. Lindner and J. C. Smith, Comput. Phys. Commun. 183 (7), 1491 (2012). https://doi.org/10.1016/j.cpc.2012.02.010
J. Y. Rho, H. Cox, E. D. H. Mansfield, et al., Nat. Commun. 10 (1), 4708 (2019). https://doi.org/10.1038/s41467-019-12586-8
B. Vestergaard, Arch. Biochem. Biophys. 602, 69 (2016). https://doi.org/10.1016/j.abb.2016.02.029
P. W. J. M. Frederix, I. Patmanidis, and S. J. Marrink, Chem. Soc. Rev. 47 (10), 3470 (2018). https://doi.org/10.1039/C8CS00040A
M. Wolff, B. Zhang-Haagen, C. Decker, et al., Sci. Rep. 7 (1), 2493 (2017). https://doi.org/10.1038/s41598-017-02370-3
Y. A. Zabrodskaya, A. V. Shvetsov, V. B. Tsvetkov, and V. V. Egorov, J. Biomol. Struct. Dyn. 37 (12), 3041 (2019). https://doi.org/10.1080/07391102.2018.1507837
R. Gallardo, M. Ramakers, F. De Smet, et al., Science 354 (6313), aah4949 (2016). https://doi.org/10.1126/science.aah4949
Y. A. Zabrodskaya, D. V. Lebedev, M. A. Egorova, et al., Biophys. Chem. 234, 16 (2018). https://doi.org/10.1016/j.bpc.2018.01.001
A. Sauter, F. Zhang, N. K. Szekely, et al., J. Phys. Chem. B 120 (24), 5564 (2016). https://doi.org/10.1021/acs.jpcb.6b03559
PIK Reactor. http://www.pnpi.nrcki.ru/ustanovki/reaktor-pik
R. Arranz, R. Coloma, F. J. Chichón, et al., Science 338 (6114), 1634 (2012). https://doi.org/10.1126/science.1228172
L. M. Tsybalova, N. E. Gorev, M. V. Potapchuk, et al., Vopr. Virusol. 57 (6), 13 (2012).
K. Luger, A. W. Mäder, R. K. Richmond, et al., Nature 389 (6648), 251 (1997). https://doi.org/10.1038/38444
J. T. Finch and A. Klug, Proc. Natl. Acad. Sci. 73 (6), 1897 (1976). https://doi.org/10.1073/pnas.73.6.1897
E. Fussner, R. W. Ching, and D. P. Bazett-Jones, Trends Biochem. Sci. 36 (1), 1 (2011). https://doi.org/10.1016/j.tibs.2010.09.002
K. van Holde and J. Zlatanova, J. Biol. Chem. 270 (15), 8373 (1995). https://doi.org/10.1074/jbc.270.15.8373
D. J. Tremethick, Cell 128 (4), 651 (2007). https://doi.org/10.1016/j.cell.2007.02.008
A. Bancaud, C. Lavelle, S. Huet, and J. Ellenberg, Nucleic Acids Res. 40 (18), 8783 (2012). https://doi.org/10.1093/nar/gks586
A. Y. Grosberg, S. K. Nechaev, and E. I. Shakhnovich, J. Phys. 49 (12), 2095 (1988). https://doi.org/10.1051/jphys:0198800490120209500
A. Bancaud, S. Huet, N. Daigle, et al., EMBO J. 28 (24), 3785 (2009). https://doi.org/10.1038/emboj.2009.340
E. Lieberman-Aiden, N. L. van Berkum, L. Williams, et al., Science 326 (5950), 289 (2009). https://doi.org/10.1126/science.1181369
A. Grosberg, Y. Rabin, S. Havlin, and A. Neer, Europhys. Lett. 23 (5), 373 (1993). https://doi.org/10.1209/0295-5075/23/5/012
L. A. Mirny, Chromosom. Res. 19 (1), 37 (2011). https://doi.org/10.1007/s10577-010-9177-0
K. Huang, V. Backman, and I. Szleifer, bioRxiv 413872 (2018). https://doi.org/10.1101/413872
A. L. Sanborn, S. S. P. Rao, S.-C. Huang, et al., Proc. Natl. Acad. Sci. 112 (47), E6456 (2015). https://doi.org/10.1073/pnas.1518552112
J. D. Halverson, J. Smrek, K. Kremer, and A. Y. Grosberg, Rep. Prog. Phys. 77 (2), 022601 (2014). https://doi.org/10.1088/0034-4885/77/2/022601
A. Guinier and G. Fournet, Small-Angle Scattering of X-Rays (Wiley, New York, 1955).
P. W. Schmidt, Modern Aspects of Small-Angle Scattering (Springer, Dordrecht, 1995), p. 1. https://doi.org/10.1007/978-94-015-8457-9_1
E. G. Yashina and S. V. Grigor’ev, JETP 129 (3), 455 (2019). https://doi.org/10.1134/s0044451019090177
D. V. Lebedev, M. V. Filatov, A. I. Kuklin, et al., Crystallogr. Rep. 53 (1), 110 (2008). https://doi.org/10.1134/S1063774508010136
D. Lebedev, M. Filatov, A. Konev, et al., FEBS J. 280 (Suppl. 1), 19 (2013). https://doi.org/10.1111/febs.12340
V. V. Isaev-Ivanov, D. V. Lebedev, Kh. Lauter, et al., Phys. Solid State 52 (5), 1063 (2010). https://doi.org/10.1134/S1063783410050379
D. V. Lebedev, Y. A. Zabrodskaya, V. Pipich, et al., Biochem. Biophys. Res. Commun. 520 (1), 136 (2019). https://doi.org/10.1016/j.bbrc.2019.09.116
A. V. Ilatovskiy, D. V. Lebedev, M. V. Filatov, et al., J. Phys. Conf. Ser. 351, 012007 (2012). https://doi.org/10.1088/1742-6596/351/1/012007
N. A. Kinney, I. V. Sharakhov, and A. V. Onufriev, Epigenet. Chromatin 11 (1), 3 (2018). https://doi.org/10.1186/s13072-018-0173-5
S. S. Roy, A. K. Mukherjee, and S. Chowdhury, Hum. Genomics 12 (1), 8 (2018). https://doi.org/10.1186/s40246-018-0140-z
D. Jost, P. Carrivain, G. Cavalli, and C. Vaillant, Nucleic Acids Res. 42 (15), 9553 (2014). https://doi.org/10.1093/nar/gku698
M. Moronta-Gines, T. R. H. Van Staveren, and K. S. Wendt, Essays Biochem. 63 (1), 167 (2019). https://doi.org/10.1042/EBC20180064
M. H. Hauer and S. M. Gasser, Genes Develop. 31 (22), 2204 (2017). https://doi.org/10.1101/gad.307702.117
S. Huet, C. Lavelle, H. Ranchon, et al., Int. Rev. Cell Mol. Biol. 307, 443 (2014). https://doi.org/10.1016/B978-0-12-800046-5.00013-8
F. Erdel and K. Rippe, Biophys. J. 114 (10), 2262 (2018). https://doi.org/10.1016/j.bpj.2018.03.011
OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Funding
The study was financially supported by the National Research Center “Kurchatov Institute” (order no. 1363 of June 25, 2019). The study of the structural and functional characteristics of ribosomal complexes was financially supported by the Russian Science Foundation (grant no. 17-14-01416, A.L.K.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by T. Safonova
The original online version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lebedev, D.V., Egorov, V.V., Shvetsov, A.V. et al. Neutron Scattering Techniques and Complementary Methods for Structural and Functional Studies of Biological Macromolecules and Large Macromolecular Complexes. Crystallogr. Rep. 66, 242–253 (2021). https://doi.org/10.1134/S1063774521020103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063774521020103